

Abstract
Biotinylation of silicon oxide surfaces, surface stability and evolution of these functionalized surfaces under bio-specific attachment of streptavidin were studied using Fourier Transform Infrared Spectroscopy. Adsorption and stability of species or changes in the resulting surfaces were monitored after each step of the attachment processes. The silicon oxide surface was initially derivatized by 3-aminopropyltriethoxysilane (APTES) and the quality of the 3-aminopropylsiloxane (APS) surface was monitored using the Si-O-Si and Si-O-C region of its vibrational spectrum. A strong correlation between surface quality and pre-silanization atmospheric moisture content was established. The vibrational fingerprint of biotinylation was determined, both for physisorption and chemisorption to the surface. A new band (i.e. not previous associated with biotin) at ~1250 cm−1 was identified as a vibrational mode of the biotin ureido group, making it possible to track changes in the biotinylated surface in the presence of streptavidin. Some of the biotin ureido at the surface was found to be affected by the protein adsorption and rinse steps while remaining chemisorbed to the surface. The stability of the APS was found to impact the behavior of the biotinylated surface (measured using the Si-O-Si/Si-O-C and ~1250 cm−1 absorption bands respectively).
Keywords: Fourier transform infrared spectroscopy, aminosilanization, biotin, biotinylation
I. INTRODUCTION
As bioassays such as Enzyme-Linked ImmunoSorbent Assays and DNA/protein microarrays are progressively scaled down to meet increasingly high throughput, small quantity and rapid scan requirements, the accuracy and sensitivity of these devices become increasingly important. If biomolecules are denatured, non-uniformly distributed or disordered at the surface, device quality at such small scales may be compromised [1–4]. The underlying surface chemistry and intermediate biolayers can significantly affect the organization and function of these biomolecules [3, 5]. Hence, control of these attachment steps to device substrates is important for device performance.
The biotin-(strept)avidin system (parentheses denote either of two analogues) is a widely used intermediate between the surface and the active biolayer. It is also employed as a model system to study biorecognition events between proteins and other biomolecules [6]. Biomolecules such as proteins and DNA can be easily biotinylated and bound to (strept)avidin coated surfaces (Scheme 1). The interaction between biotin and avidin is the most stable non-covalent biological binding couple known, with a binding affinity of 1015 M−1 for free complexes. This binding couple is highly stable under a wide range of conditions, including extreme pH, salt concentration and temperature. In a study of multiple attachment methods the biotin-streptavidin (SA) system mediated the greatest degree of homogenous binding and surface coverage of antibodies compared with other attachment methods, although antibody specific activity was not improved significantly [3].
Scheme 1.

Steps to attach biomolecules to silicon oxide surfaces. The APTES molecules are covalently bound to silicon oxide forming a 3-aminopropylsiloxane (APS) film with Si-O-Si bonds to the surface (1), biotin-NHS is covalently bound to amine-terminated surface forming amide bonds (2), SA is bio-specifically attached to biotin via the SA cleft (3), then biotinylated biomolecules can be bio-specifically attached to SA (4).
SA can be bound to a surface via biotin or covalent linkage, but attachment to biotinylated surfaces provides both increased stability and organization of the SA film at the surface [7]. A study employing both surface plasmon resonance (SPR) and quartz crystal microbalance with energy dissipation monitoring (QCM-D) demonstrated that a streptavidin layer bound to a surface via biotin contained fewer trapped water molecules and was hence more compact than a SA layer attached covalently to a surface, suggesting greater organization of the streptavidin surface bound via biotin [8].
Infrared Spectroscopy (IR) is a highly sensitive technique that yields direct information about the chemical bonds formed and broken in surface reactions. A better understanding of the underlying surface chemistry of biological assays is essential to controlling the quality and properties of assay surfaces. IR has been employed previously to characterize the chemical attachment of biotin to inorganic surfaces modified with a variety of chemical functionalities as well as to quantify the amount of SA specifically bound to such surfaces. Attempts have also been made to identify and quantify unreacted biotin derivatives physisorbed at these surface as well as SA non-specifically bound to such surfaces [9–13]. Quantification of SA at the surface was achieved through use of a metal carbonyl probe that labeled SA outside of the region of absorption by organic compounds (1400–1700 cm−1), avoiding interference [9–12].
This is the only published study that employs Fourier Transform Infrared Spectroscopy (FTIR) in transmission geometry on a planar surface to study surface biotinylation and subsequent attachment of SA. Transmission FTIR is highly sensitive at planar surfaces and is further enhanced when IR is incident on the surface at the Brewster angle, increasing transmission of the p-polarized component of infrared light. Nitrogen purging and mathematical water subtraction further enhance the clarity of vibrational modes of interest above interference and noise in this study.
Furthermore, in previous IR studies, biotinylation of surfaces has not been controlled for atmospheric moisture (critical for silane-based attachment [14, 15]), nor has physisorbed biotin derivative been completely removed from the surface in the rinse step [9–12, 16]. These factors may affect bioassay surface quality. In this study, surface aminosilanization was performed under anhydrous conditions and biotinylated surfaces were sonicated after attachment to aminosilane to remove all physisorbed biotin derivative. It is hypothesized that IR features associated with these and similar factors can be tracked at each stage of surface attachment and indicate surface quality. Various features identified at each surface modification step are correlated with each other or with initial atmospheric conditions suggesting that conditions at prior steps affect those at subsequent steps. For example, pre-silanization atmospheric moisture is taken into account as a continuous variable, not done previously for silane adsorption to surfaces.
The scope of this study is limited to the characterization of biotinylated surfaces and the effect that streptavidin attachment and subsequent rinsing procedures have on these surfaces. Hence, this study aims to characterize Steps 1–3 of Scheme 1, and in accordance with the premise that molecular underlayers affect the organization and/or function of overlayers, show a relationship between changes in the biotinylated and siloxane layers. Not noted in previous studies is the observation that surface-attached biotin may undergo changes as a result of the protein adsorption and/or rinse steps. This finding is explored quantitatively through the identification of a low frequency biotin feature to avoid interference between biotin and protein bands present in the same region of IR absorption.
II. EXPERIMENTAL METHODS
Reagents
All purchases are from Sigma-Aldrich unless otherwise noted. 3-aminopropyltriethoxysilane (APTES), Biotin 3-sulfo-N-hydroxysuccinimide ester sodium salt (biotin-NHS), biotin, Toluene, (anhydrous, 99.8%), Streptavidin from Streptomyces avidinii (SA), Dulbecco’s Phosphate Buffered Saline (PBS), Tween20®. All reagents were used as received unless otherwise noted.
Substrate
Float Zone Si(100) silicon wafers initially passivated by a layer of native oxide (~65 Angstroms) were cut to dimensions 1.5×3.8 cm.
Preparation of amine-terminated surfaces
Attachment of streptavidin to a biotinylated silicon oxide surface begins with silanization of the silicon oxide surface to prepare it for chemical modification with biotin. To prepare the oxide surface for silanization, wafer was cleaned in a solution of water:hydrogen peroxide:ammonium hydroxide (4:1:1) at 80° C for 10 minutes to remove organic contaminants (SC1). In the process, 10–15 Angstroms of silicon oxide was removed from the surface and reformed. After copious rinsing with deionized (DI) water (18 MΩ-cm), this clean is followed by a second similar clean, where the base was replaced by hydrochloric acid for the removal of metal particles from the surface, with no additional oxide formation (SC2). The wafer was then rinsed in DI water as above, and dried under a stream of nitrogen gas. All glassware was cleaned in SC1, rinsed copiously with DI water and dried under a nitrogen stream. Glassware prepared for use in the dry box was subsequently heated to 200 °C for 5–10 minutes to decrease surface water layers.
After collecting reference spectra of the oxide surface, the silicon wafers were transferred to a dry box. To silanize the oxide surface (i.e. form a 3-aminopropylsiloxane (APS) surface), the wafer was submerged in 0.1 % APTES in anhydrous toluene. Anhydrous toluene was pre-heated to 110°–120°C and APTES pipetted into the test tube, followed immediately by the wafer. After 20 or more hours (times at which the surface has reached saturation [17] ), the wafer was removed from the solution, rinsed copiously with anhydrous toluene and dried under a stream of nitrogen gas.
Biotinylation of amine-terminated surfaces
After silanization, biotin-NHS was chemically attached to the APS-terminated surface. Fresh solutions of biotin-NHS in DI water with a concentration ranging from 2.5 to 4.0 mg/ml were transferred to one side of the wafer situated on a minimally-contacting Teflon® stand, via pipette. The wafer on the stand was sealed in a humidity chamber to prevent evaporation of the NHS-biotin solution for 60 minutes. The wafer was then rinsed copiously with DI water and subsequently sonicated in DI water for ~10 to 15 minutes.
Bio-specific attachment of streptavidin to biotinylated surfaces
Bio-specific attachment of streptavidin to the biotinylated surface was performed by exposing one side of the wafer to 100 μg/ml streptavidin in phosphate buffered saline (PBS, pH 7.4) for adsorption times ranging from one to 60 minutes for various experiments. The setup to attach streptavidin to the biotinylated surface, is analogous to that for biotinylation. The wafer was subsequently rinsed about four times in a 0.05% solution of Tween20® (Polyethylene glycol sorbitan monolaurate) in PBS (PBST), one minute per rinse under constant agitation at 150–200 shakes per minute to remove non-specifically bound streptavidin from the surface, followed by an additional four rinses in PBS under the same agitation. The total rinse time was about 15 to 20 minutes. Samples were then rinsed briefly in DI water and dried under a nitrogen stream.
Data Collection
Immediately following drying by a stream of nitrogen after each adsorption step, FTIR spectra were collected on a Thermo® 760 FTIR spectrometer at a 4 cm−1 resolution for a minimum of 1000 scans (~20 min data collection).
Additional procedure for pre-heated samples
After samples were dried under a stream of nitrogen, they were placed into a clean, dried glass test tube and heated to about 200 °C for 10 to 15 minutes to reduce the amount of physisorbed water on the surfaces. The moment heating of the wafer was stopped, the wafer was cooled by blowing a stream of nitrogen gas into the test tube for one minute before immediately sealing the test tube with a screw-type cap. The wafers in the test tubes were then stored in the dry box prior to APTES adsorption.
Non-specific binding experiments
To observe non-specific binding of SA to a biotinylated surface, the active sites of SA were blocked with biotin. SA was incubated with an excess of biotin for three hours to block its active sites or “clefts”. The solution was then filtered to remove free biotin using gel chromatography, by pouring the solution into an Econo-Pac10DG desalting column (Biorad Laboratories, Hercules, California) packed with Bio-Gel® P-6DG gel, excluding solutes greater than 6000 daltons, which come out first. The first 3 ml fraction was collected at the bottom of the column containing only biotin-blocked SA. This solution was then adsorbed to the biotinylated side of a silicon wafer for 60 minutes, followed by the same rinsing protocol as for active SA (PBST/PBS/DI water) and dried under a stream of nitrogen gas.
Physisorption of biotin-NHS
To obtain a complete spectrum of unreacted biotin-NHS, biotin-NHS was dissolved in water and adsorbed onto the surface of an SC1/SC2-cleaned oxide surface of a silicon wafer. After 30–60 minutes, the liquid was removed by tilting the wafer on its side and drying it under nitrogen, leaving a film of biotin-NHS.
III. RESULTS
Step 1: Characterization of amine-derivatized silicon oxide surfaces; observation and control of silane growth and stability
Following Scheme 1, the silicon oxide surfaces are first modified with APTES. Representative FTIR spectra of silicon oxide surfaces exposed to APTES and referenced to the respective pre-treated surfaces show bands peaking near 1040 cm−1 and 1140 cm−1. These bands correspond to a combination of Si-O-Si vibrational modes including those of bonds formed between the silane and oxide surface, crosslinking between silane molecules at the surface, polymerization of silane and unreacted Si-O-C (ethoxy) groups (Figure 1) [17]. Bands in the region of 2800–2990 cm−1 (centered at 2930 cm−1) and ~1500–1700 (centered near 1580 cm−1) are assigned to CH2 stretching and NH2 bending, respectively [17, 18]. Surprisingly, the Si-O-Si band area in APS spectra (Figure 1) increases significantly from winter to summer indicating that more APS is adsorbed at the surface during the summer. The intensity of most components of the Si-O-Si band region is dependent on humidity.
Figure 1.

Representative FTIR spectra of APTES chemically attached and referenced to silicon dioxide surfaces in winter and summer.
Using the area under the Si-O-Si absorbance bands (integrated over the 900–1280 cm−1 region) as a measure of the amount of APTES adsorbed at the surface, the growth of the APS is first investigated. APS growth was monitored as a function of outdoor atmospheric moisture content prior to exposure of the silicon oxide surface to APTES in anhydrous solvent in a dry box. Outdoor humidity and temperature values are obtained from a website that collects data from a local weather center (http://weathersource.com) unless otherwise stated. These data are processed to obtain values of air moisture content using an online psychrometric calculator (WebPsycH Calculator®, Linric Company, Bedford, NH) at http://www.envirochex.com/psychro.htm. At low moisture content (< 3 g H2O/m3 air), Si-O-Si area is constant over a range of values(Figure 2). In contrast, at high moisture content (> 7 g H2O/m3 air) both Si-O-Si area and the fluctuation in this area increase with increasing moisture content.
Figure 2.

Amount of APS adsorbed to silicon oxide surface (Si-O-Si region, 900–1280 cm−1 band area) versus pre-silanization atmospheric moisture content (g H2O/m3 air, outdoor humidity reading with indoor experiment). Black squares indicate APTES adsorption in anhydrous solvent. Additional samples were pre-heated to 100–200 °C, flushed with dry nitrogen gas, then sealed from atmospheric moisture prior to exposure to APTES in anhydrous solvent (circled triangles).
To explore the ability to limit APS film growth under humid pre-silanization conditions, samples were heated prior to entry into the dry box to lessen the effects of atmospheric moisture. Two of the data points in Figure 2 (triangles) are for samples heated for 5 to 10 minutes between 100 and 200 °C and kept under nitrogen continuously through sample entry into the dry box and subsequent APTES adsorption. Although these adsorptions took place under humid outdoor conditions, the Si-O-Si absorption areas were near those at low outdoor moisture values, demonstrating that it is possible to limit the amount of silane molecules attached to the surface.
The stability of the APS layer is the second measure of surface quality at this stage of attachment. It is determined by measuring the change in Si-O-Si band area after exposure of the APS surface to biotin-NHS in deionized (DI) water for one hour, followed by 10–15 minutes of sonication in DI water. A more stable surface shows less percent change in intensity (e.g. percent loss in APS band intensity after rinsing and sonication). Figure 3 shows that the intensity of the Si-O-Si band is more stable when the moisture content (exposure of the sample to atmosphere prior to silanization in the dry box) is lower.
Figure 3.

APS Si-O-Si band area percentage loss (taken as a measure of surface instability) after APS surface was exposed to biotin-NHS in deionized water for 60 min and subsequently sonicated in deionized water for 10–15 min versus pre-silanization atmospheric moisture content (outdoor humidity reading with indoor experiment). Least squares fit; R2 =0.69.
Step 2: Biotinylation of the surface
A representative spectrum of the amine-terminated surface exposed to biotin-NHS and referenced to the pre-exposed surface is shown in Figure 4, spectrum A. Here biotin-NHS is chemically attached to the amine-terminated surface.. Bands unique to reacted biotin-NHS are present at 1540 cm−1 and ~1660 cm−1, assigned to the amide II (N-H bend + C-N stretch) and amide I (C=O stretch) vibrational modes respectively and indicate the amide bond formed between biotin-NHS and the surface [9–12, 16, 18].
Figure 4.

Representative FTIR spectra of: (A) an APS surface exposed to biotin-NHS (for covalent attachment) and referenced to the pre-exposed APS surface; (B) a silicon oxide surface exposed to biotin-NHS (for physisorption) and referenced to the pre-exposed silicon oxide surface; and (C) a sonicated biotin-NHS treated APS surface referenced to the same biotinylated surface before sonication (amplified 5x).
To separate the vibrational modes of biotin-NHS moieties reacting with the surface from those not reacting with the surface, a representative spectrum of unreacted biotin-NHS physisorbed to a silicon oxide surface is obtained (Figure 4, spectrum B, reduced 2.7x) for comparison to spectrum A. Bands unique to unreacted biotin-NHS (not present in spectrum A) are at 1741, 1788 and 1818 cm−1 assigned to the asymmetric and symmetric stretch of the succinimide carbonyls and the ester of the NHS moiety respectively [19, 20]. The NHS bands present in this spectrum are not present in the chemisorbed spectrum A, indicating loss of the NHS moiety in the reaction of biotin-NHS with the surface.
The features that are common to spectra A and B are the band complex centered near 1240 cm−1, the band at 1465 cm−1 and the band at 1700 cm−1. These bands are present in both chemisorbed and physisorbed biotin-NHS, suggesting that they correspond to modes of the non-chemically reactive part of biotin-NHS, the alkyl chain and the biotin fused rings. The band at 1465 cm−1 is assigned to the CH2 scissor mode of the alkyl chain linking the biotin fused rings to the NHS moiety [18], while the band at 1700 cm−1 is assigned to the ureido carbonyl stretch mode [16, 18, 21] of biotin-NHS. The low frequency band complex at 1240 cm−1 has not been noted in the literature previously for biotin and will be discussed in greater detail in the following results and discussion sections.
The effects of sonication after biotinylation are examined in spectrum C showing bands removed by sonication (amplified 5x for clarity). Spectrum C was generated by referencing a spectrum of the biotinylated surface before sonication (spectrum A before sonication, not shown) to the same surface after sonication (spectrum A). Spectrum C shows that bands of both chemisorbed biotin (amide bonds to surface at 1527 and 1637 cm−1) and physisorbed biotin (at 1741, 1787 and 1815 cm−1) have been removed from the surface after sonication. Since sonication cannot break chemical bonds at a surface, the origins of these bands will be examined further in the discussion section.
Bands removed by sonication that are present in both chemisorbed and physisorbed spectra (at ~1240, 1465 and 1684 cm−1 in spectrum C) are due to chemically unreactive parts of the biotin-NHS. Due to amplification of spectrum C the negative band at ~1240 cm−1 extends beyond the region shown. This negative band includes both a decrease in the biotin complex at 1240 cm−1 as well as an ~5% decrease in APS after sonication of the biotinylated surface. The percent decrease in APS is calculated from the decrease in the area of the IR region of APS (~950–1320 cm−1) after sonication compared to the total area of the APS region prior to sonication.
It is interesting to note that the positions of the negative bands in spectrum C corresponding to amide and ureido bands are ~10–20 cm−1 lower in frequency than similar bands in spectrum A (covalently attached), whereas bands associated only with physisorbed biotin-NHS are at the same position in both spectra B and C. Estimating the amount of material removed by sonication, the absorption area under the bands in spectrum C from ~1500–1800 cm−1 is calculated to be about 10% of the area under the bands in the same region for covalently bound biotin in spectrum B.
Step 3: Effect of SA attachment and rinse steps on the biotinylated surface
Upon adsorption of SA to the biotinylated surface and subsequent rinsing with PBST, bands were observed at 1530 and ~1630–1690 cm−1, indicative of protein adsorption (Figure 5a). The major band at 1640 cm−1 is due to beta-sheets, the major secondary structural component of SA [22]. In certain trials of protein adsorption, negative IR bands at 1250 and 1685 cm−1 were observed in these spectra (Figure 5). To resolve the origin of these negative bands, biotinylated surfaces were exposed to biotin-blocked SA followed by rinsing or to a PBST rinse only. The same occurrence of negative bands was observed in these trials to varying degrees and was most pronounced in rinse-only trials with no protein present (Figure 5b and c). Figure 6 shows the spectrum of the same biotinylated surface as in Figure 5c before and after exposure to the PBST rinse. A part of the biotin band at 1685 cm−1 is observed to disappear after rinsing along with part of the complex at 1240 cm−1 (decreasing in intensity at 1254 cm−1), while the aliphatic C-H scissor mode at 1459 cm−1, the amide II band at 1542 cm−1 and amide I shoulder at ~1640 cm−1 do not noticeably decrease in intensity (Figure 6). The band at ~1250 cm−1 has been cited as a vibrational mode of a cyclic urea, the ring of which the ureido carbonyl is a part [18]. Hence, these data suggest that a change in the biotin fused rings has taken place due to the rinse and/or protein adsorption steps while the biotin remains attached to the surface.
Figure 5.
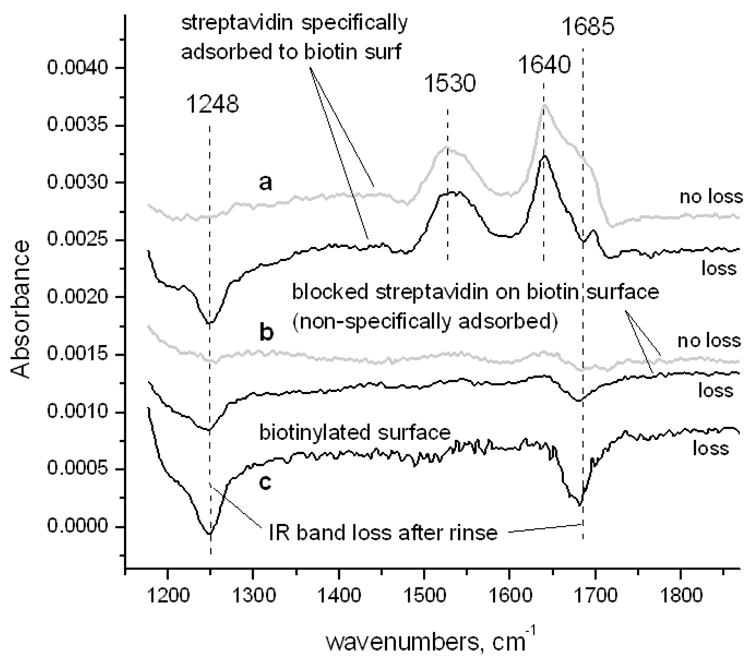
Representative FTIR spectra of: (a) biotinylated surfaces exposed to SA and referenced to pre-exposed biotinylated surfaces; (b) biotinylated surfaces exposed to biotin-blocked SA and referenced to pre-exposed biotinylated surfaces and (c) a biotinylated surface rinsed with PBST followed by PBS referenced to same surface prior to rinse. “Loss” and “no loss” indicate presence or absence of band loss in spectra of biotinylated surfaces. All samples were rinsed per methods section protocol. All biotinylated reference surfaces were sonicated prior to spectra collection.
Figure 6.

FTIR spectra of (top) an APS surface exposed to biotin-NHS, referenced to APS surface and (bottom) the same surface after rinsing with PBST and PBS, referenced to APS surface. While spectral decrease in biotin ureido is observed at 1254 and 1682 cm−1, amide and aliphatic scissor mode band areas did not decrease.
The study of biotinylated surface exposure to SA is made difficult by the fact that protein IR bands of SA in the 1400–1700 cm−1 range interfere with bands of chemisorbed biotin in the same region. This overlap prevents quantification of the change in intensity of biotin IR modes after SA adsorption and rinse steps. Only the high frequency shoulder at ~1250 cm−1 (Figure 4, spectrum A) is outside of this region and is therefore selected to track changes in the biotinylated surface upon protein attachment and rinse.
Figure 7 shows a correlation between the change in intensity in the APS Si-O-Si modes (after the biotin adsorption step) and the change in intensity of the band at ~1250 cm−1 (after SA adsorption and rinsing of the biotinylated surface). Data points for adsorption of biotin-blocked SA (open square with “X”) and surfaces subjected to the rinsing step only (circle) also fit within this correlation.
Figure 7.

Loss of biotin band absorbance area at ~1250 cm−1 after SA adsorption and PBST rinsing (or rinsing without SA adsorption) versus APS Si-O-Si band area loss after exposure of APS surface to biotin-NHS in DI water and subsequent sonication (black squares, open squares and open circle indicate SA specific adsorption, biotin-blocked SA adsorption and PBST rinse only, respectively). Least squares fit; R2 = 0.61, all points included.
IV. DISCUSSION
In this section, we consider each adsorption step sequentially and use the band assignments of all spectra shown in the results section, as displayed in Table 1.
Table 1.
IR band assignments for molecules present at each surface modification step.
| APTES and APS | |||
|---|---|---|---|
| frequency (cm−1) | assignments | references | |
| 900–1280 | Si-O-Si and Si-O-C stretching | [18, 27] | |
| 1570–1590 vb | NH2 deformation | [18] | |
| 2930 | CH2 asymmetric stretch | [18] | |
| biotin-NHS and biotinylated surface | |||
| frequency | assignments | adsorption type | references |
| 1043 | unassigned | n/a | n/a |
| 1074 | C-H deformations on NHS succinimide ring | Physi- | [18] |
| 1212 | NHS C-N-C stretching mode | Both | [28, 29] |
| 1240–1260* | Biotin ureido ring | Both | [18] |
| ~1660 | Amide I (C=O stretch) | Chemi- | [9–12, 16, 18] |
| ~1540 | Amide II (NH deformation with C-N stretch) | Chemi- | [9–12, 16, 18] |
| 1637 | Amide I (attached to physisorbed APTES) | Chemi- | [9–12, 16, 18] |
| 1527 | Amide II (attached to physisorbed APTES) | Chemi- | [9–12, 16, 18] |
| 1685–1700† | Biotin ureido carbonyl stretch | Both | [18] |
| 1741 | NHS C=O asymmetric | Physi- | [19, 20] |
| 1788 | NHS C=O symmetric | Physi- | [19, 20] |
| 1818 | NHS ester | Physi- | [19, 20] |
| Streptavidin (SA) | |||
| frequency | assignments | references | |
| ~1640 | Amide I band | [9–12, 16, 18] | |
| ~1540 | Amide II band | [9–12, 16, 18] | |
Bands for chemisorption of biotin-NHS are distinguished from those of physisorption.
Assignments not previously noted for biotin.
Bands assignments clarified. vb = very broad bands.
Step 1: Aminosilanization: Dependence of APS film growth and degradation on pre-silanization atmospheric moisture content
At the APTES stage of surface modification, we examine the surface quality in terms of growth of the Si-O-Si band area, an indicator of the amount of APTES adsorbed at the surface (Figure 2), and its percent intensity loss in the subsequent biotin attachment step (Figure 3). This section addresses the behavior of these features under a continuum of atmospheric moisture values.
Atmospheric moisture content is an important factor for the quality of aminosilanization. Although aminosilanization is performed with anhydrous solvents in a dry box, silane growth varies with exposure of the silicon oxide surface to atmospheric moisture prior to sample introduction to the dry box for silanization.
Moisture content can be divided into low and high regimes, where the growth behavior of APS films differs. Below a critical moisture of about 3 g H2O/m3 air, the Si-O-Si band area was low and remained constant with change in atmospheric moisture. This behavior suggests that film growth was self-terminating under these conditions. Above a critical moisture content of ~7 g H2O/m3 air, not only did the amount of silane at the surface increase with increasing air moisture, but the fluctuation in this amount increased as well. APS Si-O-Si absorbance area values ranged widely from small to large, in some cases even close to absorbance areas as small as those under dry conditions.
First, we examine mechanisms for high moisture conditions. As the moisture content of air increases, an increasing amount of water molecules adsorbs to the oxide surface [23]. Water can adsorb to silicon oxide when free hydroxyls are available, the maximum density of which is estimated at 5 hydroxyls/nm2 [15]. For a surface with 3 hydroxyls/nm2, the maximum density of water molecules adsorbed as a monolayer is estimated to be 2.5 to 4 water molecules/nm2. Beyond this degree of surface hydration, multilayers are believed to be present [24]. Hence, the thickness of the water layer that adsorbs to the surface is a function of both density of surface hydroxyls and moisture content of the air. Difficulty controlling these parameters in high humidity environments may explain the fluctuation in amount of APS at the surface.
In previous studies, silane film growth was shown to be dependent on atmospheric moisture, but only in terms of relative humidity and not across a continuous range of values. LeGrange et al. reported a doubling of octadecylsilane film thickness from a relative humidity of 10–30% in winter to 50–60% in summer on dehydrated silica [14]. Even though moisture was initially removed from the surface by heating, the amount that re-adsorbed to the surface from the atmosphere between the time of dehydration and the time of silane deposition affected silane growth.
In this study, the circled data points (triangles) in Figure 2 are for samples preheated for 5–10 minutes at ~200 °C then flushed with nitrogen and kept continuously in a nitrogen environment until APTES molecules are adsorbed to the surface. In contrast to the report by Legrange et al., the moisture content that the sample surface is exposed to is kept low even in the high humidity of summer. It was previously reported that pre-heating silicon oxide to a similar temperature resulted in complete removal of all surface water from silicon oxide surfaces [15]. Hence, it is probable that water at the surface of our preheated samples is minimized or eliminated as well. Since surface hydration is very difficult to control, dehydration below a critical value may be an effective method to limit the amount of aminosilanization of silicon oxide surfaces.
The amount of moisture in the air prior to APTES deposition appears to have a destabilizing effect on the APS surface. As pre-silanization moisture increases, more APS desorbs from the surface after the biotinylation and sonication steps (both in DI water) as indicated by percentage loss of Si-O-Si band intensity (Figure 3). An explanation for this behavior is based on both increased polymerization and detachment of APS when the water layer at the surface is thicker. A thicker water layer may cause increased APTES polymerization at the surface and hence covalent attachment to the surface of large polymerized silane clusters. Due to packing constraints, polymerized films are less dense than self-terminating thinner films and hence may be more prone to hydrolysis [17]. Each cluster contains many Si-O-Si bonds (not related to surface bonding) and hence the detachment of increasingly larger clusters would show increasing Si-O-Si intensity loss. Alternatively, a thicker water layer may prevent the silane from attaching covalently. Therefore, more silane and silane clusters may be physisorbed at the surface and subsequently removed in the sonication step.
In summary, these results show that APS film growth and desorption are highly sensitive to atmospheric moisture prior to silanization of the silicon oxide surface. However, this sensitivity may be dependent on the preparation of the starting oxide surface. In this study, native silicon oxide surfaces of thickness of ~65 Angstroms were used. Yet in a related study, pre-silanization humidity dependence was not observed on samples from which the native oxide was removed by hydrofluoric acid treatment and regrown by wet chemical oxidation (~10–15 Angstroms thick) [17]. Such chemically oxidized surfaces differ from the former in terms of surface roughness, oxide density and number of hydroxyls available for silane attachment, factors that may affect the thickness of the water layer at the surface during silane attachment as well as the ability of the silane to bind covalently to the substrate. Both studies have value in different applications: a native silicon oxide surface may behave more closely to glass (used in biotechnology), while the study of thinner oxides may be more applicable to modification of semiconductor surfaces (used in the microelectronics industry or in sensor applications).
Step 2: Biotinylation: Distinguishing chemisorbed from physisorbed biotin-NHS and the effects of sonication
While the assignment of bands observed at ~1540 cm−1 and ~1660 cm−1 to amide vibrations indicating covalent attachment of biotin to amine-terminated surfaces is widely accepted [9–12, 16], identification of the biotin ureido carbonyl vibrational mode has been less clear in the literature [9–12, 16]. In a number of IR reflection studies, the biotin ureido band near 1700 cm−1 was not observed [9–12]. This study, however, confirms the presence and assignment of this band by comparing physisorbed to chemisorbed forms of biotin-NHS.
Furthermore, in some cases in the literature where the amide I and II bands are small or undetected, it is possible that much of the biotin-NHS is instead physisorbed to the surface. In the spectra of various surfaces exposed to biotin-NHS, the band at ~1740 cm−1 is due to the carbonyl stretch of the unreacted NHS moiety [19, 20] and thus indicates the presence of physisorbed biotin-NHS. However, in spectra exhibited in the literature, a number of references have assigned this or nearby higher wavenumber bands to the carboxylic acid moiety of unreacted biotin [9–12, 16]. In these studies, to produce biotin-NHS, biotin was reacted with a succinimide-based compound such as N,N-diisopropylethylamine (DIPEA) or O-(Nsuccinimidyl)-N,N,N,N′,N″-tetraethyluronium (TSTU), replacing the carboxylic acid of biotin with an NHS ester. Depending on the degree to which the reaction to form biotin-NHS took place, bands observed from 1740–1770 cm−1 may instead be due to NHS carbonyl stretch vibrations or due to a combination of this vibrational mode with the carboxylic acid group.
Sonication plays an important role in removal of physisorbed biotin derivative. Figure 4, spectrum C shows negative bands corresponding to removal of both the NHS moiety and ureido carbonyl, indicating that physisorbed biotin-NHS is removed from the surface. Furthermore, the absence of NHS bands in the spectrum of the biotinylated surface (Figure 4, spectrum A) indicates that no detectable physisorbed biotin remains at that surface. In addition to these features, the intensity decrease in chemisorbed biotin features after sonication (negative amide bands in spectrum C) suggests that some of the biotin chemically bound to the surface could be removed. However, such removal is not possible with sonication. A more plausible explanation is that some of the biotin-NHS is attached covalently to physisorbed APTES or physisorbed APS film and is subsequently removed along with it. The 10–20 cm−1 red-shift of the negative amide and ureido bands in spectrum C suggests instead that the environments of these species are different from that of biotin covalently bound to a silicon oxide surface. In contrast, there is no frequency shift in the NHS bands of removed physisorbed biotin in spectrum C compared with physisorbed biotin-NHS bands in spectrum B. Hence, such shifts in vibrational modes may be a way to distinguish biotin that is chemically attached to the substrate from that chemically attached to physisorbed molecules.
Step 3: Change in the biotinylated surface upon streptavidin adsorption
Changes in the organization of the molecules of the biotinylated surface may affect the SA layer and hence the organization of the layer of functioning biomolecules that can attach to the SA layer. The presence of negative bands at ~1250 and 1685 cm−1 in the spectra of specifically bound SA, blocked SA or biotinylated surfaces rinsed only (no SA adsorption) referenced to the pre-exposed biotinylated surface suggest that a change in the biotinylated surface results from this adsorption step (Figure 5). Possible explanations for such intensity decreases include screening or chemical modification of the biotin ureido. Screening could be due to the proximity of SA to the surface and/or change in the organization or environment of the biotin molecules upon rinsing of the surface. Chemical modification may be less likely as it has only been observed previously under harsher conditions [25, 26]. Complete removal of biotin is unlikely since amide bands in the spectrum, indicative of chemical attachment of biotin to the surface, do not decrease in intensity (Figure 6).
Correlation between intensity decrease in the biotin ureido band at 1250 cm−1 during SA adsorption/rinse and decrease in intensity of APS in the preceding biotinylation step may suggest that the biotinylated surface is more susceptible to disorganization when attached to less stable APS surfaces (Figure 7). The degree to which APS is removed from the surface in the presence of water and hence the stability of its attachment to the surface is likely a reflection of the degree of organization of APS chains bound to the surface [17]. Furthermore, although APS film loss was directly dependent on air moisture prior to silanization, the lack of correlation between biotin ureido band decrease and air moisture further illustrates the complexity of the relationship between these layers.
V. CONCLUSIONS
In this study, we have shown how IR spectroscopy has made it possible to identify each step of a complex process, involving both covalent and physical attachment of molecules to the surface. We have shown that the attachment of APTES is sensitive to moisture both prior to and during anhydrous adsorption to the surface. We have further shown that changes in biotinylation of the surface are dependent on APS film stability. Stability of the APS film can be controlled to an extent by pre-heating of the silicon oxide surface and employing anhydrous adsorption conditions. Sonication is effective in removing physisorbed biotin prior to SA adsorption. We have shown that the absence of features associated with the moieties of physisorbed biotin can be used to distinguish covalent attachment of biotin derivative from derivative merely physisorbed to the surface. We have further identified a low frequency IR band of biotin that is not subject to interference from protein bands and can be effectively used to monitor the biotinylated surface during SA adsorption and rinsing. Although the mechanisms responsible for the observed molecular behavior at biotinylated surfaces are not well understood, there is a clear dependence of the properties of each layer on the chemical nature and quality of previously adsorbed layers.
As devices are scaled down and the need for improved assay sensitivity increases, finer control at the molecular level will likely be critical to device performance. This study describes a method for tracking and ultimately controlling the initial stages of bioassay surface modification.
Supplementary Material
Acknowledgments
This work was supported in part by the National Science Foundation (grant CHE-0415652) and in part by the National Institutes of Health (Kirschstein-NRSA Fellowship 5-T32-GM008339-18). The authors gratefully acknowledge Robert Pasternack and Sandrine Rivillon-Amy for their insightful input into discussion of the chemistry of APTES at the SiO2 surface, Richard Ludescher for his insights into the behavior of biotin and SA at inorganic surfaces and Meng Li for discussion on APTES attachment under low moisture conditions.
Footnotes
SUPPORTING INFORMATION AVAILABLE
To add to the discussion of several points in this manuscript, supporting information is available. The supporting document adds details for the discussion on APS, namely possible reasons for the fluctuation in APS growth. There is further discussion of the orientation of the biotin ureido and the possible implications of biotin-NHS physisorbed at the surface. Furthermore, to support the selection of the band near 1250 cm−1 as an indicator of change in the biotin ureido, additional data and arguments are presented. There is also a discussion of possible mechanisms of the chemical modification of biotin in this system. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kambhampati, editor. Kambhampati. Wiley-VCH Verlag GmbH &Co; Weinheim: 2004. Protein Microarray Technology. [Google Scholar]
- 2.Kusnezow W, Jacob A, Walijew A, Diehl F, Hoheisel JD. Antibody microarrays: An evaluation of production parameters. Proteomics. 2003;3:254–264. doi: 10.1002/pmic.200390038. [DOI] [PubMed] [Google Scholar]
- 3.Vijayendran RA, Leckband DE. A quantitative assessment of heterogeneity for surface-immobilzed proteins. Analytical Chemistry. 2001;73:471–480. doi: 10.1021/ac000523p. [DOI] [PubMed] [Google Scholar]
- 4.Walter G, Bussow K, Lueking A, Glokler J. High-throughput protein arrays: prospects for molecular diagnostics. TRENDS in Molecular Medicine. 2002;8(6):250–253. doi: 10.1016/s1471-4914(02)02352-3. [DOI] [PubMed] [Google Scholar]
- 5.Spinke J, Liley M, Guder HJ, Angermaier L, Knoll W. Molecular recognition at self-assembled monolayers: The construction of multicomponent multilayers. Langmuir. 1993;9:1821–1825. [Google Scholar]
- 6.Wilchek M, Bayer EA, Livnah O. Essentials of biorecognition: The (strept)avidin-biotin system as a model for protein-protein and protein-ligand interaction. Immunology Letters. 2006;103:27–32. doi: 10.1016/j.imlet.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 7.Smith CL, Milea JS, Nguyen GH. Immobilization of nucleic acids using biotin-strept(avidin) systems. Top Curr Chem. 2005;261:63–90. [Google Scholar]
- 8.Su X, Wu YJ, Robelek R, Knoll W. Surface plasmon resonance spectroscopy & quartz crystal microbalance study of streptavidin film struct effects on biotinylated DNA assy and target DNA hybridization. Langmuir. 2005;21:348–353. doi: 10.1021/la047997u. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Amiridis MD. FT-IRRAS spectroscopic studies of the interaction of avidin with biotinylated dendrimer surfaces. Colloids and Surfaces B: Biointerfaces. 2004;35:197–203. doi: 10.1016/j.colsurfb.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Amiridis MD. FT-IRRAS quantitative analysis of specific avidin adsorption on biotinylated Au surfaces. Surface Science. 2005;596:117–125. [Google Scholar]
- 11.Liu Z, Amiridis MD. Quantitative FT-IRRAS spectroscopic studies of the interaction of avidin with biotin on functionalized quartz surfaces. Journal of Physical Chemistry B. 2005;109:16866–16872. doi: 10.1021/jp0535240. [DOI] [PubMed] [Google Scholar]
- 12.Pradier CM, Salmain M, Liu Z, Jaouen G. Specific binding of avidin to biotin immobilised on modified gold surfaces - Fourier transform infrared reflection absorption spectroscopy analysis. Surface Science. 2002;502–503:193–202. [Google Scholar]
- 13.Ye L, Pelton R, Brook MA. Biotinylation of TiO2 nanoparticles and their conjugation with streptavidin. Langmuir. 2007;23:5630–5637. doi: 10.1021/la0626656. [DOI] [PubMed] [Google Scholar]
- 14.LeGrange JD, Markham JL. Effects of Surface Hydration on the Deposition of Silane Monolayers on Silica. Langmuir. 1993;9:1749–1753. [Google Scholar]
- 15.Zhuravlev LT. Concentration of Hydroxyl Groups on the Surface of Amorphous Silicas. Langmuir. 1987;3:316–318. [Google Scholar]
- 16.Yam CM, Pradier C, Salmain M, Marcus P, Jaouen G. Binding of Biotin to Gold Surfaces Functionalized by Self-Assembled Monolayers of Cystamine and Cysteamine: Combined FT-IRRAS and XPS Characterization. Journal of Colloid and Interface Science. 2001;235:183–189. doi: 10.1006/jcis.2000.7362. [DOI] [PubMed] [Google Scholar]
- 17.Pasternack RM, Rivillon-Amy S, Chabal YJ. Attachment of 3–(aminopropyl) triethoxysilane on silicon oxide surfaces: Dependence on solution temperature. Langmuir. 2008;24:12963–12971. doi: 10.1021/la8024827. [DOI] [PubMed] [Google Scholar]
- 18.Socrates G. Infrared and Raman Characteristic Group Frequencies - Tables and Charts. 3. John Wiley & Sons Ltd; West Sussex, England: 2001. [Google Scholar]
- 19.Frey BL, Corn RM. Covalent attachment and derivatization of poly(L-lysine) monolayers on gold surfaces as characterized by polarization-modulation FT-IR spectroscopy. Analytical Chemistry. 1996;68:3187–3193. [Google Scholar]
- 20.Voicu R, Boukherroub R, Bartzoka V, Ward T, Wojtyk JTC, Wayner DDM. Formation, Characterization, and Chemistry of Undecanoic Acid-Terminated Silicon Surfaces: Patterning and Immobilization of DNA. Langmuir. 2004;20:11713–11720. doi: 10.1021/la047886v. [DOI] [PubMed] [Google Scholar]
- 21.Hegde S, Kapoor S, Joshi S, Mukherjee T. Self-assembly of Ag nanoparticle–biotin composites into long fiberlike microstructures. Journal of Colloid and Interface Science. 2006;297:637–643. doi: 10.1016/j.jcis.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez M, Bagatolli LA, Echabe I, Arrondo JLR, Argarana CE, Cantor CR, Fidelio GD. Interaction of Biotin with Streptavidin - Thermostabilty and conformational changes upon binding. The journal of biological chemistry. 1997;272(17):11288–11294. doi: 10.1074/jbc.272.17.11288. [DOI] [PubMed] [Google Scholar]
- 23.Tripp CP, Hair ML. An Infrared Study of the Reaction of Octadecyltrichlorosilane with Silica. Langmuir. 1992;8:1120–1126. [Google Scholar]
- 24.Tripp CP, Hair ML. Direct observation of the surface bonds between self-assembled monolayers of octadecyltrichlorosilane and silica surfaces: A low-frequency IR study at the solid/liquid interface. Langmuir. 1995;11:1215–1219. [Google Scholar]
- 25.Hofmann K, Melville DB, Vigneaud Vd. Characterization of the functional groups of biotin. Journal of Biological Chemistry. 1941:141. [Google Scholar]
- 26.Ruis H, McCormick DB, Wright LD. Equilibration and acid hydrolysis of biotin sulfoxides. Journal of Organic Chemistry. 1967;32(6):2010–2012. [Google Scholar]
- 27.Pasternack RM, Rivillon-Amy S, Chabal YJ. Attachment of 3–(aminopropyl) triethoxysilane (APS) on silicon oxide surfaces: Dependence on solution temperature. Langmuir. 2007 doi: 10.1021/la8024827. Accepted, in press. [DOI] [PubMed] [Google Scholar]
- 28.McKittrick PT, Keaton JE. Infrared and Raman Group Frequencies of Cyclic Imides. Applied Spectroscopy. 1990;44:812–817. [Google Scholar]
- 29.Parker SF, Mason SM, Williams KPJ. Fourier Transform Raman and infrared spectroscopy of N-phenylmaleimide and methylene. 1990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.