

Abstract
The nonproteinogenic amino acid capreomycidine is the signature residue found in the tuberactinomycin family of antitubercular peptide antibiotics and an important element of the pharmacophore. Recombinant VioG, a single-module peptide synthetase from the viomycin gene cluster cloned from Streptomyces vinaceus (ATCC11861), specifically activates capreomycidine for incorporation into viomycin (tuberactinomycin B). Insertional disruption of the putative hydroxylase gene vioQ resulted in a mutant that accumulated tuberactinomycin O, suggesting that hydroxylation at C-5 of the capreomycidine residue is a post-assembly event. The inactivated chromosomal copy of vioQ could be complemented with a wild-type copy of the gene to restore viomycin production.
The tuberactinomycins are a small family of peptide antibiotics characterized by a 2,3-dehydroureidoalanine moiety and the arginine-derived (2S,3R)-capreomycidine (L-Cap) residue.1–3 The compounds are clinically effective antibiotics, and capreomycin is used as a second-line agent for the treatment of tuberculosis. These agents act by inhibiting bacterial protein biosynthesis and interfere with a variety of ribosomal functions. The mechanism of action of viomycin (tuberactinomycin B) has been the most extensively studied, and it has been widely used as tool to study ribosome structure and function.4–7
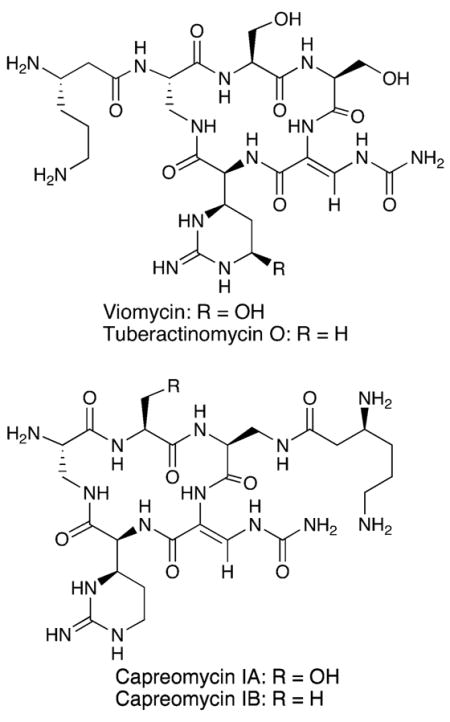
Viomycin is typical of natural product peptides assembled by nonribosomal peptide synthetases (NRPS) that commonly incorporate nonproteinogenic amino acids, such as L-Cap, into the products. The biosynthetic gene cluster for viomycin has been cloned and sequenced from Streptomyces vinaceus ATCC11861.8,9 In the first biochemical investigation of the pathway, recombinant VioC and VioD were shown to act in tandem to carry out the conversion of L-Arg to L-Cap.10–12 VioC is a non-heme iron, α-ketoglutarate-dependent dioxygenase that converts L-Arg to (3S)-hydroxy-L-Arg. This product of VioC then serves as substrate for VioD, which promotes a pyridoxal phosphate (PLP)-dependent dehydration and intramolecular cyclization to generate L-Cap. As part of our continued studies on the formation and function of the L-Cap residue in these peptides, we report here the characterization of the ATP-dependent activation of L-Cap by VioG and the VioQ-dependent hydroxylation of C-5 of the L-Cap residue in the viomycin precursor tuberactinomycin O.
Results and Discussion
VioG (see Figure 1E) is a single-module NRPS with an N-terminal adenylation (A) domain followed by a peptidyl carrier protein (PCP or T) domain, an incomplete condensation (C) domain, and an unusual C-terminal domain that exhibits significant homology only with a protein associated with an aminoglycoside cluster from S. ribosidificus.13 Analysis of the VioG A domain substrate recognition elements indicated it had a unique substrate-binding pocket that was most similar to A domains activating L-Arg or L-Orn, suggesting VioG may activate L-Cap.14–16 In order to confirm this prediction, full-length vioG and a fragment coding for the A domain were amplified by PCR from cosmid pTOV106, which contains a portion of the viomycin gene cluster.8 The fragments were then sequenced and cloned into the pET28a vector for expression as N-terminal His6-tagged proteins in E. coli. The recombinant proteins were purified by Co2+ metal affinity chromatography, and the efficiency of purification was verified by SDS-PAGE (Figure 1A,C). The production levels of VioG and VioG A domain were 2.4 and 8 mg/L, respectively.
Figure 1.

Expression, purification, and substrate specifity of recombinant VioG A domain and full-length VioG. (A) Expression and purification of VioG A domain. Lane 1, soluble protein; lane 2, flow-through; lane 3, purified His6-VioG A domain; lane 4, MW markers. (B) ATP–PPi exchange assay of VioG A domain. Activity is relative to that observed for L-Cap, which was normalized to 100%. (C) Expression and purification of VioG. Lane 1, total protein; lane 2, soluble protein; lane 3, flow through; lanes 4–6, washes; lane 7, MW marker; lane 8, purified His6-VioG. (D) ATP–PPi exchange assay of recombinant VioG. (E) Domain organization of VioG. (F) Structures of basic amino acids evaluated as substrates in B and D.
The substrate specificity of VioG was analyzed by the standard amino acid-dependent ATP–PPi exchange assay.17 In addition to L-Cap, other substrates evaluated included L-Ser and 2,3-diamino-propionate (L-Dap), the other amino acids found in viomycin. We also tested the basic amino acids L-Arg, L-Orn, and D,L-enduracididine (D,L-End) as possible substrates. The latter amino acid and its β-hydroxy derivative are found in the enduracidin and mannopeptimycin peptide antibiotics, respectively.18,19 The assay results confirmed the prediction that VioG is the NRPS in the viomycin pathway that activates L-Cap. Full-length VioG showed slightly greater specificity than the excised A domain (Figure 1B,D), but there was no significant difference in the specific activities of the two proteins. It was initially surprising that VioG was able to completely differentiate between L-Cap and enduracididine. Subsequent to performing these assays, we cloned and sequenced the enduracidin biosynthesis gene cluster.20 The substrate-specificity sequence for the enduracididine A domains (DAETDGSV) is markedly different from that found in VioG (DPQDVGIV) and helps explain why VioG does not activate enduracididine.14–16 To determine if D,L-End was simply not recognized by VioG or bound to the active site in an unproductive manner, we tested if 2 mM D,L-End could competitively inhibit the activation of L-Cap in the ATP–PPi exchange assay. The added D,L-End had no effect on L-Cap activation (data not shown), and the result suggests that enduracididine is not recognized by the VioG active site.
The activation of L-Cap by VioG suggests that hydroxylation of this residue in viomycin occurs either while an advanced precursor is attached to the NRPS or after the peptide core is assembled. The latter scenario seems most probable in light of the isolation of tuberactinomycin O (TubO) from some species.21 However, 5-hydroxy-L-capreomycidine is not available as a substrate for use in the A domain assays to validate this assumption. Interestingly, hydroxylation at the β-carbon of the enduracididine residues found in the mannopeptimycins occurs prior to incorporation into the peptide. The A domain responsible for the incorporation of the β-hydroxyenduracididine residues in this peptide showed high specificity for the hydroxylated amino acid in vitro.22 However, a mutant strain of the mannopeptimycin producer with a nonfunctional β-hydroxylase gene (mppO) will accept enduracididine as an alternate substrate and produces dideoxymannopeptimycins.23 These findings prompted us to investigate further the timing of the modification of L-Cap.
The only gene other than vioC in the viomycin cluster that is predicted to encode a hydroxylase is vioQ, which was speculated to introduce the hydroxyl group on the L-Cap residue.9 Sequence analysis of VioQ reveals the N-terminal region has a conserved [2Fe–2S] cluster binding domain found in bacterial Rieske non-heme iron dioxygenases typically involved in aromatic ring hydroxylation.24 To determine if VioQ is responsible for the C-5 hydroxylation of the capreomycidine residue in viomycin, we inactivated vioQ in S. vinaceus by insertional disruption via double-crossover homologous recombination. The disruption of the target gene was confirmed by Southern blot analysis (Figure 2A,B). The metabolite profile of the vioQ− mutant (SvdQ13) revealed that viomycin production was abolished and a new compound appeared with a retention time of 19.13 min (Figure 2C, trace iii) and a mass of m/z 670.33 ([M + H]+), which is 16 mass units less than viomycin and matches the molecular weight of tuberactinomycin O. To further confirm the role of VioQ, we complemented the disrupted chromosomal copy of the gene by expressing a wild-type copy of vioQ integrated into the chromosome of mutant SvdQ13. The wild-type vioQ was amplified by PCR and cloned into the integrative vector pXY152, and the resulting plasmid was introduced to the mutant strain SvdQ13 by conjugation.25 This recombinant strain, SvdQ13::pXY152-vioQ, was cultured under viomycin production conditions, and LC-MS analysis revealed the restoration of viomycin production and the disappearance of tuberactinomycin O (Figure 2C, trace iv).
Figure 2.

Disruption and complementation of vioQ and metabolite analysis. (A) Insertion of the 1 kb apramycin resistance marker aac-(3)IV into vioQ via double-crossover homologous recombination. (B) Southern blot analysis of genomic DNA digested with BglII from wild-type S. vinaceus (lane 4) and disruptants SvdQ7, 10, and 13 (lanes 1, 2, and 3, respectively). The blot was probed with DIG-labeled vioQ. (C) LC-MS metabolite analysis: (i) viomycin standard. Extracted fermentation broths of (ii) wild-type S. vinaceus, (iii) vioQ mutant strain SvdQ13; (iv) complemented mutant SvdQ13::pXY152-vioQ.
The finding that the vioQ disruption did not affect formation of the pentapeptide core of viomycin is consistent with the observed activation of L-Cap by VioG and indicates that hydroxylation of the L-Cap residue probably occurs after the amino acid is loaded onto the NRPS or the nascent peptide is released from the enzyme complex. Demonstrating that in trans complementation of vioQ results in the conversion of tuberactinomycin O to viomycin also suggests that hydroxylation is a post-assembly modification. The attachment of the β-lysine residue to the pentapeptide core is also very likely a tailoring event. Various tuberactinamines and tuberactinomycins have been isolated that exhibit all possible β-lysine and capreomycidine hydroxylation combinations, suggesting the order of these two tailoring events may be arbitrary.26
The results presented here, along with our earlier reports on the roles of VioC and VioD in the biosynthesis of L-Cap, delineate the complete route for the transformation of L-Arg into the 5-hydroxy-capreomycidine moiety of viomycin. This knowledge should help with applications to engineer the production of novel tuberactinomycins and their semisynthetic derivatives as leads for new agents to treat tuberculosis.
Experimental Section
Bacterial Strains, Plasmids, Cosmids, and Culture Conditions
Streptomyces vinaceus (ATCC11861) and E. coli S17-1 (ATCC47055) were purchased from American Type Culture Collection (ATCC). E. coli EPI300 (Epicentre) was routinely used as host for E. coli plasmids, cosmids, and E. coli–Streptomyces shuttle vectors, and E. coli Rosetta (DE3) (Novagen) was used for protein expression. Standard media and methods were used to culture Streptomyces.25 All DNA and protein manipulations in E. coli were performed following standard protocols.27 Plasmid pSET152 was obtained from Prof. Keith Chater (Norwich), plasmid pWHM860 was obtained from Prof. Bradley Moore (UC, San Diego), the pGEM-T easy cloning vector was purchased from Promega, and the pET28a expression vector was from Novagen.
DNA Isolation and Manipulation
QIAprep spin miniprep kits (Qiagen) were used to prepare plasmids and cosmids from E. coli strains. Restriction enzymes, T4 DNA ligase, and DNA polymerase were purchased from various suppliers and used following the manufacturers’ protocols. QIAprep spin miniprep and QIAquick gel extraction kits (Qiagen) were used for DNA purification. DNA sequencing was conducted at the Center for Genome Research and Biocomputing at Oregon State University.
Construction of E. coli Expression Plasmids for vioG and vioG A Domain
PCR primers were designed to amplify vioG and a fragment of vioG coding for the A domain. The forward PCR primer used to amplify both the vioG and vioG A domain fragment was vioGf (5′-CTGACATATGACCACCACGTCCCA-3′) (NdeI site is italic). The respective reverse primers were vioGr (5′-TCGCTCGAGTACTGCTTCTCCTCCGCT-3′, XhoI site is italic) and vioG-Ar (5′-GACCTCGAGTAAGATCGCCGATTCCACTGG-3′, XhoI site is italic). Cosmid pTOV106 contains a portion of the viomycin biosynthesis gene cluster and was used as template for the PCR.8 Reactions were carried out in a total volume of 50 μL containing 100 ng of template, 1× buffer A (AccuPrime GC-rich polymerase system, Invitrogen), 100 pmol of each primer, and 2.5 units of DNA polymerase (Expand Long Template PCR System, Roche). Gel-purified PCR products were ligated with pGEM-T easy vector (Promega). The correct plasmid constructs pGEMT-vioG and pGEMT-vioGA were confirmed by sequencing, and the inserts were excised by digestion with NdeI and XhoI and ligated with similarly prepared pET28a vector (Novagen) for expression in E. coli as His6-tagged proteins.
Expression and Purification of VioG and VioG A Domain in E. coli
The plasmids pET28a-vioG and pET28a-vioGA were used separately to transform E. coli Rosetta (DE3) cells. Single colonies were picked to inoculate 5 mL of LB medium seed cultures containing 50 μg/mL kanamycin. After growing at 37 °C overnight, the seed cultures were used to inoculate 500 mL of 2xYT medum containing 50 μg/mL kanamycin. Cells were grown at 20 °C without induction until the A600 = 1.8–2.0, harvested by centrifugation at 2000g for 15 min at 4 °C, and washed with phosphate-buffered saline (PBS; 100 mM NaCl, 50 mM Na2HPO3 NaH2PO3, pH 8.0). The washed cell pellets were stored at −80 °C until used. Frozen cells were thawed on ice, resuspended in PBS, and lysed by sonication in a Microson ultrasonic cell disruptor. The lysate was centrifuged at 18000g for 30 min at 4 °C to remove cell debris. The supernatant was used as the cell-free extract for further purification. The His6-VioG and His6-VioG A domains were purified using BD Talon metal affinity (Co2+) resin (BD Biosciences) following the manufacturer’s protocol. Purified proteins were dialyzed twice against 4 L of dialysis buffer (50 mM Tris, 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, glycerol 10%, pH 8.0) at 4 °C overnight. Bradford assay (Bio-Rad) was used to quantify the protein concentrations. The efficiency of the purification was verified by SDS-PAGE, and purified proteins were stored at −20 °C.
ATP–[32P]PPi Exchange Assay
The assay was carried out essentially as described.17 All commercially available substrates were purchased from Sigma. The D,L-enduracididine was a gift from Wyeth Research, and (2S,3R)-capreomycidine was from previous studies carried out in our laboratory.11,28 Tetrasodium [32P]-pyrophosphate was purchased from NEN-Perkin-Elmer. Assays (100 μL) were conducted by incubating 2 mM enzyme in reaction buffer (50 mM Tris, 100 mM NaCl, 10 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine, 5 mM DTT, pH 8.0) containing 4 mM ATP, 0.5 μCi tetrasodium [32P]-pyrophosphate, and 2 mM amino acid substrate for 3 h at 30 °C. The reaction was terminated by the addition of stop mix (500 μL, 1.2% w/v activated charcoal, 0.1 M tetrasodium pyrophosphate, and 0.35 M HClO4). Free [32P]-pyrophosphate was removed by centrifugation of the sample and washing the charcoal pellet three times with wash buffer (0.1 M tetrasodium pyrophosphate and 0.35 M HClO4). The final wash solution was aspirated and 1 mL of deionized H2O was added to each tube before the level of bound radioactivity was determined by scintillation counting on a Beckman LS 6800.
Disruption of vioQ
To disrupt vioQ, a 2.8 kb fragment carrying the entire vioQ gene and flanking regions was amplified by PCR from cosmid pTOV106 using primers vioQDpf, 5′-ATACATATG CCGACGTGTCGAGTC-3′, and vioQDpr, 5′-AGCGAATTCCGGGTGTGTAAAGCGA-3′. This fragment was cloned into pGEM-T-easy vector to yield the intermediate plasmid pGEMTE-vioQ. An internal native BamHI site in vioQ was used as the site to introduce the BamHI-restricted apramycin-resistance marker (AmR) into pGEMTE-vioQ. The apramycin resistance gene was amplified by PCR from pSET152 using primers apraRf, 5′-CACGGATCCAAGCTTGGTTCATGTGCA-3′, and apraRr, 5′-ATCGGATCCAAGCTTCACGTGTTGC-3′ (BamHI sites are italic). The insert of the resulting plasmid was excised by EcoRI digestion and ligated with the EcoRI linearized vector pXY3008 to deliver the final gene disruption construct pXY300-vioQ-AmR. The disruption plasmid was introduced into S. vinaceus by conjugation, and double-crossover mutants were identified as previously described.8 Southern blot analysis to confirm incorporation of inactivated vioQ into the chromosome was conducted using digoxigenin-labeled vioQ as probe, and hybridization was revealed using a digoxigenin-DNA detection kit (Roche).
Complementation of Mutant SvdQ13
The integrative expression plasmid pXY152-vioQ was used to introduce a wild-type copy of vioQ into the chromosome of SvdQ13. The vector pXY152 is a derivative of the commonly used integrative conjugal vector pSET152,29 which was modified by introducing the strong constitutive Streptomyces promoter ermE*p in front of the multicloning site and replacing the apramycin resistance marker with a hygromycin resistance gene (Yin and Zabriskie, unpublished data). Primers vioQf (5′-ATACATATGCCGACGTGTCGAGTC-3′, NdeI site italic) and vioQr (5′-AATGAATTCTCACCGGCTTTCCTTGAAATT-3′, EcoRI site italic) were used to amplify vioQ from cosmid pTOV106.8 The amplicon was sequenced and then cloned immediately downstream of the ermE*p promoter region of pXY152 to give pXY152-vioQ. This plasmid was introduced into SvdQ13 by conjugation.30 The recombinant strain SvdQ13:: pXY152-vioQ, in which pXY152-vioQ was integrated into the chromosome of mutant SvdQ13, was selected by the phenotype of resistance to both apramycin (50 μg/mL) and hygromycin (100 μg/mL) on ISP2 agar plates. Ten randomly selected colonies showing AmR and HygR were individually cultured and analyzed for viomycin production.
Extraction of Viomycin and Tuberactinomycin O
Wild-type S. vinaceus, SvdQ13, and SvdQ::pXY152-vioQ were grown under conditions previously described for the production of capreomycin in S. capreolus.31 Cells from 500 mL of 7-day production culture were harvested by centrifugation at 2000g for 30 min. The broth was mixed with an equal volume of MeOH and the volume reduced almost to dryness by rotary evaporation. The concentrated solution was dissolved in 20 mL of deionized H2O and centrifuged at 4500g for 10 min, and the pellet was discarded. The pH of the supernatant was adjusted to 3.5 using H3PO4 and then centrifuged at 4500g for 10 min. The pellet was discarded, and the pH was adjusted to 7.5 using KOH. The sample was centrifuged at 4500g for 10 min, and the supernatant was used for further analysis.
LC/MS Analysis
Metabolite analysis was performed on a ThermoFinnigan LCQ Advantage LC/MS system (ThermoElectron), equipped with an autosampler and photodiode array detector and controlled by a PC running Xcalibur 1.3 software. Separation was done with a LUNA SCX column (5 μm, 150 × 4.6 mm, Phenomenex). Samples were passed through a 0.45 μm syringe filter, and injection volumes were 15 μL. Elution buffers were (A) 25 mM NH4OAc, (B) 500 mM NH4-OAc, and (C) acetonitrile. Gradient elution from 80% buffer (A) to 60% buffer (B) in 10 min then to 80% (B) over 10 min was carried out while buffer (C) was kept constant at 20%. Flow rate was 1.0 mL/min. The effluent was monitored at 266 nm and scanned from 200 to 350 nm with the PDA detector. Positive mode electrospray ionization with full scan mode from m/z 150 to 900 was used for MS detection.
Acknowledgments
We thank Prof. K. Chater for providing plasmid pSET152, Dr. E. Graziani for a gift of D,L-enduracididine, Dr. M. Jackson for preparing L-capreomycidine, and Prof. B. Moore for plasmid pWHM860. This work was supported by National Institutes of Health grant GM69320 (T.M.Z.).
References and Notes
- 1.Haskell TH, Fusari SA, Frohardt RP, Bartz QR. J Am Chem Soc. 1952;74:599–602. [Google Scholar]
- 2.Herr EB., Jr Antimicrob Agents Chemother. 1962:201–212. [Google Scholar]
- 3.Nagata A, Ando T, Izumi R, Sakakibara H, Take T, Hayano K, Abe J. J Antibiot. 1968;21:681–687. doi: 10.7164/antibiotics.21.681. [DOI] [PubMed] [Google Scholar]
- 4.Liou YF, Tanaka N. Biochem Biophys Res Commun. 1976;71:477–483. doi: 10.1016/0006-291x(76)90812-3. [DOI] [PubMed] [Google Scholar]
- 5.Marrero P, Cabanas MJ, Modolell J. Biochem Biophys Res Commun. 1980;97:1047–52. doi: 10.1016/0006-291x(80)91481-3. [DOI] [PubMed] [Google Scholar]
- 6.Modolell J, Vazquez D. Eur J Biochem. 1977;81:491–7. doi: 10.1111/j.1432-1033.1977.tb11974.x. [DOI] [PubMed] [Google Scholar]
- 7.Johansen SK, Maus CE, Plikaytis BB, Douthwaite S. Mol Cell. 2006;23:173–82. doi: 10.1016/j.molcel.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 8.Yin X, O’Hare T, Gould SJ, Zabriskie TM. Gene. 2003;312:215–224. doi: 10.1016/s0378-1119(03)00617-6. [DOI] [PubMed] [Google Scholar]
- 9.Thomas MG, Chan YA, Ozanick SG. Antimicrob Agents Chemother. 2003;47:2823–30. doi: 10.1128/AAC.47.9.2823-2830.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin X, Zabriskie TM. ChemBioChem. 2004;5:1274–1277. doi: 10.1002/cbic.200400082. [DOI] [PubMed] [Google Scholar]
- 11.Yin X, McPhail KL, Kim K-j, Zabriskie TM. ChemBioChem. 2004;5:1278–1281. doi: 10.1002/cbic.200400187. [DOI] [PubMed] [Google Scholar]
- 12.Ju J, Ozanick SG, Shen B, Thomas MG. ChemBioChem. 2004;5:1281–1285. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- 13.Aboshanab K, Schmidt-Beissner H, Wehmeier U, Piepersberg W, Welzel K, Vente A. Direct Submission to GenBank (CAG34050) 2006. [Google Scholar]
- 14.Stachelhaus T, Mootz HD, Marahiel MA. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 15.Challis GL, Ravel J, Townsend CA. Chem Biol. 2000;7:211–24. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 16.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Nucleic Acids Res. 2005;33:5799–808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stachelhaus T, Mootz HD, Bergendahl V, Marahiel MA. J Biol Chem. 1998;273:22773–22781. doi: 10.1074/jbc.273.35.22773. [DOI] [PubMed] [Google Scholar]
- 18.Horii S, Kameda Y. J Antibiot. 1968;21:665–7. [PubMed] [Google Scholar]
- 19.He H, Williamson RT, Shen B, Graziani EI, Yang HY, Sakya SM, Petersen PJ, Carter GT. J Am Chem Soc. 2002;124:9729–36. doi: 10.1021/ja020257s. [DOI] [PubMed] [Google Scholar]
- 20.Yin X, Zabriskie TM. Microbiology. 2006;152:2969–83. doi: 10.1099/mic.0.29043-0. [DOI] [PubMed] [Google Scholar]
- 21.Izumi R, Noda T, Ando T, Take T, Nagata A. J Antibiot. 1972;25:201–7. [PubMed] [Google Scholar]
- 22.Magarvey NA, Haltli B, He M, Greenstein M, Hucul JA. Antimicrob Agents Chemother. 2006;50:2167–77. doi: 10.1128/AAC.01545-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haltli B, Tan Y, Magarvey NA, Wagenaar M, Yin X, Greenstein M, Hucul JA, Zabriskie TM. Chem Biol. 2005;12:1163–8. doi: 10.1016/j.chembiol.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. Nucleic Acids Res. 2005;33:D192–6. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich: 2000. p. 613. [Google Scholar]
- 26.Morse BK, Brown MS, Gagne JW, McArthur HA, McCormick EL, Murphy TK, Narrol MH, Perry DA, Smogowicz AA, Wax RG, Wong JW. J Antibiot. 1997;50:698–700. doi: 10.7164/antibiotics.50.698. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J, Russel DW. Molecular Cloning. A Laboratory Manual. 3. Cold Spring Harbor Laboratory; New York: 2001. [Google Scholar]
- 28.Jackson MD, Gould SJ, Zabriskie TM. J Org Chem. 2002:2934–2941. doi: 10.1021/jo016182c. [DOI] [PubMed] [Google Scholar]
- 29.Bierman MR, Logan R, O’Brien K, Seno ET, Rao RN, Shoner BE. Gene. 1992;116:43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 30.Mazodier P, Peter R, Thompson C. J Bacteriol. 1989;171:3583–3585. doi: 10.1128/jb.171.6.3583-3585.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gould SJ, Minott DA. J Org Chem. 1992;57:5214–5217. [Google Scholar]