

Abstract
The protein targets and sites of modification by 4-hydroxy-2(E)-nonenal (HNE) in human monocytic THP-1 cells after exogenous exposure to HNE were examined using a multi-pronged proteomic approach involving electrophoretic, immunoblotting and mass spectrometric methods. Immunoblot analysis using monoclonal anti-HNE antibodies showed several proteins as targets of HNE adduction. Pretreatment of THP-1 cells with ascorbic acid resulted in reduced levels of HNE-protein adducts. Biotinylation of Michael-type HNE adducts using an aldehyde-reactive hydroxylamine-functionalized probe (aldehyde-reactive probe, ARP) and subsequent enrichment facilitated the identification and site-specific assignment of the modifications by LC-MS/MS analysis. Sixteen proteins were unequivocally identified as targets of HNE adduction and eighteen sites of HNE modification at Cys and His residues were assigned. HNE exposure of THP-1 cells resulted in the modification of proteins involved in cytoskeleton organization and regulation, proteins associated with stress responses and enzymes of the glycolytic and other metabolic pathways. This study yielded the first evidence of site-specific adduction of HNE to Cys-295 in tubulin α-1B chain, Cys-351 and Cys-499 in α-actinin-4, Cys-328 in vimentin, Cys-369 in D-3-phosphoglycerate dehydrogenase and His-246 in aldolase A.
Keywords: THP-1, HNE, ascorbic acid, aldehyde-reactive probe, HNE-protein adducts, tandem mass spectrometry
Introduction
Reactive oxygen species initiate lipid peroxidation resulting in the formation of lipid peroxides which further decompose to reactive intermediates, such as aldehydic end products. 4-Hydroxy-2-nonenal (HNE) is a major lipid peroxidation product generated by oxidation of lipids containing polyunsaturated ω-6 fatty acids, such as linoleic and arachidonic acid (1, 2). HNE has been linked to the cytotoxic effects of oxidative stress by mechanisms involving adduct formation with DNA (3) and proteins, including protein constituents of the cytoskeleton (4, 5), signaling pathways (6, 7), protein folding (8) and degradation machinery (9), and the mitochondrial electron transport chain complexes (10). As an α,β-unsaturated hydroxyalkenal, HNE reacts predominately with nucleophilic sites in proteins yielding Michael-type adducts. In some cases, a Schiff base and pyrrole formation with Lys residues has been reported (11). Intramolecular or intermolecular protein cross-links occur when HNE forms a Michael adduct involving the electrophilic carbon at position 3 and a Schiff base with the ε-amino group of a Lys residue (11). Elevated levels of HNE, as observed under conditions of oxidative stress, have been associated with the impairment of protein function, initiation of cell death pathways, disruption of cellular, organelle and organ functions (12, 13). However, more recently, it has also been recognized that HNE and related α,β-unsaturated aldehydes seem to promote cell proliferation, induction of antioxidant genes and cellular stress responses that reflect the importance of electrophiles in modulating diverse cell signaling pathways (7, 14).
Because the modification of proteins by HNE (and other α,β-unsaturated aldehydic lipid peroxidation products) has been linked to the etiology and progression of various diseases (12, 13) as well as degenerative processes associated with aging (15), an extensive repertoire of analytical methods has been described to detect HNE-modified proteins in complex biological matrices. Routinely, protein carbonyls are measured by derivatization of the carbonyl groups with 2,4-dinitrophenylhydrazine (DNPH) and the DNP-modified protein carbonyls are subsequently detected by immunochemical methods, such as ELISAs or Western blots. These methods provide the total protein carbonyl content, but lack specificity for distinct modifications by lipid peroxidation products or other oxidative processes that lead to the introduction of aldehyde/keto groups into proteins (10, 16). A hallmark for HNE-related research was the introduction of anti-HNE antibodies. These antibodies show only minor reactivity with other protein-bound aldehydes, but are not necessarily specific for the site of adduction (8, 10, 17, 18). Consequently, anti-HNE immunostaining protocols based on gel-electrophoretic methods and tandem mass spectrometry provide putative identifications of protein targets of modification by HNE, but site-specific assignment of the HNE adduction is rarely possible with gel-based approaches.
More recently, mass spectrometry-based proteomics approaches have become available that allow the unequivocal identification of protein targets of modifications and the residue-specific determination of the site of adduction by reactive lipid peroxidation products (19-24). Because of the low abundance of oxidatively modified proteins in complex biological samples, enrichment protocols have been developed that make these distinct sub-groups of proteins amenable to detailed mass spectrometric analyses. Some analytical strategies employ hydrazine-functionalized beads for the selective capture and release of HNE-modified peptides (23, 24) and others are based on chemical tagging of the carbonyl group by aldehyde-specific probes that enable biotin-avidin affinity enrichment or selective chromatographic separations (19-22). An alternative approach uses HNE analogs which retain the electrophilic and hydrophobic properties of HNE and have a reactivity group allowing for the subsequent derivatization with a biotinylation reagent via click chemistry (25). Our group has explored the use of hydrazine-functionalized isotope-coded affinity probes (HICATs) (21) and a biotinylated hydroxylamine derivative, N′-aminooxymethylcarbonylhydrazino D-biotin (aldehyde-reactive probe, ARP) for chemical tagging, enrichment and tandem mass spectrometric (MS/MS) characterization of carbonyl-modified proteins (19).
In the present study, we applied the ARP labeling strategy to identify protein targets of HNE in the human monocytic cell line THP-1, a cell line which has properties similar to human monocyte-derived macrophages and is widely used for studying mechanisms of foam cell formation associated with atherosclerosis (26). Our previous studies have demonstrated that exposure of THP-1 cells to HNE resulted in apoptosis, necrosis, and protein carbonylation. THP-1 cells have the ability to accumulate ascorbic acid (Asc) in millimolar concentrations and Asc pretreatment of these cells enhanced the efflux of GSH-HNE phase I metabolites and lessened the formation of protein carbonyls detected by ELISA using an anti-DNPH antibody (27). In order to gain a deeper understanding of the mechanisms that link the protein modifications caused by HNE to the observed cytotoxic effects of HNE exposure and prevention by Asc pretreatment in human THP-1 cells, we combined gel-based approaches, anti-HNE immunostaining and a targeted mass spectrometry-based proteomics strategy to unequivocally identify the protein targets and sites of adduction of HNE in THP-1 cells.
Experimental Section
Materials
Fetal bovine serum, phenol red-free RPMI 1640, penicillin, streptomycin and trypsin-EDTA were purchased from Invitrogen (Carlsbad, CA, USA). HNE (10 mg/mL in ethanol) was obtained from Cayman Chemical (Ann Arbor, MI). Monoclonal anti-HNE IgG and monoclonal anti-β actin IgG were purchased from Oxis International, Inc. (Foster City, CA) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA), respectively. Goat anti-mouse IgG-HRP conjugate and SuperSignal West Pico Chemiluminescent Substrate were obtained from Pierce Biotechnology, Inc. (Rockford, IL). Aldehyde-reactive probe (ARP, N-aminooxymethylcarbonylhydrazino-D-biotin) was purchased from Dojindo Laboratories (Kumamoto, Japan). UltraLink-immobilized monomeric avidin was obtained from Pierce (Rockford, IL).
Cell culture and treatment
THP-1 cells (American Type Culture Collection (Manassas, VA) were routinely cultured in 75 cm2 flasks in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin and 0.05 mM 2-mercaptoethanol. The cells were incubated in humidified atmosphere of 5% carbon dioxide at 37 °C.
THP-1 cells were incubated in 6-well plates (2 mL/well) with or without 1 mM Asc for 18 h in phenol red-free RPMI 1640 medium with 10% FBS and 50 μM 2-mercaptoethanol. After an 18-h incubation at 37 °C in 5% CO2, the cells were centrifuged at 500 × g. The cells were washed with PBS and then resuspended in HBSS, 2 mL/well (3 × 106 cells/mL). HNE (or ethanol for control wells) was added to the wells to a final concentration of 100 μM. After 3 h of incubation with HNE, the cells were pelleted by centrifugation. The cell pellet was resuspended in 250 μl of PBS, lysed by sonication and frozen at -80 °C prior to analysis by Western blotting and mass spectrometry.
SDS-PAGE, Western blotting and putative assignment of HNE-modified proteins by mass spectrometry
The protein samples (20 μg), mixed with Laemmli sample buffer (Bio-Rad cat. no. 161-0737) and heated at 95 °C for 5 min, were loaded onto a 10-well 12% Tris-HCl gel (Bio-Rad Cat. no. 161-1102). After 1-dimensional SDS-PAGE (1-DE), proteins were transferred to a nitrocellulose membrane at 0.3 A for 2 h. Membranes were blocked with 5% non-fat milk in TBS containing 0.1% (v/v) Tween 20 (TBS-T) at room temperature for 2 h. For detection of protein-HNE adducts, the membranes were incubated with anti-HNE antibody for 1 h. After washing the blots 6 times in TBS-T for 5 min each, the membranes were incubated with 1:3000 diluted goat anti-mouse HRP-conjugated IgG in 40 mL TBS-T for 1 h. Subsequently, the blots were washed 6 times in TBS-T for 5 min each and then incubated in SuperSignal West Pico Chemiluminescent substrate (Pierce). Protein bands were visualized by exposing the membranes to X-ray film.
For identification, an identical duplicate was stained with Bio-Safe Coomassie-staining solution (Bio-Rad, 161-0787) for 2 h at room temperature. The protein bands that were found to correspond to those in the Western blot to be immunoreactive with anti-HNE antibody were selected, excised, digested with trypsin (28) and the resulting peptides were separated using a nano-LC system (Dionex Corporation, Sunnyvale, CA) using a PepMap C18 column (75 μm i.d. × 150 mm, 3 μm 100 Å, Dionex), spotted onto a MALDI target plate and subjected to MALDI tandem mass spectrometry as described in detail below.
Two-dimensional electrophoresis (2-DE) and Western blotting
2-D electrophoresis followed by Western blotting was performed as described by Chung et. al. (5) to aid in the identification of HNE-modified proteins in THP-1 cells. Proteins (100 μg), dissolved in rehydration buffer [7 M urea, 2 M thiourea, 2% CHAPS, 0.2% (v/v) biolytes, 100 mM dithiothreitol (DTT), and 0.001% bromophenol blue], were applied to a Ready Strip IPG (pH 3–10). After covering with mineral oil, the strips were loaded in a Protean IEF isoelectric focusing cell (BioRad, Hercules, CA) and rehydrated overnight at 50 V. Isoelectric focusing (IEF) was performed at 20 °C by conditioning the strips at 250 V for 15 min and then ramped to 8000 V over 2.5 h and held at 35,000 V·h. The strips were removed and stored at −80 °C. Prior to SDS-PAGE, the strips were equilibrated for 10 min in 375 mM Tris–HCl (pH 8.8) containing 6 M urea, 2% (w/v) sodium dodecyl sulfate, 20% (v/v) glycerol, and 2% DTT. The strips were then equilibrated for 10 min in the same buffer containing 2.5% iodoacetamide instead of DTT. Subsequently, the strips were mounted on Gradient criterion Tris–HCl gels (8-16%, Bio-Rad) to perform the second dimension electrophoresis at 130 V for 115 min. The separated proteins were then transferred to a nitrocellulose membrane before probing with anti-HNE IgG. The membrane was incubated with goat anti-mouse IgG-HRP conjugate and then incubated with SuperSignal West Pico Chemiluminescent Substrate for detection of HNE-positive spots on X-ray film.
To identify the HNE-positive spots in the Western blot, a second dimension electrophoresis gel was performed in parallel. After electrophoresis, the gel was placed in a gel fixative (10% methanol, 7% acetic acid) for 1 h at room temperature. The gel was then rinsed with MilliQ water and stained with Bio-Safe Coomassie-staining solution for 2 h at room temperature. Protein spots corresponding to the HNE-positive bands on the Western blot were excised, digested with trypsin and the extracted peptides analyzed by nanoLC-MALDI-MS/MS (29).
Identification of HNE-modified proteins in THP-1 cells following ARP labeling and tandem mass spectrometry
THP-1 cell proteins were labeled with ARP and enriched as described by Chavez et al. (19). Briefly, THP1 cells (1 mg protein) were lysed by incubation with 1% Triton X-100 in 50 mM sodium phosphate buffer, pH 7.4, for 10 minutes. The lysate was then incubated with 5 mM ARP in 50 mM sodium phosphate buffer (pH 7.4) for 1 h at room temperature and excess ARP was removed by buffer exchange using Millipore BioMax centrifugal filters (10 kDa MWCO). The protein mixture was digested overnight with a 1:50 ratio of trypsin at 37 °C in 100 mM ammonium bicarbonate (pH 8.0). Peptides were then filtered (Amicon Microcon centrifugal filters, 10 kDa MWCO) to remove trypsin and membrane fragments. Peptides containing the ARP label were enriched using a 100 μL monomeric avidin affinity column (Ultralink monomeric avidin, Thermo). After removing non-labeled peptides with extensive washing, ARP-labeled peptides were eluted from the affinity column with 0.4% formic acid in 30% aqueous acetonitrile. The enriched ARP-labeled peptide fraction was concentrated using vacuum centrifugation before being submitted to LC-MS/MS analysis.
The ARP-labeled peptide samples were fractionated by reversed-phase (RP) liquid chromatography and spotted to a MALDI stainless steel target plate using a Dionex/LC Packings Ultimate nanoLC system coupled to a Probot™ target spotter. Peptides were trapped on a 300 μm i.d. × 1 mm PepMap™ C18 trap and washed at 30 μL/min for 3 minutes with solvent A (0.1% trifluoroacetic acid (TFA) in 5% aqueous acetonitrile). A PepMap C18 column (75 μm i.d. × 150 mm, 3 μm particles, 100 Å pore size; LC-Packings, Sunnyvale, CA) was used for the chromatographic separations. Separation was achieved by performing a 60-minute linear gradient from 10% B to 60% B (0.1% TFA in 80% aqueous acetonitrile). Flow rate was 260 nL/min. The eluate from the column was transferred to the Probot™ target spotter, where it was mixed with the MALDI matrix (2 mg/mL α-cyano-4-hydroxycinnamic acid in 50% aqueous acetonitrile acidified with 0.1% TFA) and spotted to a 144-spot MALDI target plate. Fraction collection began after a delay period of 20 minutes. The fraction collection/spotting time was set to 25 seconds.
Mass spectrometric analysis was performed using an Applied Biosystems 4700 MALDI-TOF/TOF instrument equipped with a Nd:YAG laser operating at a wavelength of 355 nm. All data were acquired in the positive reflector mode. External mass calibration was performed prior to analysis using the ABI 4700 calibration mixture consisting of the following peptides: des-Arg1-bradykinin ([M+H]+ at m/zcalc 904.4675), angiotensin 1 ([M+H]+ at m/zcalc 1296.6847, Glu1-fibrinopeptide B ([M+H]+ at m/zcalc 1570.6768), and ACTH 18-39 ([M+H]+ at m/zcalc 2465.1983). For peptide analyses, the instrument was operated in the data-dependent acquisition mode in which for each spot a survey mass spectrum (m/z 700-4000) was obtained followed by MS/MS analysis on the 10 most abundant ion signals detected. Tandem mass spectral data was acquired by accelerating the precursor ions to 8 keV and selecting them with the timed gate set to a nominal precursor mass window of 50 ppm. Collision-induced dissociation was performed with a collision energy of 1 keV and a collision gas pressure of 6 × 10-7 Torr. Fragment ions were accelerated to 14 keV before entering the reflector.
MALDI-MS/MS data were processed into peak list files using 4000 Series Explorer v3.0. Default settings were used except for the peak density for MS/MS spectra which was set to a maximum of 10 peaks per 200 Da. The minimum signal-to-noise (S/N) ratio was set to 10, with a minimum peak area of 500. A setting of 40 was used for the maximum number of fragment ion peaks per precursor.
Mascot was used to search the MS/MS data against the Swiss Prot Database v56.8 (410518 sequences; 148080998 residues) limited to human taxonomy (20401 sequences). The Mascot search settings were as follows: the digesting enzyme was set to Trypsin/P and 2 missed cleavage sites were allowed. Both precursor and fragment ion tolerances were set to 0.2 Da with the instrument type set to MALDI-TOF/TOF. Dynamic modifications included ARP-HNE (+469.2359 Da) for Cys, His, and Lys residues, and oxidation (+15.9949 Da) for Met residues. Tandem mass spectra of potential ARP-HNE modified peptides were then manually inspected for verification of the sequence and modification.
ESI-MS/MS was performed using a ThermoFinnigan LTQ-FT Ultra (Thermo Scientific, San Jose, CA) equipped with a Michrom ADVANCE electrospray ionization source (Bio Resource, Inc., Cotati, CA) and coupled to a capillary HPLC (CapLC, Waters). The ADVANCE source was operated at 1.8 kV. Peptide mixtures were separated on a C18 column (Agilent Zorbax 300SB-C18, 250 × 0.3 mm, 5 μm) using a flow rate of 4 μL/min with a binary solvent system consisting of solvent A, 0.1% formic acid in water, and solvent B, 0.1% formic acid in acetonitrile. Peptides were trapped on a Michrom ‘captrap’ and washed for 3 min using 0.1% aqueous formic acid containing 1% acetonitrile at a flow rate of 6 μL/min. After trapping, peptides were separated on the analytical column using a linear gradient starting at 10% solvent B and rising to 30 % B over 50 minutes.
The LTQ-FT mass spectrometer was operated in the data-dependent MS/MS acquisition mode. Preview FTMS option was enabled. ESI-MS acquisitions were performed in the ICR cell over the range of 400-1800 m/z. The resolving power was set to 100,000 at m/z 400. MS scans were acquired using 1 microscan with a maximum ion accumulation time of 1000 ms. Centroided MS/MS spectra were acquired in the linear ion trap on the five most abundant doubly or triply charged precursor ions. Precursor ions were selected with an isolation width of 2 Th and subjected to collision-induced dissociation (CID) in the linear ion trap using a normalized collision energy of 35% with an activation Q of 0.250 and activation time of 30 ms. MS/MS scans were performed using 3 microscans with a maximum ion accumulation time of 50 ms. MS/MS data was recorded using a dynamic exclusion period of 60s for previously analyzed precursor ions. Prior to analysis, the mass spectrometer was tuned with Glu1-fibrinopeptide B (EGVNDNEEGFFSAR) using the following tune parameters: source voltage of 1.80 kV, a capillary temperature of 180 °C, a capillary voltage of 49.00 V, and a tube lens voltage of 155 V. For FT-MS scans the automatic gain control (AGC) target value was set to 1 × 106, while an AGC setting of 30,000 was used for MS/MS scans in the ion trap. Mass calibration was performed using the Supelco MSCAL5 ProteoMass™ LTQ/FT hybrid ESI Pos. Mode Calibration Mixture (Sigma-Aldrich).
Sequence Database Searching
MALDI-MS/MS
Mascot version 2.2.04 was used to search the MALDI-MS/MS data against the Swiss Prot Database v56.8 (410518 sequences; 148080998 residues) limited to human taxonomy (20401 sequences). The Mascot search settings were as follows: the digesting enzyme was set to Trypsin/P and 2 missed cleavage sites were allowed. Both precursor and fragment ion tolerances were set to 0.2 Da with the instrument type set to MALDI-TOF/TOF. Dynamic modifications included ARP-HNE (+469.235890 Da) for Cys, His, and Lys residues, and oxidation (+15.994915 Da) for Met residues. MALDI tandem mass spectra of potential ARP-HNE modified peptides were then manually inspected for verification of the sequence and modification.
LTQ-FT MS/MS data
Thermo RAW data files were processed with Proteome Discoverer v1.0 using default parameters except for a S/N threshold setting of 10. For grouping spectra with the same measured m/z, a precursor tolerance of 10 ppm and a maximum retention time difference of 1.5 min was applied. A Mascot search of the Swiss Prot Database v56.9 (412525 sequences; 148809765 residues) was launched from Proteome Discoverer with the following search parameters: the taxonomy was limited to Homo sapiens (20401 sequences), the digesting enzyme was set to trypsin/P and two missed cleavage sites were allowed; peptide cut-off score was 10 and the protein cut-off score was 20. The instrument type was set to ESI-TRAP. The precursor ion mass tolerance was set to 10 ppm, while a fragment ion tolerance of 0.5 Da was used. Dynamic modifications included ARP-HNE (+469.235890 Da) for Cys, His, and Lys residues, and oxidation (+15.994915 Da) for Met residues. The tandem mass spectra of ARP-HNE modified peptides were visually inspected to verify (1) peptide fragment ions and (2) ions originating from the fragmentation of the ARP tag and (3) ions resulting from the loss of the ARP tag and the ARP-HNE moiety from the precursor ions. The proposed mechanisms for ARP and ARP-HNE loss fragmentations as well as proposed structures for the ARP-specific fragment ions are provided in the supporting information in Figure S26.
Mascot uses a decoy database, containing randomized sequences with the same average amino acid content as the target database (Swiss Prot, human), to obtain false positive (FP) matches. The false discovery rate (FDR) is calculated by dividing the number of FP matches by the number of peptide identifications above the identity or extensive homology threshold (p<0.05). This ratio is expressed as a percentage. The average false discovery rate of our datasets (1 MALDI-MS/MS; 2 ESI-MS/MS datasets) was 6.4% for peptide matches above the homology or identity threshold (p<0.05). However, it should be noted that not all of the identified ARP-HNE modified peptide sequences were above the significance threshold of p<0.05, and are therefore not included in the calculation of the false discovery rate. A point of caution, in targeted proteomics experiments, such as the ones described in this study, the datasets are often too small to provide an accurate determination of a false discovery rate.
Results
Western blotting and tandem mass spectrometric analysis of putative protein-HNE adducts in Asc-deficient and Asc-adequate THP-1 cells
Assignments of immunoreactive bands were made by aligning the protein bands in the Western blot with the protein bands in the parallel gel stained with Coomassie (Figure 1). Protein bands of interest were then excised, and subjected to in-gel trypsin digestion followed by nanoLC-MALDI-MS/MS analysis to identify the proteins by mass spectrometric peptide sequencing (Supplementary material Table S1). The protein assignments of the immunoreactive bands are at best tentative since in some cases, multiple proteins were detected in the excised gel piece. For example, immunoreactive bands 7 and 13 were composed of 3 proteins (protein disulfide isomerase A3, adenylyl cyclase-associated protein 1, and vimentin) and 2 proteins (carbonic anhydrase 2 and 14-3-3 protein β/α), respectively. Using the alignment procedure, other proteins in THP-1 cells recognized by anti-HNE antibody were tentatively assigned as follows: heat shock protein HSP 90-β, heat shock cognate 71 kDa protein, α-enolase, cytoplasmic 1 actin (β-actin), carbonic anhydrase 2, 14-3-3 protein β/α, A, peroxiredoxin-6, and histone H2B type 1-M.
Figure 1.
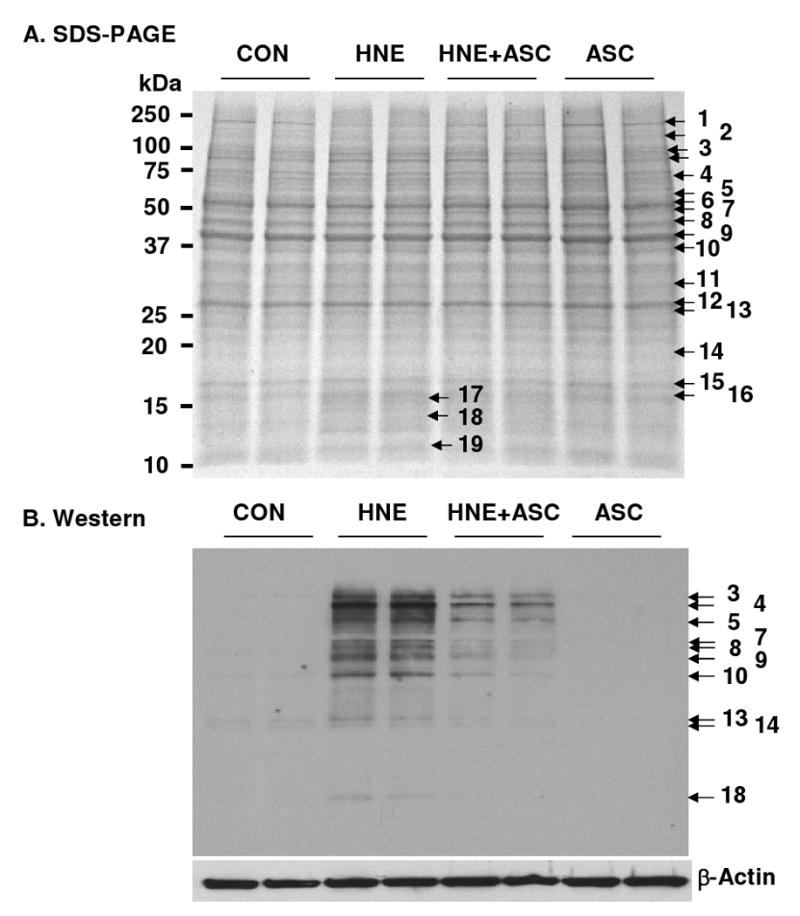
SDS-PAGE (A) and corresponding Western blot probed with monoclonal anti-HNE antibody (B) of cell lysates from THP-1 cells pretreated with 0 or 1 mM ascorbic acid for 18 h before exposure to 0 or 0.1 mM HNE for 3 h. The proteins were tentatively identified as follows: 1: Myosin-9 (MYH9-Human, MW 226,392); 2: ATP-citrate synthase (ACLY_Human, MW 120,762); 3: Heat shock protein HSP 90-β (HS90B_Human, MW 83,212); 4: Heat shock cognate 71 kDa protein (HSP7C_Human, MW 70,854); 5-1: Protein disulfide-isomerase precursor (PDIA1_Human, MW 57,081); 5-2:Pyruvate kinase isomerase M1/M2 (KPYM_Huamn, MW 57,900); 5-3: 60 kDa heat shock protein (CH60_Human, MW 61,016); 6-1: Protein disulfide isomerase A3 (PDIA3_Human, MW 56,747); 6-2: Adenylyl cyclase-associated protein 1 (CAP1_Huamn, MW 51,823); 6-3: Vimentin (VIME_Human, MW 53,619); 7: Tubulin α-1B (TBA1B_Human, MW 50,120); 8: α-Enolase (ENOA_Human, MW 47,139); 9: β-Actin (ACTB_Human, MW 41,710); 10: Fructose-bisphosphate aldolase A (ALDOA_Human, 39,395); 11: Voltage-dependent anion selective channel protein (VDAC1_Human, MW 30,754); 12-1: Carbonic anhydrase 2(CAH2_Human, MW 22,298); 12-2: 14-3-3 protein β/α (1433B_Human, MW 28,065); 13: Peroxiredoxin-6 (PRDX6_Human, MW 25,019); 14: 60S ribosomal protein L18a (RL18A_Human, MW 20,749); 15: Histone H3.1t (H31T_Human, MW 15,499); 16,17: Histone H2B type 1-M (H2B1M_Human, MW 13,981); 18: Histone H2A type 2-C (H2A2C_Human, MW 13,980); 19: Histone 4 (H4_Human, MW 11,360).
The intensity of immunoreactive bands varied according to treatment of cells. HNE-treated cells showed the darkest bands, and the intensity of these bands was reduced by Asc pretreatment of THP-1 cells. Control THP-1 cells or cells treated with Asc alone showed no or only weak staining for HNE-modified proteins (Figure 1B).
2-D Gel electrophoresis, Western blotting and MALDI tandem mass spectrometry of HNE-protein adducts
Since the SDS-PAGE did not show a clear separation of some proteins, a 2-DE separation was performed followed by Western blotting to determine the identity of the HNE-adducted proteins with higher confidence (Figure 2). Anti-HNE immunoreactive spots in the Western were aligned with corresponding Coomassie-stained spots from the 2-D gel and the respective protein spots were excised, subjected to tryptic digestion and protein identities established by nanoLC-MALDI-MS/MS analysis. The following proteins were identified: ribonuclease inhibitor; protein disulfide-isomerase A1; tubulin beta-2C; β-actin*; 14-3-3 protein β/α; keratin, type II cytoskeletal 1; coronin-1A; stress-induced-phosphoprotein 1; α-enolase*; purine nucleoside phosphorylase; and carbonic anhydrase 2*. β-actin and α-enolase were found in 4 spots and 2 spots, respectively, implying post-translational modification or different protein isoforms. Four of the 11 immunoreactive proteins were also detected in the 1-DE blot and these are marked by an asterisk (see also Table 1). However, even with the 2-DE approach, the identification of the HNE-modified proteins remains putative at best due to the known difficulties associated with gel-based approaches that are based on matching gel spots detected by a visual staining method with spots detected on an immunoblot. Visual staining methods are commonly biased toward the most abundant proteins while immunochemical detections on membranes are exquisitely sensitive and specific. The limitation of the 2D gel-based approach becomes apparent by comparing the shapes of some of the protein spots detected on the immunoblot with the supposedly corresponding spots on the 2D gel (Figure 2). Discrepancies in the shape of some of the protein spots detected by Western blotting and Coomassie-stained 2D-gel may indicate that, with the 2D-PAGE protocol used in this study, separation of the cellular protein mixture into a single protein per gel spot was not consistently achieved. Consequently, LC-MS/MS-based analyses of tryptic peptides extracted from gel spots containing multiple proteins are skewed towards identifying preferentially the most abundant protein(s). For that reason, unambiguous identification of HNE-modified proteins relies on the detection of the HNE-peptide adduct and the site-specific localization of the residue modified by the lipid aldehyde.
Figure 2.

Coomassie Blue G250-stained gel (A) after 2-dimensional electrophoresis of HNE-treated THP-1 cells and the parallel Western blot (B) probed with anti-HNE IgG. The identities of the spots are as follows: 1, Ribonuclease inhibitor (RINI_Human); 2, Protein disulfide-isomerase* (PDIA1_Human); 3, Tubulin beta-2C (TBB2C_Human); 4, β-Actin* (ACTB_Human); 5, 14-3-3 protein β/α* (1433B_Human); 6, Keratin, type II cytoskeletal 1 (K2C1_Human); 7, Coronin-1A (COR1A_Human); 8, Stress-induced-phosphoprotein 1 (STIP1_Human); 9, α-enolase (ENOA_Human)*; 10, Purine nucleoside phosphorylase (PNPH_Human); and 11, Carbonic anhydrase 2 (CAH2_Human)*. Proteins marked with asterisks (*) were also detected by anti-HNE immunostaining on the 1-D blot (Figure 1B).
Table 1.
Summary of THP-1 proteins modified by Michael-type adduction of HNE based on tandem mass spectral data (MS/MS), proteins that are putative (based on anti-HNE 2D blots) and tentative (based on anti-HNE 1D blots) targets of HNE modifications. Proteins marked by $ and #, respectively, co-migrated in the 1D GE analysis. LTQ-FT: tandem mass spectral data acquired with the linear ion trap of the ESI LTQ-FT system; TOF/TOF: tandem mass spectral data obtained with the MALDI TOF/TOF instrument. Biological and functional assignments were made on the basis of information from the NCBI (www.ncbi.nlm.nih.gov/Pubmed) and the UniProt Knowledgebase (http: http://www.uniprot.org) websites.
| Protein (Swiss Prot ID, residue modified if available) | ARP-HNE Modified Peptides (modified residue) | MS/MS | 2D | 1D |
|---|---|---|---|---|
| Proteins linked to cytoskeleton structure, organization and regulation | ||||
| α-actinin-1 (ACTN1_Human) | IC*DQWDNLGALTQK (Cys-480) | LTQ-FT | ||
| α-actinin-4 (ACTN4_Human) | IC*DQWDALGSLTHSR (Cys-499) | LTQ-FT TOF/TOF |
||
| C*QLEINFNTLQTK (Cys-351) | LTQ-FT | |||
| Adenylyl cyclase associated protein 1 (CAP1_Human)$ | ALLVTASQC*QQPAENK (Cys-93) | LTQ-FT | x | |
| β-actin (ACTB_Human) | H*QGVMoxVGMoxGQK (His-40) | LTQ-FT | x | x |
| Cofilin 1 (COF1_Human) | HELQANC*YEVKDR (Cys-139) | LTQ-FT TOF/TOF |
||
| Coronin-1A (COR1A_Human) | x | |||
| Dynein light chain Tctex-type 3 (DYLT3_Human) | H*CDEVGFNAEEAHNIVK (His-7) | LTQ-FT | ||
| F-actin capping protein (CAPZB_Human) | GC*WDSIHVVEVQEK (Cys-93) | LTQ-FT TOF/TOF |
||
| Keratin, type II cytoskeletal 1 (K2C1_Human) | x | |||
| Vimentin (VIME_Human)$ | QVQSLTC*EVDALK (Cys-328) | LTQ-FT | x | |
| Tubulin α-1B chain (TBA1B_Human) | SIQFVDWC*PTGFK (Cys-347) | LTQ-FT TOF/TOF |
x | |
| AYHEQLSVAEITNAC*FEPANQMVK (Cys295) | LTQ-FT | |||
| Tubulin β-2C (TBB2C_Human) | x | |||
| Proteins associated with stress and immune responses | ||||
| Heat shock protein HSP90β (HSB90B_Human) | x | |||
| Heat shock protein 71kDa (HSP7C_Human) | x | |||
| Peroxiredoxin-6 (PRDX6_Human) | x | |||
| Protein disulfide isomerase A1 (PDIA1_Human) | x | |||
| Protein disulfide isomerase A3 (PDIA3_Human)$ | x | |||
| Purine nucleoside phosphorylase (PNPH_Human) | x | |||
| SAM domain and HD domain binding protein (MOP-5) (SAMH1_Human) | NPIDHVSFYC*K (Cys-522) | TOF/TOF | ||
| Stress-induced-phosphoprotein 1 (STIP1_Human) | x | |||
| Proteins involved in glycolysis | ||||
| Fructose-bisphosphate aldolase A (ALDOA_Human) | FSH*EEIAMATVTALR (His-246) | LTQ-FT | ||
| α-Enolase (ENOA_HUMAN) | x | x | ||
| Proteins involved in other metabolic processes | ||||
| Carbonic anhydrase (CAH2_Human)# | x | x | ||
| D-3-phosphoglycerate dehydrogenase (SERA_Human) | NAGNC*LSPAVIVGLLK (Cys-369) | LTQ-FT | ||
| Proteins involved in (negative) regulation of translation and RNA binding | ||||
| Plasminogen activator inhibitor 1 RNA-binding protein (PAIR_Human) | PGHLQEGFGC*VVTNR (Cys-11) | LTQ-FT TOF/TOF |
||
| Ribonuclease inhibitor (RINI_Human) | x | |||
| Signal recognition particle 9 kDa protein (SRP09_Human) | VTDDLVC*LVYK (Cys-48) | LTQ-FT TOF/TOF |
||
| Others | ||||
| 14-3-3 protein β/α (1433B_Human)# | x | x | ||
| Histone H2B type 1-M (H2B1M_Human) | x | |||
| Leucine-rich repeat-containing protein 59 (LRC59_Human) | RH*EILQWVLQTDSQQ (His-294) | LTQ-FT | ||
| Voltage-dependent anion-selective channel protein 2 (VDAC2_Human) | SC*SGVEFSSGSSNTDTGK (Cys-47) | LTQ-FT TOF/TOF |
Detection and characterization of ARP-labeled peptides by LC-MS/MS analysis
Employing the ARP-labeling strategy, enrichment and LC-MS/MS analysis, 18 peptides belonging to 16 proteins were identified with ARP-HNE modifications to distinct cysteine or histidine residues (Cys, His with asterisk, Table 1). Ten peptides were detected and characterized using the LTQ-FT mass spectrometer, one peptide was identified exclusively by MALDI-TOF/TOF tandem mass spectral analysis and seven peptides were detected and sequenced by both instruments. Fourteen of the identified peptides contain a Michael-type adduct of HNE to cysteine residues while the remaining four have Michael-type adducts to histidine residues. The four peptides containing modified histidine residues were only detected by the LTQ-FT mass spectrometer. Annotated tandem mass spectra for the peptides identified with the ARP-HNE modification are available in the supporting information (Figures S1-S25).
Identification of post-translationally modified peptides in targeted proteomics approach remains a challenging undertaking. Peptides containing PTMs often have complex or atypical fragmentation patterns, and although traditional proteomics search engines, such as Mascot, include the ability to search for PTMs, they often struggle to assign peaks resulting from neutral loss of the PTMs, or fail to annotate non-peptide ions related to the modification. The tandem mass spectra of ARP-labeled peptides display several unique characteristics that, although generate problems for the database search software used in this study, aid in the manual inspection to unequivocally identify ARP-labeled peptides. Diagnostic ions frequently observed in our laboratory include the ions observed at m/z 227.1 (F1) and m/z 332.2 ([ARP+H]+) which originate from the ARP tag. Additionally, ions resulting from the loss of the ARP and HNE-ARP moiety from the precursor ion were observed. Several fragmentation pathways seem to be possible. We noticed that the charge state of the precursor ion, the ionization technique and CID conditions used may at least partly determine which of the fragmentation mechanisms prevails (Supplementary Material, Figure S26). In the MALDI-MS/MS analyses, the precursor ions are singly charged with the charge residing primarily on the C-terminal Lys or Arg residue, therefore we observe ions resulting from the neutral loss of ARP (-331.2 Da) and ARP-HNE (-469 Da) from the precursor ion. In the ESI-MS/MS analyses reported the precursor ions were multiply charged, allowing for a higher probability that a protonation site resides on the ARP tag. Therefore, in the ESI-MS/MS spectra of ARP-labeled peptides we observed frequently ions resulting from a) the loss of protonated ARP (-332.2 Da) and b) the loss of protonated ARP followed by the neutral loss of the dehydrated HNE moiety (additional loss of -138.10 Da) from the multiply charged precursor ion. This proposed mechanism would account for the reduction of the charge state observed for these diagnostic ions compared to the precursor ion used for the CID experiment (Supporting Information, Figure S26). Occasionally, ESI-MS/MS spectra did also display ion peaks which indicate neutral loss of the ARP-HNE moiety from the precursor ion, e.g. the [M+3H-(ARP-HNE)]3+ ion at m/z 668.3 in the ESI-MS/MS spectrum of the [M+3H]3+ ion of the dynein light chain peptide (Figure S13).
Figure 3 depicts tandem mass spectra of the tubulin α-1B peptide encompassing the partial sequence 340-352 in which Cys-347 was identified as being modified by the ARP-HNE moiety. The low energy CID fragment ion mass spectrum of the doubly protonated molecular ion of the HNE-modified peptide SIQFVDWC*PTGFK obtained with the linear ion trap of the LTQ-FT instrument is depicted in Figure 3A. In this peptide, Cys-347 was identified as being modified by the ARP-HNE moiety. Modification of the cysteine residue results in a m/z-difference of 572.2 Th between the y5 (m/z 549.3) and y6 ion (m/z 1121.4). Unequivocal assignment of the site of modification was further supported by additional y-type fragment ions, y6 and higher, and b-type fragment ions, b8 (m/z 1448.5) and b12 (m/z 1850.6), which show a mass shift of 469.2 Da due to the ARP-HNE modification. In addition, the singly charged ions at m/z 1665.6 and 1527.5 were characteristic for the loss of a protonated ARP tag (-332 Da) and the loss of the protonated ARP tag followed by neutral loss of the dehydrated HNE moiety (-470 Da) from the doubly charged precursor ion, respectively. The facile loss of the protonated ARP tag and subsequent loss of the dehydrated HNE moiety were frequently observed in tandem mass spectra acquired with the LTQ for peptides containing a HNE-modified cysteine residue. The use of the LTQ-FT mass spectrometer enabled exact mass measurements of modified peptides with high resolution which significantly increased the confidence of the peptide assignments. For example, high resolution mass analysis of the [M+2H]2+ ion of the HNE-modified peptide SIQFVDWC*PTGFK enabled the observation of the monoisotopic ion at m/z 998.9862 (expected m/z 998.9866) with an accuracy of 0.4 ppm (Supplemental information, Figure S1).
Figure 3.
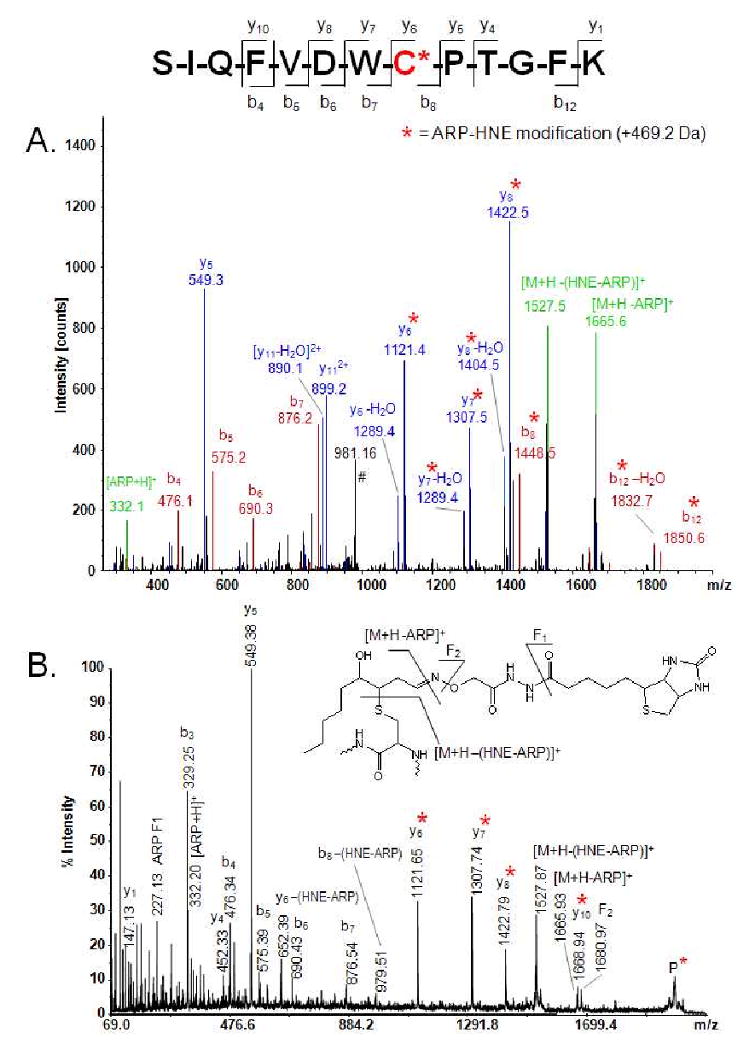
Tandem mass spectral analysis of the ARP-labeled HNE-modified peptide SIQFVDWC*PTGFK from tubulin α-1B chain. A. ESI-MS/MS spectrum of its doubly protonated precursor ion, [M+2H]2+ (m/z 999.0) obtained with the linear ion trap of the LTQ-FT mass spectrometer. The peak at m/z 981.2 (marked with “#”) indicates neutral loss of two H2O molecules from the doubly protonated precursor ion. The prominent ions with m/z 1665.6 and 1527.5 reflect loss of the protonated ARP tag and loss of the protonated ARP tag followed by a neutral loss of the dehydrated HNE moiety, respectively, from the doubly protonated precursor ion. Fragment ions marked with an asterisk (*) retained the ARP-HNE moiety during collision-induced fragmentation and allowed the unequivocal localization of the ARP-HNE moiety to the Cys residue at position 8 in this peptide. FTICR full-scan mass analysis of the doubly protonated molecular ion ([M+2H]2+) yielded m/z 998.9862 (m/zcalc 998.9866 (monoisotopic ion); accuracy Δ(m/z) -0.4 ppm) and supported the identification of this peptide as the partial sequence encompassing the residues 340 to 352 of tubulin α-1B chain. B. MALDI-MS/MS spectrum of the same ARP-labeled HNE-peptide adduct, SIQFVDWC*PTGFK. High-energy collision-induced fragmentation of the singly protonated precursor ion, [M+H]+ at m/z 1997.1 (marked with P), resulted in b- and y-type fragment ions. The short y-fragment ion series, y6-y8 and y10, highlighted by an asterisk (*), confirmed the localization of the ARP-HNE modification to the Cys residue at position 8. P, precursor ion; F1 and F2, non-peptide fragment ions related to the ARP-tag (insert and Figure S26)
Figure 3B depicts the MALDI-TOF/TOF tandem mass spectrum of the same HNE-modified peptide, SIQFVDWC*PTGFK, obtained by high energy (1 keV) collision-induced fragmentation of the singly protonated molecular ion ([M+H]+, m/z 1997.1). HNE adduction of the Cys residue in this peptide is confirmed by the y6, y7 and y8 ions observed at m/z 1121.7, 1307.7 and 1422.8, respectively. Neutral loss of the ARP-HNE moiety (-469.2 Da) from the singly protonated precursor ion results in the ion at m/z 1527.9. Additional fragment ions related to the ARP label were observed at m/z 227.1 (ARP-F1) and 332.2 ([ARP+H]+). Fragmentation of the oxime bond (-O-N=CH-) resulted in the formation of the fragment ion F2 at m/z 1681.0.
As an example of a tandem mass spectrum that permitted the identification of a His residue as the site of ARP-HNE modification, the low energy CID-MS/MS spectrum of the peptide, FSH*EEIAMATVTALR, is shown in Figure 4A. This peptide represents the partial sequence 244-258 of fructose-bisphosphate aldolase A containing His-246 modified by HNE and ARP. CID was performed on the triply protonated molecular ion [M+3H]3+ (m/z 715.7). The resulting tandem mass spectrum was dominated by doubly charged b-type ions, b5-b7, b9-b11, b13 and b14, which indicated that the ARP-HNE modification is located in the N-terminal region of the peptide. Because there is only one nucleophilic residue in this region we concluded that the His residue must be the site of modification by the ARP-HNE moiety. This assignment was in line with the observed y-type ions, y3-y6, y8, and y9, which were observed at the predicted m/z values without a mass shift related to the modification. In addition, the exact mass determination of the triply charged molecular ion [M+3H]3+ with m/z 715.7001 (m/zcalc 715.6992; accuracy Δ(m/z) 1.2 ppm) using the FT-MS capabilities of the LTQ-FT instrument supported the identification of this HNE-peptide adduct with high confidence.
Figure 4.
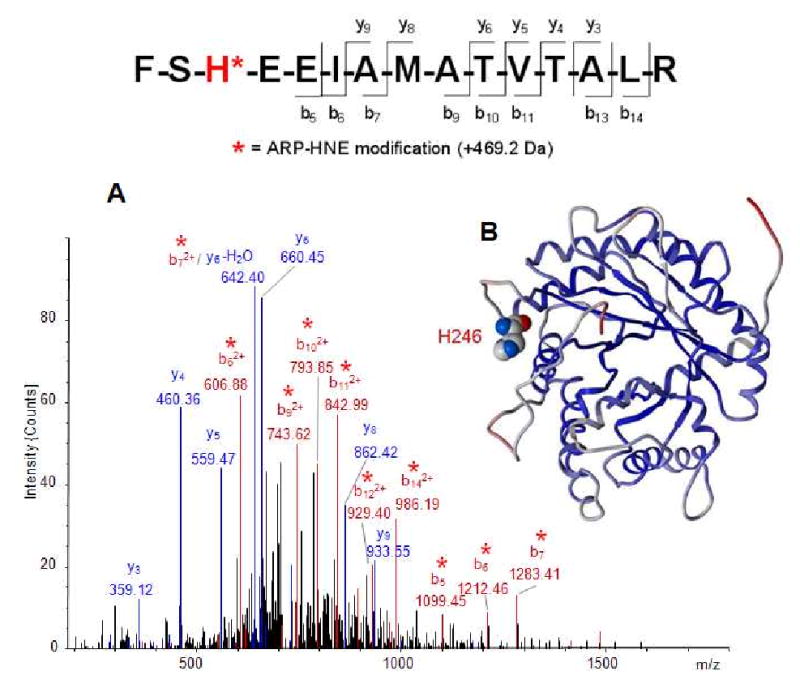
A. Tandem mass spectral analysis of the ARP-labeled HNE-modified peptide, FSH*EEIAMATVTALR, from fructose-bisphosphate aldolase A. The triply protonated molecular ion [M+3H]3+ (m/z 715.7) was used for collision-induced fragmentation in the linear ion trap of the LTQ-FT MS instrument. The ion peak marked with (#) at m/z 703.8 corresponds to the ‘neutral loss’-derived ion [M+3H - 2H2O]3+. The b-type fragment ions marked with an asterisk (*) enabled the localization of the ARP-HNE moiety to the His residue at position 3 of this peptide, which corresponds to the partial sequence 244-258 of aldolase A (ALDOA_Human). Full-scan FTICR analysis yielded for the monoisotopic [M+3H]3+ ion m/z 715.7001 (m/zcalc 715.6992; accuracy Δ(m/z) 1.2 ppm). B. Crystal structure of fructose-bisphosphate aldolase A (pdb:2ald) with the protein backbone displayed as ribbon and colored according to the B-factors reported for the structure pdb:2ald. The residue His-246, which was identified as site of the ARP-HNE modification, is displayed in CPK style and colored by atom type with the carbon atoms in white, nitrogen in blue and oxygen in red.
Discussion
Protein carbonylation may contribute to the cytotoxicity of HNE observed in THP-1 cells. We have chosen for the present study an exposure concentration of 100 μM, because our previous work indicated that at this concentration HNE triggers apoptotic and necrotic processes associated with elevated levels of protein carbonyls as determined by anti-DNP ELISA (27). Indeed, exposure of THP-1 cells at 100 μM HNE resulted in the formation of covalent protein-HNE adducts that were readily detected by anti-HNE immunostaining (Figure 1B). The attenuating effect of Asc on protein-HNE adduct levels was demonstrated by the lower intensity of the HNE-positive bands (Figure 1B). Thus, it would be conceivable that Asc attenuates HNE toxicity by preventing or inhibiting protein-HNE adduct formation. Our previous work has indicated a possible route for the observed diminishing effect of Asc on protein carbonylation. Asc promotes detoxification by enhancing HNE metabolite formation and their cellular export. Rapid inactivation of the electrophilic properties of HNE by glutathionylation and subsequent elimination of the respective HNE metabolites will result in lower cellular HNE levels and reduced protein-HNE adduct formation (27).
So far, only a few studies have attempted to identify protein targets of modification of HNE in complex biological systems. In our present study we combined gel-based and gel-free mass spectrometry-based approaches to detect, identify and characterize protein targets of HNE in THP-1 cells (summarized in Table 1). Immunostaining using an anti-HNE antibody in combination with mass spectrometry enabled the tentative assignment of 18 immunoreactive proteins (12 in 1-DE and seven additional ones in 2-DE). The four immunoreactive proteins that were assigned in both methods were β-actin, α-enolase, the 14-3-3 protein β/α and carbonic anhydrase (Table 1). In the 1-DE, protein disulfide isomerase A3 co-migrated with adenylyl cyclase-associated protein 1 and vimentin, and carbonic anhydrase co-migrated with 14-3-3 protein β/α. The subsequent 2-DE analysis resolved some of the ambiguities and confirmed the 14-3-3 protein β/α and carbonic anhydrase as proteins modified by HNE.
In the current study, mass spectrometric analysis of the immunoreactive proteins resolved on 1-DE or 2-DE gels did not yield the actual amino acid residues modified by HNE. Therefore, a gel-free strategy was employed, in which the aldehyde/keto-reactive reagent, ARP, was used for the biotinylation of Michael-type protein-HNE adducts, followed by trypsin digestion and subsequent enrichment using monomeric avidin. Using this targeted approach, 18 HNE-modified peptides belonging to 16 proteins were successfully identified by LC-MS/MS analysis, enabling the assignment of the ARP-HNE modification to specific Cys or His residues (Table 1). Of the 16 ARP-HNE modified proteins identified, four matched with proteins tentatively detected in the 1D and 2D immunoblotting analyses, namely β-actin, vimentin, adenylyl cyclase associated protein 1 and tubulin α-1B. These proteins are all highly abundant structural proteins and it is therefore not too surprising that these proteins were identified by different approaches.
In this study THP-1 cells were exposed to 100 μM HNE. In our previous study, this HNE concentration has been shown to induce apoptosis and necrosis in this cell line (27). At 100 μM, HNE may induce necrosis and apoptosis by adducting to proteins that are involved in the regulation of cell death or are important for cell survival. We found proteins associated with the cytoskeleton comprised the major group of HNE-modified proteins which are important for cellular survival (30), namely β-actin, vimentin, α-actinin-1, α-actinin-4, cofilin-1, F-actin-capping protein, one of the dynein light chains and tubulin α-1B chain. All of these proteins were detected using the ARP labeling strategy in combination with tandem mass spectrometry and as site of adduction distinct Cys and His residues were identified (Table 1). Additional cytoskeletal proteins were found by Western blotting analysis using a monoclonal anti-HNE antibody and these were tentatively assigned as: coronin-1A, type II cytoskeletal 1 keratin and tubulin beta-2C. However, the site of HNE modification in these immunoreactive proteins was not detected by mass spectrometry.
Cytoskeletal proteins are important in maintaining cell shape and intracellular transport. These proteins contribute to cellular organization and differentiation. Agents that specifically disrupt the cytoskeleton have been shown to stimulate apoptosis (31). β-Actin is a component of the actin cytoskeleton and serves as a mediator of internal cell motility (32). β-Actin is a target of carbonylation in Alzheimer's disease brain (4). Alpha-actinin binds to actin filaments and associates with cytoskeletal and signaling molecules, cytoplasmic domains of transmembrane receptors and ion channels (33). Cofilins promote cytoskeletal dynamics by depolymerizing actin filaments (34). Coronins are conserved actin binding proteins that help regulate cellular processes associated with cytoskeletal remodeling, endocytosis and cell motility (35). F-actin-capping proteins bind to the fast growing ends of actin filaments (barbed end) thereby blocking the growth of the actin filament at the barbed ends (36). Depolymerization of F-actin and degradation of intermediate filaments has been shown to be an important in the early stages of apoptosis (37). Vimentin is an intermediate filament protein and found in THP-1 derived macrophages (38). Tubulin α and tubulin β are components of the microtubule cytoskeleton.
Changes in cytoskeletal proteins could be one of the molecular bases for morphological changes occurring in the cell induced by oxidative stress and toxic chemicals (5). The adduction of Cys residues in α- and β-tubulins may have important implications on the structure of the microtubule cytoskeleton. Microtubules are in a dynamic state, alternately assembling and disassembling on the basis of the changing needs of the cell. Apoptotic phenomena, such as cell rounding, contraction and compaction of cellular organelles, require microtubule reorganization (31). Microtubule assembly can be impaired by HNE and destruction of microtubule structure has been observed in certain cells exposed to HNE (39). Modification of tubulin at Cys residues by lipid aldehydes could lead to the inhibition of polymerization because Cys residues are critical for tubulin polymerization (40). α-Tubulin has been identified as a major target of oxidative modification by HNE in rat primary neuronal cells, PC12 cells, and rat fibroblasts (39).
Two proteins of the glycolytic pathway, α-enolase and fructose-bisphosphate aldolase A (aldolase A), were identified by anti-HNE 2D Western blot and LC ESI-MS/MS analysis, respectively. In certain cancer cells glycolytic enzymes are upregulated due to the Warburg effect (41) which may at least partially explain why these proteins were identified as two of the major protein targets of HNE adduction in THP-1 cells. α-Enolase can bind cytoskeletal and chromatin structures and may serve as a cell surface plasminogen receptor and as a heat-shock protein (42). Increased carbonylation of α-enolase has been found in the brain of patients with mild cognitive impairment (43) and Alzheimer's disease (44). Several studies have identified fructose-bisphosphate aldolase (aldolase A) as a target of oxidative stress-related modifications (45, 46). However, the site of modification in aldolase A has not been previously identified. Our results suggest that His-246 is a susceptible target site for Michael-type addition by HNE. The location of His-246, at the start of the 9th α-helix, can be seen in the protein ribbon structure of aldolase A (pdb:2ald) shown in Figure 4B. Aldolase A has been described of having a dual functionality as glycolytic enzyme that also interacts with monomeric or multimeric forms of actin including actin filaments (47) and the α subunit of tubulin (48).
Other proteins that were tentatively identified as targets of modification by HNE were proteins related to stress response, including heat shock proteins, protein disulfide isomerase (the A1 and A3 isoforms) and peroxiredoxin-6. Heat-shock proteins function as molecular chaperones in response to cellular stress. Protein disulfide isomerase is an endoplasmic reticulum chaperone that promotes correct disulfide formation in newly synthesized proteins and has been shown to be modified by HNE at a Cys residue at the active site of the enzyme (18). Peroxiredoxin-6, an important antioxidant protein, is at Cys-91 susceptible to modification by HNE resulting in a cross-link, involving the residues Cys-91 and Lys-209, and inactivation of the protein (49). Carbonic anhydrase, an enzyme involved in the reversible hydration of carbon dioxide and aldehydes (50), has been shown to be a major intracellular target of HNE in erythrocytes (51), ventilatory muscles (46), skeletal muscles (52) and AD brain (53).
Our ARP labeling and enrichment strategy for the mass spectrometry-based identification and characterization of HNE-adducted peptides in THP-1 cells is analogous to the Codreanu et al. method which uses biotin hydrazide and capture with streptavidin to identify HNE-adducted proteins in human colon carcinoma RKO cells treated with 50 or 100 μM HNE (20). Codreanu et al. describe distinct adduction sites for 18 proteins. A comparison of the modification site assignments made in the RKO and the current THP-1 cell exposure studies revealed that only two HNE modification sites, namely Cys-347 in the tubulin α-1B chain and Cys-47 of the voltage-dependent anion-selective protein, were found at equivalent sequence positions in both systems. The minimal overlap between the two exposure studies suggests that the protein targets of HNE may be cell-specific and/or that there might be differences in the efficacy of the ARP/monomeric avidin method and biotin-hydrazide/streptavidin method for the analysis of protein-HNE adducts.
Prokai and coworkers introduced a solely mass spectrometry-based approach employing data-dependent and neutral loss-driven MS3 acquisition for the selective detection and identification of HNE-modified proteins in rat brain mitochondria exposed to HNE ex vivo (54). In this study, the majority of HNE adducts were found to be on His residues. Codreanu et al. (20) and we used prior enrichment of biotin-tagged HNE adducts and subsequent mass spectrometric analysis. The majority of the HNE targets in the RKO cells were modified on cysteine residue; only one His residue was observed to be adducted to HNE (20). In the present study, the sites of Michael-type addition involved predominately Cys residue and to a lesser degree His residues. It is not known at this time if the analytical LC-MS/MS-based strategy employed introduces a bias for which Michael-type adducts are observed. In general, it should be noted that proteomics-type analyses of protein targets of HNE adduction are commonly skewed toward highly abundant proteins. Also, HNE exposure studies of cellular systems may not necessarily reveal the protein targets of HNE formed endogenously due to the presence of ROS-generating centers and the compartmentalization of cellular systems (55).
Conclusions
The present study describes protein targets of HNE in the human monocytic cell line THP-1, a cell line which exhibits properties similar to monocyte-derived macrophages and is used for studying foam cell formation associated with atherosclerotic processes. A multifaceted strategy, involving electrophoretic, immunochemical and mass spectrometric methods, was employed to detect and identify HNE-protein adducts in THP-1 cells after exogenous exposure to HNE. Gel-based approaches in combination with immunoblotting using monoclonal anti-HNE antibodies were used to demonstrate a diminishing effect of ascorbic acid on protein modification by HNE. In addition, a chemical proteomic analysis based on the biotinylation of Michael-type HNE adducts using an aldehyde-reactive hydroxylamine-functionalized probe (aldehyde-reactive probe, ARP), enrichment and LC-MS/MS analysis enabled site-specific assignments of Michael-type HNE adductions. A total of 16 proteins were unequivocally identified as protein-HNE adducts. HNE adduction was predominately found at Cys residues (14-times out of 18); the remaining target sites were His residues. The majority of the HNE-modified proteins were involved in cytoskeleton organization/regulation and actin binding, stress response and glycolysis. This study provides the first site-specific assignments of adduction of HNE to Cys-295 in tubulin α-1B chain, Cys-351 and Cys-499 in α-actinin-4, Cys-328 in vimentin, Cys-369 in D-3-phosphoglycerate dehydrogenase and His-246 in aldolase A.
Supplementary Material
Acknowledgments
We thank Brian Arbogast for technical assistance on the LTQ-FT instrument. This study was supported by NIH Grants R01HL081721 and R01AG025372. We acknowledge the use of the Mass Spectrometry Facility and the Cell Culture Facility of Oregon State University's Environmental Health Sciences Center (NIH Grant P30ES00210). PNNL is a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under Contract DE-AC06-76RL01830.
Abbreviations
- AD
Alzheimer's Disease
- ARP
aldehyde-reactive probe
- Asc
ascorbic acid
- capLC
capillary liquid chromatography
- CID
collision-induced dissociation
- 1-DE
One-dimensional electrophoresis
- 2-DE
Two-dimensional electrophoresis
- ELISA
enzyme-linked immunosorbent assay
- DNP
dinitrophenyl
- ESI
electrospray
- GSH
glutathione
- GSH-HNE
glutathione Michael-type adduct of HNE
- HNE
4-hydroxy-2(E)-nonenal
- HRP
horseradish peroxidase
- IgG
Immunoglobulin G
- LTQ-FT-MS
linear ion trap Fourier transform mass spectrometer
- MALDI
matrix assisted laser desorption ionization
- MS/MS
tandem mass spectrometry
- PTM
post-translational modification
- ROS
reactive oxygen species
Footnotes
Supporting Information Available: Table S1 summarizes the mass spectrometry data of proteins tentatively identified in the gel regions (Fig. 1a) that matched to the immunoreactive bands in Figure 1B. Table S2 lists the mass spectrometry data of the HNE-positive protein spots assigned in the 2D blot depicted in Figure 2. Figures S1 to S25 display the annotated MS/MS spectra of ARP-HNE-modified peptides. Figure S26 depicts the proposed mechanisms for the CID fragmentations observed for ARP-labeled HNE-peptide adducts and proposed chemical structures of the non-peptide fragment ions characteristic for the collision-induced fragmentation of the ARP-tag. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 3.Chung FL, Pan J, Choudhury S, Roy R, Hu W, Tang MS. Formation of trans-4-hydroxy-2-nonenal- and other enal-derived cyclic DNA adducts from omega-3 and omega-6 polyunsaturated fatty acids and their roles in DNA repair and human p53 gene mutation. Mutat Res. 2003;531:25–36. doi: 10.1016/j.mrfmmm.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 5.Chung WG, Miranda CL, Stevens JF, Maier CS. Hop proanthocyanidins induce apoptosis, protein carbonylation, and cytoskeleton disorganization in human colorectal adenocarcinoma cells via reactive oxygen species. Food Chem Toxicol. 2009;47:827–836. doi: 10.1016/j.fct.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008;477:183–195. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West JD, Marnett LJ. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem Res Toxicol. 2006;19:173–194. doi: 10.1021/tx050321u. [DOI] [PubMed] [Google Scholar]
- 8.Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem Res Toxicol. 2004;17:1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrington DA, Kapphahn RJ. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004;578:217–223. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Choksi KB, Boylston WH, Rabek JP, Widger WR, Papaconstantinou J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim Biophys Acta. 2004;1688:95–101. doi: 10.1016/j.bbadis.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 12.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 13.Romero FJ, Bosch-Morell F, Romero MJ, Jareno EJ, Romero B, Marin N, Roma J. Lipid peroxidation products and antioxidants in human disease. Environ Health Perspect. 1998;106 5:1229–1234. doi: 10.1289/ehp.98106s51229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HP, Zhu X, Skidmore SC, Perry G, Sayre LM, Smith MA, Lee HG. The essential role of ERK in 4-oxo-2-nonenal-mediated cytotoxicity in SH-SY5Y human neuroblastoma cells. J Neurochem. 2009;108:1434–1441. doi: 10.1111/j.1471-4159.2009.05883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals and brain aging. Clin Geriatr Med. 2004;20:329–359. doi: 10.1016/j.cger.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Toroser D, Orr WC, Sohal RS. Carbonylation of mitochondrial proteins in Drosophila melanogaster during aging. Biochem Biophys Res Commun. 2007;363:418–424. doi: 10.1016/j.bbrc.2007.08.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 18.Carbone DL, Doorn JA, Kiebler Z, Petersen DR. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 19.Chavez J, Wu J, Han B, Chung WG, Maier CS. New role for an old probe: affinity labeling of oxylipid protein conjugates by N′-aminooxymethylcarbonylhydrazino D-biotin. Anal Chem. 2006;78:6847–6854. doi: 10.1021/ac0607257. [DOI] [PubMed] [Google Scholar]
- 20.Codreanu SG, Zhang B, Sobecki SM, Billheimer DD, Liebler DC. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol Cell Proteomics. 2009;8:670–680. doi: 10.1074/mcp.M800070-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han B, Stevens JF, Maier CS. Design, synthesis, and application of a hydrazide-functionalized isotope-coded affinity tag for the quantification of oxylipid-protein conjugates. Anal Chem. 2007;79:3342–3354. doi: 10.1021/ac062262a. [DOI] [PubMed] [Google Scholar]
- 22.Mirzaei H, Regnier F. Affinity chromatographic selection of carbonylated proteins followed by identification of oxidation sites using tandem mass spectrometry. Anal Chem. 2005;77:2386–2392. doi: 10.1021/ac0484373. [DOI] [PubMed] [Google Scholar]
- 23.Rauniyar N, Stevens SM, Prokai-Tatrai K, Prokai L. Characterization of 4-hydroxy-2-nonenal-modified peptides by liquid chromatography-tandem mass spectrometry using data-dependent acquisition: neutral loss-driven MS3 versus neutral loss-driven electron capture dissociation. Anal Chem. 2009;81:782–789. doi: 10.1021/ac802015m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roe MR, Xie H, Bandhakavi S, Griffin TJ. Proteomic mapping of 4-hydroxynonenal protein modification sites by solid-phase hydrazide chemistry and mass spectrometry. Anal Chem. 2007;79:3747–3756. doi: 10.1021/ac0617971. [DOI] [PubMed] [Google Scholar]
- 25.Vila A, Tallman KA, Jacobs AT, Liebler DC, Porter NA, Marnett LJ. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem Res Toxicol. 2008;21:432–444. doi: 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banka CL, Black AS, Dyer CA, Curtiss LK. THP-1 cells form foam cells in response to coculture with lipoproteins but not platelets. J Lipid Res. 1991;32:35–43. [PubMed] [Google Scholar]
- 27.Miranda CL, Reed RL, Kuiper HC, Alber S, Stevens JF. Ascorbic acid promotes detoxification and elimination of 4-hydroxy-2(E)-nonenal in human monocytic THP-1 cells. Chem Res Toxicol. 2009;22:863–874. doi: 10.1021/tx900042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung WG, Miranda CL, Maier CS. Detection of carbonyl-modified proteins in interfibrillar rat mitochondria using N′-aminooxymethylcarbonylhydrazino-D-biotin as an aldehyde/keto-reactive probe in combination with Western blot analysis and tandem mass spectrometry. Electrophoresis. 2008;29:1317–1324. doi: 10.1002/elps.200700606. [DOI] [PubMed] [Google Scholar]
- 29.Chung WG, Maier CS. Current Protocols in Toxicology. Unit 17.19. 2008. Mass Spectrometry-based identification and characterization of oxylipid-protein conjugates. [DOI] [PubMed] [Google Scholar]
- 30.Papakonstanti EA, Stournaras C. Cell responses regulated by early reorganization of actin cytoskeleton. FEBS Lett. 2008;582:2120–2127. doi: 10.1016/j.febslet.2008.02.064. [DOI] [PubMed] [Google Scholar]
- 31.Raffray M, Cohen GM. Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol Ther. 1997;75:153–177. doi: 10.1016/s0163-7258(97)00037-5. [DOI] [PubMed] [Google Scholar]
- 32.Hunter T, Garrels JI. Characterization of the mRNAs for alpha-, beta- and gamma-actin. Cell. 1977;12:767–781. doi: 10.1016/0092-8674(77)90276-8. [DOI] [PubMed] [Google Scholar]
- 33.Sjoblom B, Salmazo A, Djinovic-Carugo K. Alpha-actinin structure and regulation. Cell Mol Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 35.Gandhi M, Goode BL. Coronin: the double-edged sword of actin dynamics. Subcell Biochem. 2008;48:72–87. doi: 10.1007/978-0-387-09595-0_7. [DOI] [PubMed] [Google Scholar]
- 36.Barron-Casella EA, Torres MA, Scherer SW, Heng HH, Tsui LC, Casella JF. Sequence analysis and chromosomal localization of human Cap Z. Conserved residues within the actin-binding domain may link Cap Z to gelsolin/severin and profilin protein families. J Biol Chem. 1995;270:21472–21479. doi: 10.1074/jbc.270.37.21472. [DOI] [PubMed] [Google Scholar]
- 37.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci. 2000;113(Pt 7):1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 38.Kang JH, Ryu HS, Kim HT, Lee SJ, Choi UK, Park YB, Huh TL, Choi MS, Kang TC, Choi SY, Kwon OS. Proteomic analysis of human macrophages exposed to hypochlorite-oxidized low-density lipoprotein. Biochim Biophys Acta. 2009;1794:446–458. doi: 10.1016/j.bbapap.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 39.Kokubo J, Nagatani N, Hiroki K, Kuroiwa K, Watanabe N, Arai T. Mechanism of destruction of microtubule structures by 4-hydroxy-2-nonenal. Cell Struct Funct. 2008;33:51–59. doi: 10.1247/csf.07038. [DOI] [PubMed] [Google Scholar]
- 40.Stewart BJ, Doorn JA, Petersen DR. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem Res Toxicol. 2007;20:1111–1119. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- 41.Unwin RD, Craven RA, Harnden P, Hanrahan S, Totty N, Knowles M, Eardley I, Selby PJ, Banks RE. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the Warburg effect. Proteomics. 2003;3:1620–1632. doi: 10.1002/pmic.200300464. [DOI] [PubMed] [Google Scholar]
- 42.Pancholi V. Multifunctional alpha-enolase: its role in diseases. Cell Mol Life Sci. 2001;58:902–920. doi: 10.1007/PL00000910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 44.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 45.Martinez A, Dalfo E, Muntane G, Ferrer I. Glycolitic enzymes are targets of oxidation in aged human frontal cortex and oxidative damage of these proteins is increased in progressive supranuclear palsy. J Neural Transm. 2008;115:59–66. doi: 10.1007/s00702-007-0800-y. [DOI] [PubMed] [Google Scholar]
- 46.Hussain SN, Matar G, Barreiro E, Florian M, Divangahi M, Vassilakopoulos T. Modifications of proteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am J Physiol Lung Cell Mol Physiol. 2006;290:L996–1003. doi: 10.1152/ajplung.00337.2005. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Morris AJ, Tolan DR, Pagliaro L. The molecular nature of the F-actin binding activity of aldolase revealed with site-directed mutants. J Biol Chem. 1996;271:6861–6865. [PubMed] [Google Scholar]
- 48.Volker KW, Knull H. A glycolytic enzyme binding domain on tubulin. Arch Biochem Biophys. 1997;338:237–243. doi: 10.1006/abbi.1996.9819. [DOI] [PubMed] [Google Scholar]
- 49.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chem Res Toxicol. 2008;21:2289–2299. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 50.Pocker Y, Dickerson DG. The catalytic versatility of erythrocyte carbonic anhydrase. V. Kinetic studies of enzyme-catalyzed hydrations of aliphatic aldehydes. Biochemistry. 1968;7:1995–2004. doi: 10.1021/bi00845a050. [DOI] [PubMed] [Google Scholar]
- 51.Uchida K, Hasui Y, Osawa T. Covalent attachment of 4-hydroxy-2-nonenal to erythrocyte proteins. J Biochem. 1997;122:1246–1251. doi: 10.1093/oxfordjournals.jbchem.a021888. [DOI] [PubMed] [Google Scholar]
- 52.Chen CN, Ferrington DA, Thompson LV. Carbonic anhydrase III and four-and-a-half LIM protein 1 are preferentially oxidized with muscle unloading. J Appl Physiol. 2008;105:1554–1561. doi: 10.1152/japplphysiol.90680.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 54.Stevens SM, Jr, Rauniyar N, Prokai L. Rapid characterization of covalent modifications to rat brain mitochondrial proteins after ex vivo exposure to 4-hydroxy-2-nonenal by liquid chromatography-tandem mass spectrometry using data-dependent and neutral loss-driven MS3 acquisition. J Mass Spectrom. 2007;42:1599–1605. doi: 10.1002/jms.1349. [DOI] [PubMed] [Google Scholar]
- 55.Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J Biol Chem. 2003;278:19587–19590. doi: 10.1074/jbc.C300135200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.