

Abstract
Plants like Arabidopsis thaliana are convenient model systems to study fundamental questions related to regulation of the stress transcriptome in response to stress challenges. Microarray results of the Arabidopsis transcriptome indicate that several genes could be upregulated during multiple stresses. High-salinity, drought, and low temperature are three common environmental stress factors that seriously influence plant growth and development worldwide. Recently, microRNAs (miRNAs) have emerged as a class of gene expression regulators that have also been linked to stress responses. However, the relationship between miRNA expression and stress responses is just beginning to be explored. Here we have computationally analyzed 123 non redundant miRNA sequences reported for Arabidopsis thaliana, including 17 miRNA sequences which were reported to be stress regulated in literature. A significant increase in the GC content of stress regulated miRNA sequences was observed which further extends the view that miRNAs act as ubiquitous regulators under stress conditions. GC content may also be considered as a critical parameter for predicting stress regulated miRNAs in plants like Arabidopsis thaliana.
Keywords: Arabidopsis thaliana, Stress, miRNAs, transcriptome, miRBase, GC content, Bioinformatics
Background
Our understanding of the regulation of functional genes responsive to stress signals is still nascent. Multiple signalling pathways regulate the stress responses of plants and there is significant overlap between the patterns of gene expression that are induced in plants in response to different stresses. Many genes induced by stress challenges, including those encoding transcription factors, have been identified and some of them have been shown to be essential for stress tolerance [1]. Many studies have also revealed some of the complexity and overlap in the responses to different stresses and are likely to lead to new ways to enhance crop tolerance to disease and environmental stress. MicroRNAs (miRNAs) are a highly conserved class of small noncoding RNAs that regulate gene expression by posttranscriptional degradation or translational repression [2]. They play important roles in multiple biological and metabolic processes, including developmental timing, signal transduction, cell maintenance, differentiation and diseases [3,4]. Recently, there has been strong evidence leading to the proposal that miRNAs are hypersensitive to abiotic or biotic stress as well as to diverse physiological processes. More and more evidence has shown that gene silencing is widely adopted in plant immunity. In the past, studies often focused on transposon or siRNA-mediated RNA silence [5,6] . Since miRNAs and siRNAs share many features in common, it is supposed that miRNAs may also be involved in silencing invaders. This was supported by the observation that siRNAs functioned as miRNAs and miRNAs interacted with mRNA in the same way as siRNAs [7]. A family of Arabidopsis mRNAs encoding SCARECROW-LIKE (SCL) transcription factors is cleaved by an RNAi-like process directed by miR171 [8]. In plant embryo extracts, an endogenous miRNA that lacks perfect complementarity to its RNA targets acts as a siRNA [9]. In other words, the data reveals an interchangeable functional role between miRNA and siRNA. Plant virus-derived small RNAs in the gene silencing (VIGS) process were generally considered to be siRNAs. The prevalence of imperfect hairpin structure prompts a reevaluation of their biochemical nature. In fact, many of these molecules might be akin to miRNAs, because their hairpins have greater similarity to miRNA precursors than to the perfect dsRNAs that produce siRNAs [10,11]. Plant virus infections resulted in a dramatic increase in miRNA whereas virus infected vertebrate cells increased siRNA content [12].
There have been many studies to identify plant miRNAs and numerous miRNAs have been discovered in Arabidopsis thaliana. Currently, a variety of biochemical, molecular, and bioinformatics approaches and technologies have been developed for miRNA analysis and detection. Using tiling path microarray analysis as a tool, it is now possible to perform high-throughput profiling of the expression of all the known miRNAs to examine their expression profiles under different environmental stresses [13]. In a recent report the effects of 117 miRNAs under high-salinity, drought, and low-temperature stress conditions were analyzed using miRNA chips representing nearly all known miRNAs cloned or identified in Arabidopsis thaliana [14]. Seventeen stress-inducible miRNAs were detected and the results were further confirmed by detecting their expression patterns and analyzing the cis-regulatory elements in their promoter sequences [14]. In our work, we analyzed the nucleotide base frequencies at each position of the 123 miRNA sequences that have been reported for Arabidopsis. These 123 sequences obtained from the miRBase include the 117 sequences that were analyzed by Han-Hua L. and coworkers [14] and 6 newly reported miRNA sequences in Arabidopsis thaliana. We also calculated the base density for each nucleotide in these 123 sequences. This same approach was then used for analyzing groups of miRNA sequences which have been reported to regulate 3 types of abiotic stresses ‐ high salinity, drought and low temperature. A graphical comparison and trend analysis was done to observe differences in frequencies of nucleotide bases between the stress-regulated miRNAs and all miRNA sequences as a whole.
Methodology
The microRNA sequence database miRBase was used to create a dataset of 123 non redundant miRNA sequences reported in Arabidopsis thaliana (see supplementary material). We grouped the miRNA sequences into 5 groups, 1st containing all the 123 sequences, 2nd containing all the sequences induced by high salinity stress, 3rd by drought stress, 4th by cold stress and 5th by all the three stresses. Further we calculated the relative frequency of each nucleotide base at each position in the miRNA sequence in each of the groups.
miRBase: the microRNA sequence database
The miRBase Sequence database is freely accessible primary repository for published microRNA (miRNA) sequence and annotation data. miRBase provides a user-friendly web interface for miRNA data, allowing the user to search using key words or sequences, trace links to the primary literature referencing the miRNA discoveries, analyze genomic coordinates and context, and mine relationships between miRNA sequences. miRBase also provides a confidential gene-naming service, assigning official miRNA names to novel genes before their publication [15].
Calculation of position specific nucleotide frequencies
All sequences were divided into no. of cells corresponding to their length with each nucleotide in each cell of an excel sheet. The sequences were then aligned in the above mentioned five groups respectively. The following function was used to count the no. of each nucleotide at each position.
COUNTIF (range, criteria)
Counts the number of cells within a range that meet the given criteria.
Calculation of position specific relative frequencies
The relative frequency of each nucleotide at each position of the sequence was calculated by dividing the absolute frequencies (position specific nucleotide frequencies) by the no. of sequences having a nucleotide base at that specific position. To find out the no. of sequences having a nucleotide base at each position we sum up the absolute frequencies of all the nucleotide bases at each position by SUM (number1, number2...) Adds all the numbers in a range of cells.
Finding out nucleotide content in a group of sequences
The absolute frequency of each nucleotide at all positions of a group of sequences was summed up using the above mentioned SUM function. The total no. of sequences at each position was also summed up using the same function. The percentage content of each nucleotide in a given group of sequences was calculated by the following formula.
Percentage content of N = 100 * Sum of absolute frequencies of N / Sum of total no. of sequences at each position
Line graphs were plotted to compare the variations in the pattern of position wise nucleotide frequency distribution between stress regulated miRNAs and all miRNAs (Figure 1). Then TREND LINEs were added to these line graphs to compare the trend of nucleotide frequency distribution for each nucleotide base in stress regulated sequences and all miRNAs (Figure 2).
Figure 1.
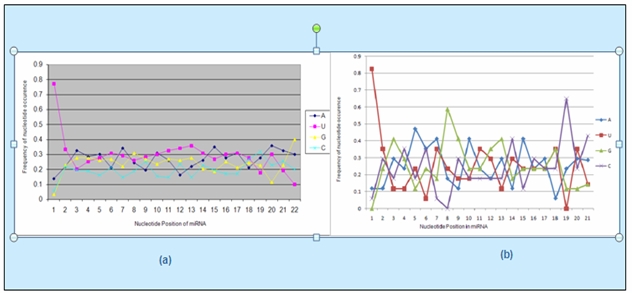
Nucleotide frequency distribution of miRNAs: (a) The nucleotide position wise AUGC distribution of all miRNAs. (b) The nucleotide position wise AUGC distribution for stress regulated miRNAs. The degree of randomness in the position wise nucleotide frequency distribution is more in the stress regulated miRNAs.
Figure 2.
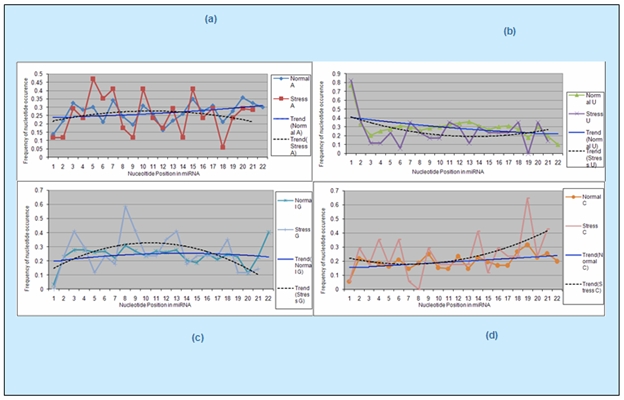
Position based trend analysis of nucleotide frequency (a), (b), (c), (d) show the comparison between stress regulated and normal miRNAs for frequency distribution of nucleotide A, U, G and C respectively at each position of the corresponding sequences. The group of normal sequences corresponds to the 1st group of sequences which includes all 123 sequences of the dataset consisting of both the stress regulated sequences as well those not regulated by stress.
Discussion
There is a significant increase in the GC content of the sequences regulated by the three stresses. The average GC content of Arabidopsis thaliana genome has been reported as just 36% due to the large intronic regions present which contain large stretches of A-T base pairs. The average GC content of all miRNAs reported for Arabidopsis thaliana is calculated to be 43% by our analysis. On the other hand the GC content of stress induced sequences calculated by our method is significantly higher 51-52% (Table 1 in supplementary material).
Within a long region of genomic sequence, genes are often characterized by having a higher GC-content in contrast to the background GC-content for the entire genome. The high GC-content content is also a feature of genes coding for transcriptional activators or repressors which regulate gene expression under various stress condition. The promoter sequences of many stress regulated genes have also been reported to have a high GC content [16]. Since the miRNAs which were induced by stress were also found to have a higher GC content like the coding regions, it may be hypothesized that the GC content may play a significant role in the interaction of these miRNAs with their target genes for regulating gene expression under stress conditions. The high GC content may be significant because of two reasons ‐ firstly the higher GC ratio gives a higher chance of complementarities with the coding regions also having a higher GC ratio, secondly since GC pair is bound by 3 hydrogen bonds, these bonds are more stable, so the miRNA binding and degradation of the target genes may be more probable.
The AUGC frequency distribution at each of the nucleotide positions was analyzed for all miRNAs and compared with that for the stress regulated miRNAs by plotting line graphs from the data calculated from these sequences (Figure 1). When these graphs were compared for the normal and stress regulated sequences, the stress graph was seen to have a considerable variation (degree of randomness) at each position and for each base pair (A,U,G,C) thus proving that under stress, cell responds in a different way which can be studied further to understand its importance.
Trend Analysis
The trend analysis graphs were plotted for normal and stress sequences to visualize the trend of frequency distribution for each nucleotide base. For nucleotide “A” the trend analysis showed to some extent a concave downward curve for stress regulated sequences, whereas a marginal linear increase graph for normal sequences (Figure 2a). The trend for nucleotide “U” showed a concave upward graph for stress regulated sequences, whereas a marginal linear decrease graph for normal sequences. Also the curve for stress lies below the trend line graph corresponding to normal sequences (Figure 2b).
Nucleotide “G” showed an altogether different trend of a significant concave downward curve for stress regulated sequences, indicating higher frequencies of this nucleotide in the mid positions of these sequences. The trend of normal sequences for this nucleotide showed a more or less constant graph (Figure 2c). For nucleotide “C” the trend of stress regulated sequences was opposite to that for G, as it showed a significant concave upward curve for stress regulated sequences, indicating higher frequencies of this nucleotide at the two ends of the sequences. Also curve for stress lies above the trend line graph corresponding to normal sequences. A slight linear increase graph was observed for the normal sequences (Figure 2d). These trends noticeably indicate that since the frequency of nucleotide “G” is higher in the mid positions and that of nucleotide “C” is higher at the two ends of the stress regulated sequences, the average GC content becomes high altogether for these sequences.
Conclusion
Our results suggest that the GC content may play a important role in the interaction of miRNAs with their target genes. The significance of the degree of randomness of nucleotide frequency distribution in stress regulated miRNAs can be identified by future studies. An overall increase in the GC content of the stress regulated miRNAs further extends the view that high GC can be a critical parameter for prediction of stress regulated miRNAs in Arabidopsis thaliana.
Supplementary material
Footnotes
Citation:Akash et al, Bioinformation 4(4): 151-154 (2009)
References
- 1.Ambika SS, et al. Bioinformation. 2008;2:431. [Google Scholar]
- 2.Sunkar R, et al. Plant Cell. 2004;16:2001. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang B, et al. Comput Biol Chem. 2006;30:395. doi: 10.1016/j.compbiolchem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Zhang B, et al. Gene. 2007;397:26. doi: 10.1016/j.gene.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 5.Kavi HH, et al. Bioessays. 2005;27:1209. doi: 10.1002/bies.20328. [DOI] [PubMed] [Google Scholar]
- 6.Zamore PD. Nat Struct Biol. 2001;8:746. doi: 10.1038/nsb0901-746. [DOI] [PubMed] [Google Scholar]
- 7.John GD, et al. Genes Dev. 2003;17:438. [Google Scholar]
- 8.Llave C, et al. Science. 2002;297:2053. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- 9.Hua T, et al. Am J Hum Genet. 2005;76:268. [Google Scholar]
- 10.Dunoyer P, Voinnet O. Curr Opin Plant Biol. 2005;8:415. doi: 10.1016/j.pbi.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 11.Yan-du L, et al. Plant Cell Rep. 2008;27:1571. doi: 10.1007/s00299-008-0584-z. [DOI] [PubMed] [Google Scholar]
- 12.Bennasser Y, et al. Immunity. 2005;22:607. doi: 10.1016/j.immuni.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Jiao Y, et al. Plant Cell. 2005;17:1641. doi: 10.1105/tpc.105.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han-Hua L, et al. RNA. 2008;14:836. [Google Scholar]
- 15. http://microrna.sanger.ac.uk/
- 16.Keisuke M, et al. Nucleic Acids Res. 2009;37:1438. doi: 10.1093/nar/gkn1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.