

Abstract
Oral squamous cell carcinoma (OSCC) has 50% 5-year survival rate, highlighting our limited understanding of the molecular events that contribute to disease progression. Microarray analyses of primary oral tumors have identified urinary type plasminogen activator (uPA) and its receptor (uPAR) as key genes associated with human OSCC progression. The uPAR functions both as a proteinase receptor and an integrin ligand, modifying proteolysis, migration, integrin signaling and cellular transcription. In the current study, uPAR expression levels were modified in OSCC cells, followed by analysis of tumor growth in an in vivo orthotopic xenograft model and by transcriptional profiling. Overexpression of uPAR resulted in more infiltrative and less differentiated tumors, with ill-defined borders, cytologic atypia, and enhanced vascularity. Analysis of serial sections of both murine experimental tumors and microarrayed human OSCC demonstrated a statistically significant association between uPAR and α3 integrin co-localization in areas exhibiting ERK phosphorylation, suggesting that uPAR/α3 integrin interaction potentiates ERK signaling in vivo. This is supported by cDNA microarray analysis which showed differential expression of 148 genes (113 up, 35 down). Validation of gene expression changes in human OSCC using immunohistochemistry and quantitative real-time PCR showed increased growth factors, proteinases/inhibitor and matrix components in uPAR-overexpressing tumors. Together these results support a model wherein increased uPAR expression promotes α3β1 integrin association, resulting in increased MAPK signaling and transcriptional activation, leading to the formation of more aggressive tongue tumors. This combined approach has efficacy to identify additional biomarkers and/or prognostic indicators associated with aggressive human OSCC.
Keywords: oral cancer, squamous cell carcinoma, uPAR, α3β1 integrin, tongue cancer
INTRODUCTION
Oral cavity cancer ranks among the top ten most frequently diagnosed cancers worldwide, resulting in over 200,000 deaths annually [1]. Squamous cell carcinoma (OSCC) is the most common malignancy of the oral cavity, with 30,000 new cases detected each year in the U.S alone [2]. Of these, 25–40% occur in the tongue (squamous cell carcinoma of the oral tongue, SCCOT) [3]. In the U.S., the incidence of oral tongue cancer has actually increased in the 20–44 age group in the last three decades [4]. However, the 5-year survival rate has remained at a low 50% for the past 20 years, highlighting our limited understanding of the molecular events that govern OSCC initiation, progression and metastasis [5]. Tongue tumors frequently remain asymptomatic until advanced stage, due in part to anatomic location, and several studies suggest a poorer survival rate than other anatomic sites in the oral cavity [3,6]. As the high mortality from OSCC is attributed to regional and distant metastasis, a more detailed analysis of the molecular events that potentiate growth and dissemination is a necessary prerequisite to the development of novel early detection and treatment strategies [7].
Recent studies have used cDNA microarray analysis for genome-wide monitoring of genetic changes associated with primary oral tumors compared to normal oral mucosa or lymph node metastases [8,9]. These studies have identified the proteinase urinary type plasminogen activator (uPA, urokinase) as one of 25 genes that comprise an “OSCC gene signature” for molecular classification of oral tumors and as a key candidate biomarker for prediction of poor disease outcome [8,9]. OSCC cell lines display enhanced uPA expression relative to cells derived from normal oral mucosa and exhibit increased uPA-dependent invasion in vitro [10–12]. Explants of OSCC tissues also show higher levels of uPA relative to adjacent non-malignant tissues [13,14].
Similarly, human OSCC tumors with high levels of uPA and its cell surface receptor (uPAR) are more invasive, exhibit enhanced lymph node metastasis [15–17] and more frequent tumor relapse [18]. These data suggest the potential utility of uPA/R as a prognostic indicator and/or therapeutic target. In addition to regulation of pericellular proteolysis, uPAR can also function as a lateral, non-matrix ligand for α3β1 integrin, and thereby affect α3β1 integrin function and integrin-dependent cellular signal transduction pathways [19, 20]. We have previously reported a functional link between adhesion and proteolysis, showing that matrix engagement of the α3β1 integrin exerts multifunctional control on the uPA system by inducing upregulated uPA activity together with redistribution of uPAR to sites of clustered integrins [19]. Further mechanistic analyses demonstrated that uPAR/α3β1 integrin interaction initiates a Src/MEK/ERK-dependent signaling pathway, leading to transcriptional activation of the uPA promoter [20]. A functional requirement for uPAR/α3β1 integrin interaction was confirmed using pharmacologic approaches, siRNA knockdown of uPAR, and cells from α3 integrin integrin knockout mice [20]. The impact of uPAR regulation of integrin function in OSCC has not been explored in vivo.
As spontaneous OSCC are very rare in domestic laboratory animals, orthotopic murine tumor models have utility for investigating cellular and molecular mechanisms in tumor progression and metastasis [21]. Orthotopic implantation of cell lines or primary tumor explants into the tongue has been shown to result in both expansive and invasive growth [22–23] and has demonstrated efficacy in preclinical testing of potential therapeutics [24]. To examine how uPAR modulation of α3β1 integrin function may alter progression of SCCOT in vivo, the effect of modulated uPA/R expression on SCCOT growth was evaluated in an orthotopic murine model [23]. Changes in gene expression as a result of uPAR/integrin signaling were identified using cDNA microarray analysis and validated by quantitative real-time PCR (qPCR) and immunohistochemical analysis of murine tumors. The results support a model wherein uPAR/α3β1 integrin binding leads to growth of more aggressive, invasive tumors with concomitant changes in gene expression that may contribute to SCCOT progression in vivo.
RESULTS
Formation of SCCOT Tumors in Nude Mice
Genome-wide monitoring of genetic changes associated with OSCC primary tumors and lymph node metastases have identified the proteinase uPA and the cell-matrix adhesion molecule α3 integrin as key candidate biomarkers for the prediction of poor disease outcome [9,11,25,26]. To evaluate the potential contribution of uPAR/α3β1 integrin binding to tumor growth and invasion in vivo, cells with modified uPAR levels were generated for in vivo analysis. Pooled clones of SCC25 cells with enhanced (SCC25-uPAR+) or reduced (SCC25-uPAR-KD) levels of uPAR (suppl. Fig. 1A) were evaluated using FACS for changes in expression of other cell surface markers. Neither α3 integrin nor E-cadherin levels were altered by modulation of uPAR expression and no change in proliferation was observed in vitro (data not shown). However uPAR overexpression resulted in a substantial increase in in vitro invasive activity (suppl. Fig. 1B, C). These results also show the contribution of α3 integrin to invasion, as a 66% decrease in invasion was observed in the presence of α3 integrin function blocking antibodies. Similarly, use of an antibody strategy to block uPAR decreased invasion by 70%. As was previously observed (suppl. Fig. 1B), knockdown of uPAR expression using siRNA also decreased invasion substantially (90%). In addition, the contribution of ERK activity to the invasive process was demonstrated using a MEK inhibitor (PD98059), resulting in an 54% inhibition of invasion. Together these functional data implicate α3 integrin as well as uPAR and pERK in the invasive process.
To investigate the effect of uPAR modulation on tumor growth at the orthotopic site in vivo, pooled clones of SCC25-uPAR+ or SCC25-uPAR-KD cells were injected submucosally into the anterior tongue and mice were examined visually for the development of tongue tumors. Eight mice per group were injected and the experiment was repeated three times; representative images are shown. Visual tongue tumors developed by 5–6 weeks and tumors were allowed to progress for 9 weeks. No lymph node or lung metastases were observed in the time frame of this study. Examination of H&E and immunohistochemically-stained sections shows neoplastic cells arranged in cysts, nests, and packets frequently extending between and through muscle bundles and surrounded by inflammatory infiltrate (Fig. 1A–F). Neoplastic cells are markedly pleiomorphic but display all stages of development seen in keratinized stratified squamous epithelium with flattened to cuboidal basophilic cells and scant cytoplasm. The cells become increasingly eosinophilic and develop abundant cytoplasm toward the center. In tumors formed by SCC25-uPAR-KD cells, low power evaluation demonstrates multi-centric well-circumscribed tumor nests with a pushing margin of invasion (not shown). Higher power examination (Fig. 1A,C,E) reveals that all tumors formed from SCC25-uPAR-KD cells have features of a well-differentiated squamous cell carcinoma, including numerous aggregates of keratin (keratin pearls, arrowheads), some dystrophic calcification, low mitotic index and minimal pleomorphism. Most tumor cells showed abundant eosinophilic cytoplasm and a lower nuclear to cytoplasmic ratio than that seen in the uPAR overexpressor group. Mitoses were present, but infrequent. Prominent desmoplasia, characterized by loose mesenchymal tissue, was also present (Fig. 1A, asterisk). No lymphovascular or peri-neural invasion was seen. In contrast, low power examination of uPAR-overexpressing tumors (SCC25-uPAR+) reveals cords of tumor with ill-defined borders and high vascularity (not shown). Higher power evaluation shows that all tumors formed from SCC25-uPAR+ cells were moderately to poorly differentiated squamous cell carcinomas. These were poorly circumscribed and deeply, diffusely, and raggedly infiltrative with thin cords of tumor cells dissecting through skeletal muscle (Fig. 1B,D,F).
Figure 1. Histological and immunohistochemical examination of oral tongue tumors.
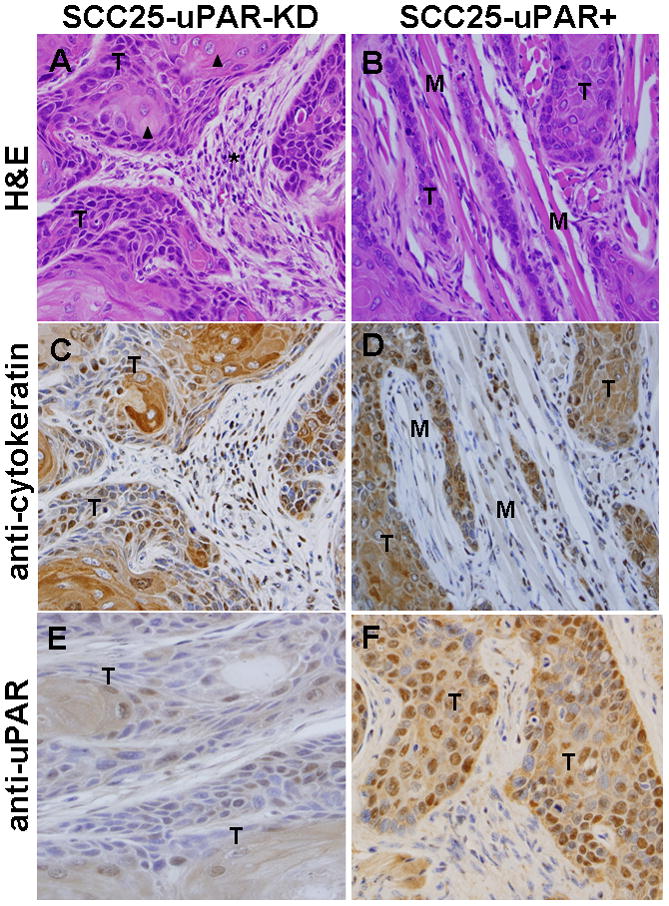
Tumors generated from pooled clones of SCC25-uPAR-KD and SCC25-uPAR+ cells were (A,B) stained with H&E or immunostained with (C,D) anti-cytokeratin AE1/AE3 (1:25 dilution) or (E,F) anti-uPAR (1:25 dilution) followed by a biotinylated secondary antibody and detection of avidin-biotin with DAB chromagen and substrate as in Experimental Procedures. 400X magnification. (T) – designates areas of tumor cells; (M) designates host tongue muscle; (arrowheads) – focal keratinization (‘keratin pearls’ are highlighted); (arrows) – example of well-circumscribed tumor nests; (asterisk) – desmoplastic host response.
Tumors are composed of clusters of hyperchromatic cells with a high nuclear to cytoplasm ratio and cytologic atypia, with only focal keratin production (Fig. 1B,D,F). Rare keratin pearls were seen. This is supported by qPCR analysis of keratin mRNA expression levels, showing loss of keratin 13 and keratin 19 expression in SCC25-uPAR+ relative to SCC25-uPAR-KD (suppl. Fig. 2A). No quantitative differences in overall tumor area or degree of inflammation were apparent (suppl. Fig. 2B). Furthermore, numerous foci of peri-neural invasion and focal vascular invasion were evident in tumors formed from SCC25-uPAR+ cells (Fig. 2A-D, Suppl. Fig. 3) and higher magnification H&E images clearly show cords of tumor cells wrapping around nerves (Fig. 2B,C; Suppl. Fig. 3B) or invading small vessels (Fig. 2D; Suppl. Fig. 3A). Studies of human OSCC have identified tumors with diffuse spread, small cords, vascular and perineural invasion as associated with a more aggressive tumor and a poor prognosis [27–29].
Figure 2. Perineural and vascular invasion in SCC-25-uPAR+ tumors.
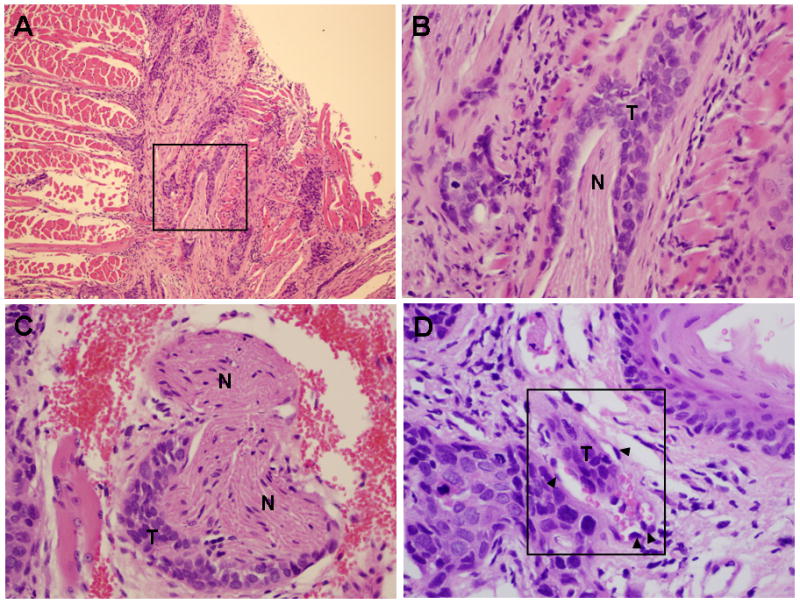
(A–C) Representative H&E sections showing tumor cords surrounding nerve bundle. (T) – tumor, (N) – nerve. Panel A – 100X magnification, panels B,C – 400X magnification. (D) Representative H&E section showing tumor localization within a small vessel (boxed area). Magnification 600X. (T) – tumor tissue; (arrowheads) – vascular endothelial cells. Note red blood cells also present in the vessel.
Colocalization of uPAR,α3 integrin and phospho-ERK in murine and human SCCOT
Lateral complex formation between uPAR and α3β1 integrin induces src/MEK/ERK signaling and transcriptional activation in vitro [19, 20]. To assess whether these molecular players are co-localized in vivo, uPAR, α3 integrin and phospho-ERK staining was assessed in murine SCCOT tumors using both immunohistochemical analysis of serial sections and 4-color immunofluorescence analysis of single tumor sections. Multiple areas of co-localization between uPAR and α3 integrin were evident in all SCC25-uPAR+ tumors in areas also exhibiting ERK phosphorylation (Fig. 3A) and this is supported by immunofluorescence analysis showing fluorescence overlap between uPAR, α3 integrin and phospho-ERK (Fig. 3B, white areas). Furthermore quantitation of ERK activation, by enumerating cells with positive nuclear phospho-ERK staining in a minimum of 10,000 cells/group, showed an highly significant difference between the groups (p=.0001), with 67.9 +/− 15.0 % of tumors generated from uPAR overexpressors exhibiting ERK activation, while only 14.1 +/− 8.2% of tumors from uPAR knockdown cells exhibited activated ERK (Fig. 3C). Similar results were obtained from analysis of microarrayed human SCCOT serial sections, demonstrating co-localization of uPAR, α3 integrin and phospho-ERK (not shown). In human oral tumors, the bivariate relationship between α3 integrin positivity and pERK was significantly positive, with 82.5% of the variance in the latter explained by variation in the former (r=0.909; p<0.0001). As uPAR and α3 integrin positivity are also significantly positively correlated (r=0.763; p<0.0001), a multiple regression was performed to investigate if this interaction had a significant influence on pERK expression beyond the singular influence of α3 integrin. Such an analysis indicates a significant positive affect of the interaction between uPAR and α3 integrin on pERK (p<0.0001). This multifactorial model explains 91.9% of the variance in pERK positivity, which is 9.4% greater than a more simple bivariate model where only the influence of α3 integrin on pERK positivity is considered. Thus, incorporation of the relationship between uPAR and α3 better models the variance in pERK staining positivity than does a model that omits uPAR/α3 integrin interaction.
Figure 3. Co-localization of uPAR, α3 integrin and phospho-ERK immunostaining in murine tongue tumors.
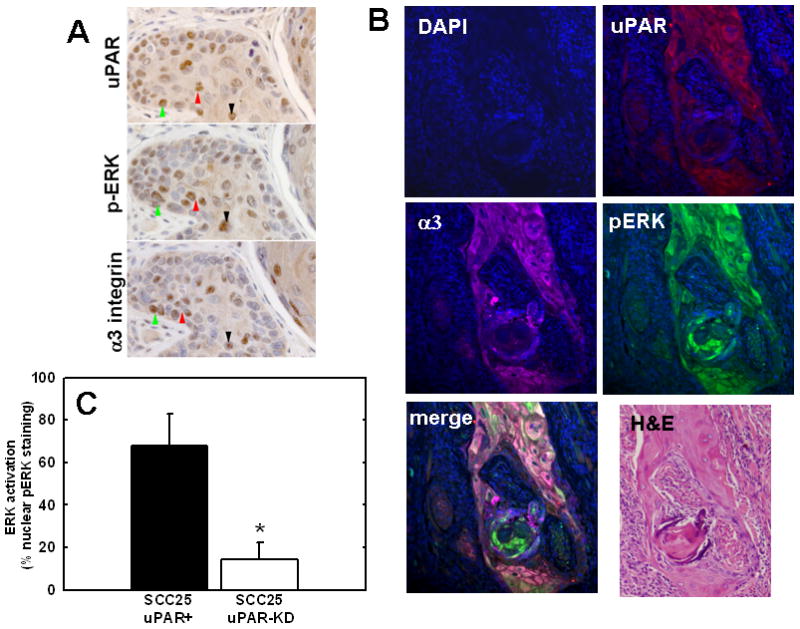
(A) Serial sections of tongue tumors generated from SCC25-uPAR+ cells were immunostained with anti-uPAR (3936 antibody, 1:25 dilution), anti-phospho-ERK (9101S antibody, 1:25 dilution) or anti-α3-integrin (1952 antibody, 1:25 dilution), as indicated, followed by biotinylated secondary antibody and detection as described in Experimental Procedures. 400X magnification. Colored arrows denote areas of co-localization of staining for activated (phospho-)-ERK in areas with prevalent uPAR and α3 integrin staining (brown stain). (B) Four color immunofluorescence analysis of uPAR, α3 integrin and phospho-ERK co-localization. Tumor sections were co-incubated with the primary antibodies goat anti-human uPAR (1:10 dilution), rabbit anti-phosphorylated ERK-1/2 (Thr202/Tyr204) (1:25 dilution), and mouse anti-α3 integrin (P1B5) (1:25 dilution) in blocking solution as described in Experimental Procedures, followed by fluophor-conjugated secondary antibodies (donkey anti-Goat IgG-Alexa Fluor-546, donkey anti-Rabbit IgG-Alexa Fluor-488, donkey anti-Mouse IgG-Alex Fluora-647, all from Invitrogen). Slides were mounted in DAPI-containing mounting medium. All stained slides were examined with an Olympus IX-80 with DSU spinning disk confocal system equipped with Hamamatsu EM-CCD camera ImagEM and the following 4 set of emission and excitation filters: for DAPI, D350/50x and ET455/50m (blue); for Alexa Fluor-488, 490/20x and ET525/36m(green); for Alexa Fluor-546, 555/25x and ET605/52m (red); and for Alexa Fluor-647 (magenta), 645/30x and ET705/72m. Image acquisition and normalization were all via the controlling software Slidebook 4.1 (Olympus). No additional image manipulations were performed. (C) Quantitation of active ERK (nuclear phospho-ERK) staining. Positive staining was evaluated by enumerating nuclei staining positively for nuclear phospho-ERK in a minimum of 85 distinct high powered tumor fields, and counting a minimum of 10,000 cells. ANOVA analysis demonstrates *p<.0001.
cDNA Microarray Analysis and Validation by qPCR and Immunohistochemistry
To examine additional changes in gene expression that result from uPAR/α3β1 integrin interaction, gene expression profiles of SCC25-uPAR-KD and SCC25-uPAR+ cells were compared using cDNA microarray analysis. An array containing oligonucleotides corresponding to 12,500 known human genes was used to analyze changes in gene expression. Statistical analysis of triplicate microarray data yielded 148 genes that were greater than 2-fold differentially expressed between SCC25-uPAR+ and SCC25-uPAR-KD (p<.05) including 35 downregulated and 113 upregulated genes [Suppl. Tables 1 and 2]. Genes were annotated and biological processes were analyzed using the Functional Annotation Clustering Tool of DAVID, that organizes redundant annotation terms into a clustered format [30]. This simplified annotation condenses the data into modules, enabling a focus on biological processes [Table 1]. A number of biological processes were altered in SCC25-uPAR+ cells, including developmental processes, inflammation, proliferation, and adhesion.
Table 1.
Functional Annotation Clusters Identified by the DAVID Functional Annotation Clustering Tool1
| Annotation Cluster | Enrichment Score | Functional Annotation | # of genes |
|---|---|---|---|
| 1 | 12.34 | Extracellular Region | 47 |
| Secreted | 43 | ||
| 2 | 10.10 | Signal | 65 |
| Secreted | 43 | ||
| Signal Peptide | 63 | ||
| Glycoprotein | 64 | ||
| 3 | 8.46 | Developmental Process | 66 |
| Anatomical Structure Development | 50 | ||
| 4 | 6.61 | Extracellular Region Part | 32 |
| Proteinaceous Extacellular Matrix | 15 | ||
| Extracellular Matrix | 15 | ||
| 5 | 6.24 | Cell Proliferation | 27 |
| 6 | 5.77 | Response To External Stimulus | 25 |
| Response To Wounding | 19 | ||
| Response To Stress | 29 | ||
| 7 | 5.66 | Tissue Development | 16 |
| 8 | 5.19 | Receptor Binding | 23 |
| Cytokine Activity | 13 | ||
| 9 | 5.07 | Positive Regulation of Biol. Process | 29 |
| 10 | 4.51 | Locomotory Behavior | 12 |
| 11 | 4.47 | Neg. Regulation of Biol. Process | 30 |
| 12 | 4.0 | Neg. Regulation of Biol. Process | 30 |
| Cellular Developmental Process | 38 | ||
| Cell-Cell Signaling | 17 | ||
| Cell Death | 21 | ||
| Apoptosis | 20 | ||
| 13 | 3.98 | Cell Adhesion | 21 |
| 14 | 3.94 | Cytokine Activity | 13 |
| Inflammatory Response | 13 | ||
| 15 | 2.56 | Epidermolysis bullosa | 4 |
| 16 | 2.53 | Growth Factor Binding | 4 |
| Regulation of Cell Growth | 8 | ||
| 17 | 2.48 | Cell Motility | 13 |
| 18 | 2.16 | Leukocyte Migration | 4 |
| Cell Migration | 8 | ||
| 19 | 2.14 | Regulation of Prog. Through Cell Cycle | 14 |
| Regulation of Cell Cycle | 14 | ||
| Cell Cycle Process | 16 | ||
| 20 | 2.06 | EGF-like Region | 12 |
Genes from Tables 1 and 2 were analyzed using the DAVID Functional Annotation Clustering Tool (http://david.abcc.ncifcrf.gov). The top 20 annotation clusters are shown.
Several candidate genes with known relevance to oral cancer progression were validated using real time RT-PCR and immunohistochemical analysis of murine tumors generated from orthotopic injection of SCC25-uPAR+ or SCC25-uPAR-KD cells. For example, a significant increase in expression of VEGF-C is demonstrated in SCC25-uPAR+ cells (Fig. 4A), consistent with the enhanced vascularity observed in SCC25-uPAR+ tumors (Fig. 2). This is supported by qPCR analysis of VEGF-C mRNA expression levels, showing a 5-fold increase in SCC25-uPAR+ relative to SCC25-uPAR-KD cells (Fig. 4A). Similarly, qPCR analysis of a distinct uPAR-overexpressing cell line, SCC1-uPAR+, exhibited a similar 2.9-fold increase in VEGF-C mRNA relative to vector controls (not shown). As we have previously demonstrated that uPAR/α3β1 integrin interaction initiates a Src/MEK/ERK-dependent signaling pathway resulting in transcriptional activation [20], control experiments were performed to evaluate the role of uPAR/α3β1 integrin interaction in VEGF-C induction. Cells treated with microsphere-immobilized integrin antibodies in the presence of the Src inhibitor PP2 showed a 4.4-fold reduction in VEGF-C mRNA relative to vehicle (DMSO)-treated controls. Similar results were obtained by pre-incubation with peptide α325, previously demonstrated to block uPAR/α3β1 binding [20, 48], leading to a 2.3-fold decrease in VEGF-C mRNA relative to cells treated with a scrambled peptide control. This result is supported by further quantitation of serial tumor sections stained for active ERK (nuclear phospho-ERK) or VEGF-C, which showed that 66% of cells with strong positive nuclear phospho-ERK also exhibited strong positive VEGF-C staining (not shown).
Figure 4. Validation of cDNA microarray results using immunohistochemical analysis of murine tongue tumors and qPCR.
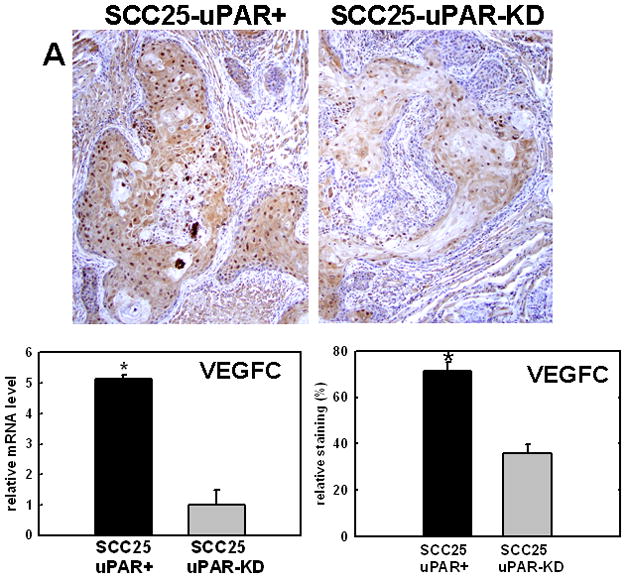
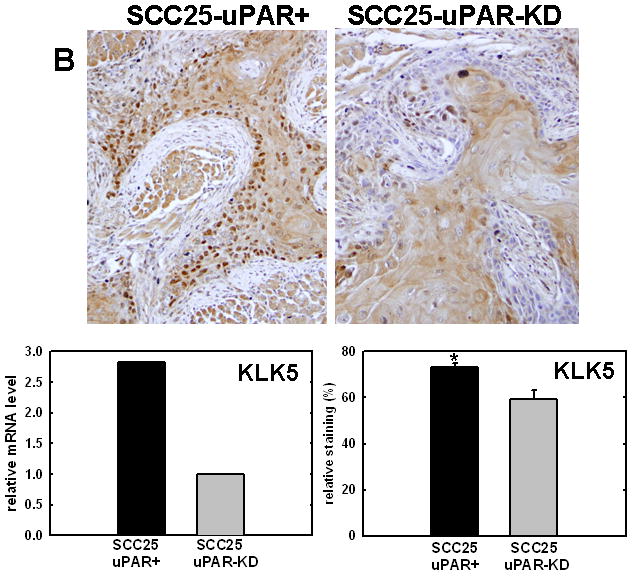
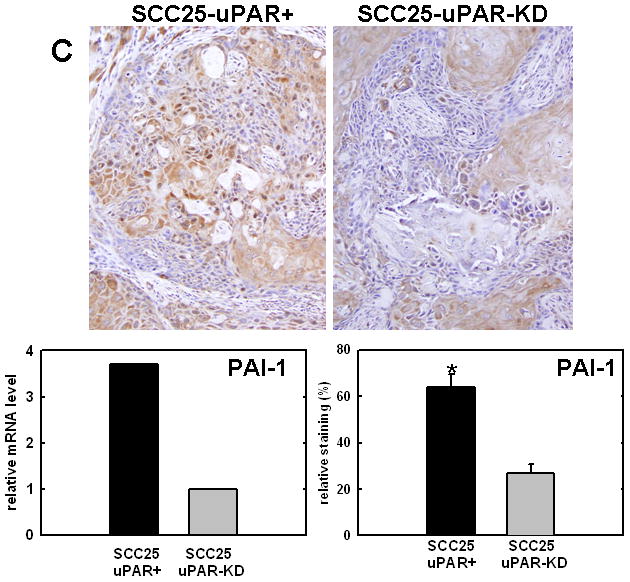
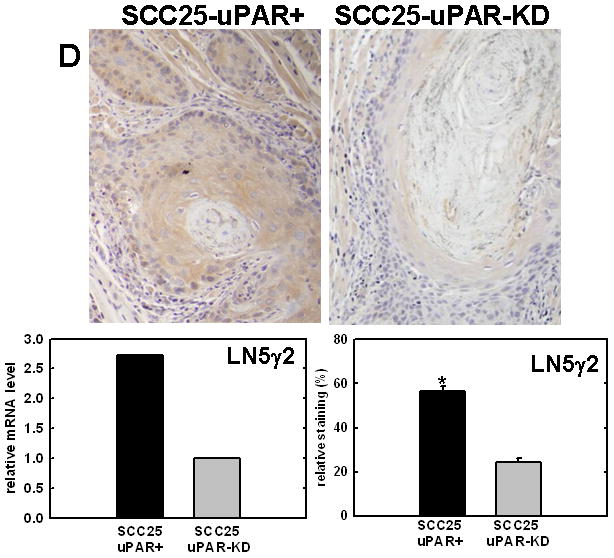
Representative sections from SCC25-uPAR+ or SCC25-uPAR-KD tumors were immunostained with (A) anti-VEGF-C (1:20 dilution), (B) anti-kallikrein-5 (1:25 dilution), (C) anti-PAI-1 (1:20 dilution) or (D) anti-laminin-γ2 chain (1:20 dilution) followed by biotinylated secondary antibody as in Experimental Procedures. 100X magnification. Corresponding qPCR analyses of gene expression in SCC25-uPAR+ and SCC25-uPAR-KD cell lines (left) and quantitation of immunohistochemical staining in murine tumors (right) are shown in the bar graphs. For qPCR, relative expression levels were normalized to housekeeping gene PGK. Each bar depicts the mean of replicate values expressed as fold-difference in mRNA level relative to SCC25-uPAR-KD cells (designated as 1, grey bars). (black bar) - SCC25-uPAR+, (grey bar) – SCC25-uPAR-KD. For quantitation of tumor staining, 2500–6000 cells from 10 tumor sections were scored and results are presented as relative staining (% of total cell number scored).
In addition to uPA, other proteinases associated with tumor progression and/or invasion were also elevated in SCC25-uPAR+ cells. For example, the serine proteinase kallikrein 5 is overexpressed in SCC25-uPAR+ tumors (Fig. 4B), while the related enzyme kallikrein 3 (aka prostate specific antigen) was not detected [31]. Kallikreins have been widely implicated as cancer biomarkers as well as potential therapeutic targets in a number of malignancies [32–33]. The serpin plasminogen activator inhibitor-1 (PAI-1) was also elevated in SCC25-uPAR+ cells and tumors (Fig. 4C). In addition to blocking uPA activity, PAI-1 is known to stimulate tumor cell motility and survival [34–36]. Expression of a number of ECM proteins was upregulated in SCC25-uPAR+ cells, including LAMC2, encoding the γ2 chain of laminin-332 (aka laminin-5) (Fig. 4D). LAMC2 was increased 2.7-fold in SCC25-uPAR+ cells and 1.9-fold in SCC1-uPAR+ cells. This epithelial basement membrane component has been implicated as a promoter of tumor invasion and cell survival [37–38].
DISCUSSION
Squamous cell carcinoma of the oral cavity is one of the most common cancers worldwide, with a poor 5-year survival rate of approximately 50% due to local recurrence, regional or distant metastasis, and/or second primary tumors [5,39]. Late presentation combined with the lack of early detection markers and poor chemotherapeutic response leads to poor outcome of oral cancer patients. Thus, a more detailed understanding of factors that regulate tumor progression and metastasis may result in advances in diagnostics or therapeutics. Approximately 25–40% of oral cancers occur on the tongue [3] and increased rates of tongue squamous cell carcinoma have been observed in young adults (<45 years old) in both the US and Europe [4]. Patients with tongue cancer have poorer survival rates relative to patients with tumors on other anatomic subsites in the oral cavity [3,6,40]. This poor prognosis has been attributed to multiple factors including lack of early detection due to the relative inaccessibility of the tumor to inspection, as well as anatomic factors including proximity to bone and the rich lymphatic network of the tongue [3,5].
Numerous studies have utilized cDNA microarray analysis to profile patterns of gene expression in primary oral tumors relative to metastases or normal oral mucosa. Interestingly, principal components analysis of expression profiling data demonstrated a distinct separation of tongue from non-tongue specimens, indicative of site-specific gene expression in the oral cavity [8]. Enhanced expression of uPA was consistently observed in many studies [8,41–43] and uPA was listed as one of 25 genes that comprise an OSCC gene signature to distinguish tumor versus normal samples [8]. Similarly, in vivo selection of cell lines for enhanced metastatic propensity in mice, coupled with cDNA microarray analysis, identified uPAR as a key metastasis-related gene [44]. This is consistent with immunohistochemical analysis of primary oral cancers, showing a correlation between uPA or uPAR expression and both cancer invasion and regional lymph node metastasis [15]. Immunohistochemical localization of uPAR at the invasive front of OSCC lesions has also been reported, suggesting a role for uPAR in early cancer invasion [16]. This is supported by results of the current study, showing the development of poorly differentiated, aggressive infiltrative lesions with perineural and/or vascular invasion from uPAR-overexpressing cells. These features are well associated with aggressive clinical behavior and poor prognosis in humans [27–29]. In contrast, tumors seen in the uPAR-KD group showed histologic features associated with less aggressive tumors, including pushing borders of invasion and absence of perineural or vascular invasion. [27–29]. Although loss of uPAR expression has been previously associated with tumor dormancy [45], no evidence for dormancy was observed in the current study.
Interaction of uPAR with transmembrane integrins regulates cellular adhesion, invasion, and motility [19, 45–47], but transcriptional activation downstream of α3β1 integrin/uPAR complex formation has not been extensively evaluated. Association of uPAR with α3β1 integrin is via theα3 subunit β-propeller, in a region distinct from the matrix binding site [48]. Binding of uPAR and α3β1 integrin results in activation of a Src/MEK/ERK-dependent signaling pathway, leading to transcriptional upregulation of uPA expression [20]. Complex formation between α3β1 integrin and uPAR is necessary for transcriptional activation, as MAPK signaling is substantially attenuated by several blocking strategies that inhibit uPAR interaction with α3β1 integrin [20]. Furthermore α3β1 integrin engagement in cells in which uPAR levels were downregulated with siRNA failed to activate MAPK signaling and uPA transcription [20]. Data from the current study support these results and show statistically significant co-localization of uPAR and α3 integrin in foci also exhibiting ERK activation in both murine and human tongue tumors, supporting the hypothesis that uPAR/α3β1 association may also regulate gene expression in vivo.
Altered transcriptional profiles of SCC25-uPAR+ cells are also apparent from cDNA microarray analysis as well as immunohistochemical analysis of murine tumors. Many of the genes transcriptionally activated in uPAR-overexpressing cells, including VEGF-C, kallikrein-5, PAI-1, and laminin-γ2 chain, have been previously implicated in OSCC progression. For example, VEGF-C is a major modulator of lymphatic vessel density and microvessel density, is elevated in human oral tumors, and expression correlates with enhanced regional lymph node metastases of tongue tumors [49–53]. While staining for enhanced lymphatic endothelium was negative in the current study, it should be noted that lymph node metastases are also not present in our model, suggesting that upregulation of VEGFC may be a precursor to altered lymphangiogenesis and metastasis. As a recent pre-clinical study has shown that anti-vascular therapy targeting tumor microvessels with a small-molecule inhibitor of VEGFR-2 blocks tumor growth in vivo, these data suggest that vascular targeting of lymphatic and angiogenic vessels may represent an effective therapeutic strategy for oral SCC. Kallikrein-5 is a member of the human tissue kallikrein family of serine proteinases. Although the physiological roles of many kallikreins are not clearly defined, aberrant kallikrein expression patterns have been reported in many carcinomas and multiple kallikreins have been proposed as biomarkers in a number of malignancies [32,33,54]. Kallikrein activity has been linked to malignant behavior at multiple stages of tumor progression including proliferation, invasion, metastasis and angiogenesis [32,33,54,55]. Further, kallikrein-5, as well as the related proteases kallikrein-7, -8, and -10 are abundantly expressed in human OSCC and may be implicated in malignant progression [31]. PAI-1 is a serpin that blocks the activity of uPA, but also regulates adhesion and motility by virtue of its ability to bind vitronectin. Somewhat paradoxically, PAI-1 levels are highly elevated in many tumor types and correlate with poor prognosis [36]. Recent studies indicate that PAI-1 transcription is initiated in wounded keratinocytes as a component of the wound repair program [34]. PAI-1 stimulates adhesion and migration and blocks anoikis, suggesting a role as a pro-survival, pro-migratory factor. Elevated PAI-1 correlates with poor outcome (relapse-free survival) in patients with oral cavity cancer [56] and is a biomarker for early oncogenesis and invasion [57,58]. The ECM component laminin-γ2 chain also correlates with early invasive events in OSCC [58]. Laminin-γ2 is a subunit of laminin-332 (aka laminin-5), a major basement membrane laminin isoform [38]. Laminin-332 has been implicated in promotion of tumor cell adhesion, migration and invasion [38] as well as in activating PI3-kinase and RAC1 to induce pro-survival pathways [37]. Laminin-γ2 staining identified dysplastic epithelia in oral brush biopsies, suggesting the utility of evaluating laminin-γ2 expression as an initial diagnostic step [59]. This is supported by studies showing that laminin-γ2 expression may be an effective biomarker for early detection of invasive OSCC [60,61]. Aside from these validated examples, the majority of the gene products listed in Table 1 have not been previously associated with aggressive OSCC, indicating the potential efficacy of this strategy for identification of novel OSCC-related genes.
In summary, orthotopic murine models of OSCC have demonstrated efficacy for analysis of factors that modulate tumor progression and metastasis as well as for pre-clinical testing of potential therapeutics [23,24,31,39,62,63]. The current results together with available clinical data support a role for uPAR/α3β1 in progression of tongue tumors, indicating that inhibition of uPAR/α3β1 integrin interaction may represent a novel therapeutic approach to block Src/MEK/ERK signaling and the downstream functional consequences to OSCC progression. Furthermore, our results provide proof-of-concept that cDNA microarray analysis together with examination of orthotopic murine tumors can identify factors associated with aggressive human OSCC. It thereby follows that this approach may also provide a strategy for biomarker discovery to reveal novel diagnostic or prognostic indicators. As the vast majority of OSCC patients succumb due to complications of metastasis, identification of additional markers of aggressive disease is necessary to improve patient survival.
MATERIALS AND METHODS
Cell Lines
SCC25 cells, originally derived from OSCC of the human tongue, were the generous gift of Dr. James Rhinewald (Brigham & Women’s Hospital, Harvard Institutes of Medicine, Boston, MA). SCC25 cells are well-characterized with respect to molecular properties and display characteristics representative of those commonly found in human SCC25 [8]. Cells were routinely maintained in DMEM/Ham’s F-12 1:1 media containing 10% fetal calf serum and supplemented with 100 units/ml penicillin. An siRNA knockdown approach was used to generate cells with reduced levels of surface uPAR as previously described [20]. The paired oligonucleotides indicated below were annealed and ligated to BbsI-cut vector (psiRNAhH1neo from Invivogen) and transformed into HB101 competent cells. Target seq2 oligonucleotide 4A (5′-tcccaagccgttacctcgaatgcatttcaagagaatgcattcgaggtaacggctttt-3′) and target seq2 oligo-nucleotide 4B (5′-caaaaaaagccgttacctcgaatgcattctcttgaaatgcattcgaggtaacggctt-3′) DNA was isolated (Qiaprep spin miniprep kit; Qiagen), and the identities of the clones were confirmed by restriction digestion and sequencing with primer OL381 (sequencing primer oligonucleotide OL381, 5′-ccctaactgacacacattcc-3′). Selected clones were then grown in 500-ml cultures, and DNA isolations were done using a nuclease-free DNA isolation kit (Qiagen). SCC25 cells were transfected by electroporation using the human keratinocytenucleofector kit and device (Amaxa) following the recommended protocol. Briefly, the cells were cultured to ~65% confluence, trypsinized, and resuspended at a density of 500,000 cells/100 μl nucleofector solution. DNA (1.5 μg in less than 5 μl) was added to each aliquot of cells and gently mixed, and each aliquot was electroporated. After 24 h growth under nonselective conditions, the medium was replaced with medium containing 850 μg/ml G418. During outgrowth, the cells were subcultured before reaching 70% confluence, and the selection medium was replaced every 2 days. Colonies were picked at the 100cell stage and grown until they were numerous enough to test for knockdown of uPAR by flow cytometry. Loss of uPAR surface expression and purity of clonal cell lines was assessed by FACS (see below).
To generate cells that overexpress uPAR (SCC25-uPAR+), uPAR was cloned into the expression vector pcDNA 3.1(+) by rtPCR with primer 1: gcgaagcttgggatgggtcacccgccgctg and primer 2: gaattccggtcaggtccagaggagagt. The cDNA was gel-purified using a Qiaquick gel extraction kit (Qiagen), and the cDNA was cut with EcoRI and Hind III and ligated into EcoRI and Hind III – cut vector. The DNA was used to transform HB101 host cells. Cells carrying the pcDNA 3.1-uPAR plasmid were grown in LB medium containing 100 μg/ml ampicillin and plasmid DNA was isolated using a Qiagen DNA Isolation kit. After the uPAR sequence was verified, the DNA was used as template with the following primer sets: PCR Rx 1A: T7 promotor primer + primer 3: cttgtcatcgtcgtccttgtagtcgccccaagaggctgggacgca; and Rx 1B: BGH reverse primer + primer 4: gactacaaggacgacgatgacaagctgcggtgcatgcagtgtaag. The gel-purified cDNA products from Rx’s 1A and 1B were used as template for the final PCR Rx 2 using T7 and BGH Rev primers. The resulting uPAR/flag cDNA was cut with EcoRI and Hind III and ligated into EcoRI and Hind III – cut pcDNA 3.1(+) vector. The transformation and DNA purification was carried out as above, and the DNA was used to transfect SCC25 cells. The resulting clonal cell lines were routinely maintained in the medium described above supplemented with G418 and were assessed for expression of uPAR by FACS (see below). Changes in uPAR expression were also confirmed by quantitative RT-PCR (below). Alternatively, lentiviral transduction was used to generate UM-SCC1 cells (generous gift of Dr. Ernst Lengyel, University of Chicago) that overexpress uPAR (designated SCC1-uPAR+). Briefly, a retroviral uPAR expression vector was first created by subcloning into the EcoRI and SalI sites in pBabe-puro system (Plasmid 1764 from Addgene, Inc.) with a full length human uPAR from human cDNA clone product SC319092 (OriGene Technologies, Inc.). Retroviral particles were made by liposome-based transfection in HEK293 cell-derived retroviral packaging cell line, Phoenix Amphotropic (originally created in Dr Garry Nolan’s lab in Stanford University) and sublines expressing full length of uPAR or harboring empty retroviral vector with puromycin resistance gene only were correspondingly created by spinfection and puromycin selection (2.5ug/ml). Quantitative real-time PCR analysis demonstrated a 46-fold increase in uPAR mRNA relative to parental UM-SCC1 cells with control viral vector.
Antibodies
For immunohistochemistry, the following antibodies were purchased: mouse anti-human cytokeratins clone AE1/AE3 (DAKO M3515; 1:25 dilution); mouse anti-human PCNA clone PC10 (DAKO M0879; 1:12.5 dilution); mouse anti-human vimentin clone V9 (DAKO M0725; 1:25 dilution); mouse anti-human uPAR clone 3/B10 (American Diagnostica 3936; 1:25 dilution); mouse anti-human integrin α3 clone P1B5 (Chemicon MAB 1952; 1:25 dilution); rabbit anti-human phospho-p44/42 MAP Kinase (Thr202/Tyr204, pERK1/2) (Cell Signalling Technology #9101S; 1:25 dilution); rabbit anti-human VEGF-C clone H-190 (Santa Cruz #sc9047), rabbit anti-human kallikrein-5 (Abcam #ab28565, 1:20 dilution), anti-plasminogen activator inhibitor-1 (PAI-1) (Santa Cruz Biotechnology sc-8979, 1:20 dilution), and anti-laminin-5-γ2 chain (Dakocytomation M7262 ab4G1, 1:20 dilution). Mouse anti-human E-cadherin clone HECD-1 (Zymed 13-1700), mouse anti-human integrin α3 and mouse anti-human uPAR clone 13.1 (American Diagnostica #3937) were purchased for use in flow cytometric analyses.
Flow Cytometry
Surface expression of uPAR, integrin α3, and E-cadherin were determined using flow cytometry as previously described [11,20]. Briefly, cells were trypsinized and 1.8 × 105 cells were incubated in 100 μl of medium containing specific antibodies against the respective protein for 45 min at room temperature. Antibodies were used at the following dilutions: anti-uPAR (American Diagnostica #3937; 1:100), anti integrin α3 (Chemicon MAb #1952; 1:100), anti-E-cadherin (Zymed HECD-1, #13-1700; 1:100). The cells were then washed twice with PBS and incubated with the corresponding fluorescein isothiocyanate-conjugated secondary antibody (Molecular Probes #GM488, Eugene, OR; 1:500 dilution) for 30 min in the dark at room temperature. The cells were then washed twice with PBS and resuspended in medium for fluorescence analysis on an Epics XL-MCL flow cytometer (Beckman Coulter, Hialeah, FL). Control experiments contained only the appropriate secondary antibody.
Analysis of Invasion
Invasive activity was quantified using a Boyden chamber (8-μm pore size) coated with Matrigel(10 μg for 1 h at room temperature) as described earlier [20]. The cells (2 × 105) were added to the upper chamber in 500 μl of serum-free medium containing the selection drug. Following 24–48 h of incubation at 37 °C, the non-invading cells were removed from the upper chamber with a cotton swab, the filters were fixed and stained with Diff-Quik stain, and the invading cells adherent to the underside of the filter were enumerated using an ocular micrometerand counting a minimum of 10 high powered fields. The data are expressed as relative invasion over control (number of cells/field). Some experiments contained antibodies directed against uPAR (American Diagnostica 3936, 20 ug/ml), α3 integrin (Millipore MAB1952P, 20 ug/ml) or control IgG (Chemicon, 20 ug/ml) or the small molecule MEK inhibitor PD98059 (10 uM) or DMSO control, added at the time of cell seeding.
Orthotopic Sublingual Injections
Murine tongue tumors were generated as previously described [23]. At least 8 mice per group were injected, and the experiment was repeated three times over the course of two years for a total of 24 mice/group. Briefly, six week old female athymic nu/nu mice were purchased from Charles River (Wilmington, MA) and housed in a specific pathogen-free animal facility at the NIDCR (Bethesda, MD). All animal protocols were performed with approval of the NIDCR Animal Care and Use Committee. Cells were cultured as described above, collected using trypisn and resuspended in PBS. Mice were anesthetized using 2.5% isofluorane. A 1-ml syringe (Becton Dickenson) with a twenty-five gauge needle was used to inject 30 μl of cells (pooled clones of SCC25-uPAR-KD or SCC25-uPAR+; 6.25X 106, in sterile PBS) into the lateral border of the tongue just anterior to the junction of the anterior 2/3 and posterior 1/3 of the tongue. The tongue of the anesthetized mouse was held with a small tooth forceps in the median raphe of the tongue while the cells were injected just under the mucosa of the tongue in an anteropostero direction. Mice were examined every day for the development of tongue tumors and weight loss. At approximately 9 weeks mice were sacrificed using CO2 when they began to show adverse signs of disease including weight loss. The tongue and cervical lymph nodes were dissected from each mouse (n=24 for each group), fixed in freshly prepared 4% paraformaldehyde (4°C), paraffin embedded, sectioned (4 μm), and stained with H&E. For each tumor in each group, multiple sections were prepared and that with the greatest cross-sectional tumor area, as determined using ImageJ software, was used for histopathological analysis. All tumors were analyzed by a board-certified human pathologist (S.F.) and a veterinary pathologist (A.E.). Tumor invasiveness was subjectively graded on a scale of 1–5, based loosely on the number of distinct cross-sections of one tumor on one slide and which ranged from 1 to approximately 50. Inflammation was also graded on a scale from 1–5, with 1 indicating absence of inflammation and 5 indicating marked diffuse inflammation. Tumors were placed in increasing order based on cross-sectional area, invasiveness (1–5) and inflammation (1–5), and an ANOVA on ranks was performed for each criterium. The mitotic index was determined by counting a minimum of 1000 cells/slide and scoring for the presence or absence of visible mitoses. Data were analyzed using a non-parametric ANOVA (Mann-Whitney U test; Systat 11.0, Systat Inc., CA, USA).
Immunohistochemical Analysis
Immunohistochemical analysis was performed on paraformaldehyde fixed, paraffin-embedded sections (4 μm) from all murine tongue tumors in each group and the experiment was repeated in triplicate, as indicated above (total n=24 for each group) [31]. Images that most clearly represent the average group phenotype are shown. Quantitation of uPAR immunohistochemical staining shows staining of 50.2 +/− 3.47% of cells in SCC25-uPAR-KD tumors and 71.1 +/− 2.2% in SCC25-uPAR+ tumors (p<.005, ANOVA). Quantitation of staining for active ERK (nuclear phospho-ERK) was performed by scoring a minimum of 10,000 cells/group from at least 85 high powered fields. Analysis of human tongue tumors was performed using microarrayed human tumor tissue (Biomax #OR601) containing 29 cases of tongue tumor of varying grade. Sections were de-paraffinized with Xylene and rehydrated in a series of ethanol washes. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide in methanol for 30 min. Antigen retrieval was enhanced by microwaving in 10mM sodium citrate pH 6.0 (or 0.01% citric acid for cytokeratins AE1/AE3). Non-specific binding was blocked with 3% normal horse serum in PBS for 30 min. Sections were incubated for 1 hour at room temperature with primary antibody in 1% BSA in PBS. Staining was detected using an avidin-biotin horse radish peroxidase system (Vectastain Universal Elite ABC kit, #PK-6200, Vector Laboratories, Burlingame, CA, USA), with positive cells staining brown using diaminobenzidine chromogen and hydrogen peroxide as substrate (Liquid DAB substrate #HK153-5K from BioGenex, San Ramon, CA, USA). For quantitation of tumor staining, 2500–6000 cells from 10 tumor sections were scored and results are presented as relative staining (% of total cell number scored). In studies to evaluate co-localization of uPAR, α3 integrin and pERK, serial sections were used. To quantify human TMA staining, a minimum of 1600 cells from 22 tumors were scored as positive or negative. The percentages for positive staining results were then used to evaluate a potential relationship between antigen (uPAR, pERK or α3 integrin) and tumor grade via a Spearman’s rank correlation analysis (p<0.05; Systat 11.0, Systat Inc., CA, USA). Following from the biochemical results suggesting that pERK staining is positively linked to α3 integrin and that the positive influence of α3 integrin on pERK staining is accentuated by uPAR [19,20], multiple and bivariate regression analysis were used to evaluate the potential contribution of α3 integrin staining and uPAR/α3 integrin interaction to pERK positivity (p<0.05; Systat 11.0).
Immunofluorescence Analysis
Paraffin sections (5μm thick) were de-paraffinized in xylene and rehydrated in decreasing concentration serial ethanol solutions and re-distilled water. Antigen retrieval was performed by heating in citrate buffer (pH6.0) for 20 minutes. Blocking for non-specific binding was done in 10% normal donkey serum at room temperature for 1 hour. Then the primary antibodies goat anti-human uPAR (399G) from American Diagnostica at 1:10 dilution, rabbit anti-phosphorylated ERK-1/2 (Thr202/Tyr204) (9101) from Cell Signaling at 1:25 dilution, and mouse anti-α3 integrin (P1B5) from Millipore at 1:25 dilution were co-incubated in blocking solution at 4C overnight. PBS containing 0.5% Tween-20 (PBS-T) was used to rinse slides for 5 minutes and repeated 3 times. Fluophor conjugated secondary antibodies (donkey anti-Goat IgG-Alexa Fluor-546, donkey anti-Rabbit IgG-Alexa Fluor-488, donkey anti-Mouse IgG-Alex Fluora-647, all from Invitrogen) were incubated together in a dilution of 1:400 at room temperature for 30 minutes. After appropriate wash in PBS-T and air drying, slides were mounted with ProLong Gold antifade medium containing DAPI (4′,6-diamidino-2-phenylindole). All stained slides were examined with an Olympus IX-80 with DSU spinning disk confocal system equipped with Hamamatsu EM-CCD camera ImagEM and the following 4 set of emission and excitation filters: for DAPI, D350/50x and ET455/50m (blue); for Alexa Fluor-488, 490/20x and ET525/36m(green); for Alexa Fluor-546, 555/25x and ET605/52m (red); and for Alexa Fluor-647 (magenta), 645/30x and ET705/72m. Image acquisition and normalization were all via the controlling software Slidebook 4.1 (Olympus). No addition image manipulations were performed.
cDNA Microarray
All DNA microarray gene expression studies used human oligonucleotide arrays custom-printed by a dedicated core facility within the Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center (Dr. David Kelly, Omaha, NE). Arrays were constructed from a set of 12,140 sense oligonucleotide (60-mers) probes designed for each human target gene by Compugen Inc. (Rockville, MD) and manufactured by Sigma-Genosys, Inc. (The Woodlands. TX). Individual arrays contain 12,288 spot features, including 12,107 different genes, 28 replications of GAPDH, and negative controls. In brief, approximately 40 μg of experimental and reference RNA samples (i.e., from SCC25-uPAR+ vs SCC-25 uPAR-KD) were reverse-transcribed with anchored oligo-dT primer. cDNA was labeled with either Cy3 or Cy5 monofunctional NHS-ester (Amersham Pharmacia) and dye-swap experiments were performed. Labeled cDNA in hybridization solution was applied to DNA microarrays and incubated at 42°C for 16–20 h. After hybridization, microarray slides were washed, dried, and scanned immediately with a ScanArray 4000 confocal laser system (Perkin-Elmer). Fluorescent intensities were extracted using the QuantArray software package (Perkin-Elmer). The experiment was repeated three times.
Statistical Analysis of Microarray Data
Analysis of microarray gene expression data, accumulated from three independent experiments, was performed using the limma package [64], available through the Bioconductor project [65] for use with R statistical software [66]. Data quality was examined by looking for spatial effects across each microarray with image plots of raw log 2 ratios and examining MA-plots of the M-values (log 2 ratios) versus A-values (average log 2 intensities). Both views indicated no large-scale systematic effects indicative of technical problems with the arrays. Background correction was carried out using a normal plus exponential convolution model [65] and print-tip (within-array) loess normalization was used to reduce systematic dye-related bias in the intensity values [67]. After pre-processing, the analysis of differential gene expression was based on moderated t-statistics on the replicated log 2 ratios for each gene. Statistical significance was assessed using an Empirical Bayes approach [64]. Adjustment for multiple comparisons according to the false discovery rate method of Benjamini and Hochberg [68] was performed, and genes with adjusted p-values less than 0.05 were selected as differentially expressed. Log 2 ratios were transformed back to fold change values for interpretation purposes in this report (Tables 1 and 2). Genes were annotated and biological processes were analyzed by the Database for Annotation, Visualization and Integrated Discovery (DAVID, Table 3; http://david.abcc.ncifcrf.gov). The Functional Annotation Clustering Tool organizes redundant annotation terms into a clustered format, enabling the comprehensive pooling of all related genes associated with a functional annotation cluster consisting of many related terms [30].
Quantitative Real Time PCR (qPCR) Analysis
Quantitative real time PCR was used to monitor the changes in mRNA expression level in transfected SCC cells. Total RNA was extracted with Trizol Reagent (Invitrogen). Reverse transcription was performed with 10 ug of the total RNA from each specimen using SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer’s protocol. The cDNA products were then diluted 1:10 and 5 ul of each were used for PCR templates. The following primer sequences were used at final concentrations of 167 nM for each: VEGF-C primers forward 5′ – GTG TCC AGT GTA GAT GAA CTC – 3′ and reverse 5′-ATC TGT AGA CGG ACA CAC ATG – 3′; for endogenous control gene phosphoglycerate kinase (PGK) forward, 5′-GGG CTG CAT CAC CAT CAT AGG-3′ and reverse 5′-GAG AGC ATC CAC CCC AGG AGG-3′; keratin 13 primers forward 5′ - AAC GTG GAG ATG GAT GCC A - 3′ and reverse 5′ - TGG CGT GGA ACC ATT CCT - 3′; keratin 19 primers forward: 5′-TGA GTG ACA TGC GAA GCC AAT AT - 3′ and reverse: 5′ - GCG ACC TCC CGG TTC AAT - 3′; kallikrein-5 primers forward 5′ – GCA GGT AGA GAC TCC TGC CA -3′ and reverse 5′ – CAC AAG GGT AAT CTC CCC AG – 3′; PAI-1 forward 5′ – CTC CTG GTT CTG CCC AAG TT – 3′ and reverse 5′-TCG TGA AGT CAG CCT GAA AC – 3′; laminin-5-g2 chain forward 5′ – AGG CTG TCC AAC GAA ATG GG – 3′ and reverse 5′ – GGA GCT GTG ATC CGT AGA CCA – 3′. DNA oligos were custom synthesized (integrated DNA Technologies).
Real-time PCR was performed with SYBR green Master Mix (Aplied Biosystems, Foster City, CA). PCR cycling conditions were 95 °C for 10 min followed by 40 cycles of 94 °C for 15s and 60 °C for 1 min. All reactions were carried out on an iCycleriQ real-time PCR detection system (Bio-Rad Laboratories). Each sample was in quadruplicate for each PCR measurement. Melting curves were checked to ensure specificity. Quantification of mRNA expression was calculated using the delta delta Ct method with the endogenous housekeeping gene PGK level as normalizer and control sample calibrator.
In control experiments, cells were serum starved in the presence of the Src inhibitor PP2 (10 uM, Calbiochem, Temecula, CA), DMSO vehicle control, the uPAR/α3β1 blocking peptide α325 (160 mM, Accurate Chemical & Scientific, Westbury, NY), or scrambled peptide control (160 mM) for 3 hr prior to incubation with microsphere-immobilized β1 integrin antibodies (β1-beads) for 3 hr as previously described [20]. RNA was extracted and quantitative real-time PCR performed using VEGF-C specific primers as described above.
Supplementary Material
Supplemental Figure 1. Characterization of SCC25-uPAR-KD and SCC25-uPAR+ cells. (A) Relative surface expression levels of uPAR were measured by flow cytometric analysis using primary antibody 3937, as described in Experimental Procedures. Mean fluorescence index values are shown. (vec) – SCC25 parental cells expressing empty vector; uPAR-KD – SCC25 cells with downregulated uPAR; uPAR+ - SCC25 cells that overexpress uPAR. (B) Invasive activity was determined by seeding cells in the top of a Boyden chamber (8 um pore size, coated with Matrigel). After 24 h, cells in the upper chamber were removed with a cotton swab and invading cells adherent to the bottom chamber were fixed and stained with Diff-quik prior to enumeration. Bars, mean +/− SEM. *p<.05. (C) Invasive activity was determined as in (B) above in the presence of function blocking antibodies directed against uPAR or a3 integrin (20 ug/ml), the MEK inhibitor PD98059 (10 uM) or DMSO (vehicle control). Bars, mean +/− SEM *p<.05.
Supplemental Figure 2. Analysis of tumor parameters. (A) Loss of differentiation of SCC25-uPAR+ cells, observed from analysis of H&E stained sections, was confirmed using qPCR analysis of keratin 19 (K19, black bar) and keratin 13 (K13, white bar) expression. Relative expression levels were normalized to housekeeping gene PGK. Each bar depicts the mean +/− SD of replicate values expressed as fold-difference in keratin mRNA level relative to SCC25-uPAR-KD cells (designated as 1). *p<.05 relative to corresponding gene in SCC25-uPAR+. (B) Quantitation of relative tumor area and level of inflammation as described in Experimental Procedures. Bars, mean +/− SEM. (open bar) – SCC25-vec, (grey bar) – SCC25-uPAR-KD, (black bar) – SCC25-uPAR+.
Supplemental Figure 3. Quantitation of blood vessel density and perineural invasion. (A) Blood vessel density and (B) perineural invasion was quantified from H&E stained sections by counting a minimum of 140 high powered fields from three biologic replicate experiments. Bars, mean +/− SEM. (black bar) – SCC25-uPAR+; (grey bar) – SCC25-uPAR-KD.
LITERATURE CITED
- 1.Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA: A Cancer Journal for Clinicians. 1999:33–64. doi: 10.3322/canjclin.49.1.33. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics. CA: A Cancer Journal for Clinicians. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 3.Gorsky M, Epstein BJ, Oakley C, Le ND, Hay J, Stevenson-Moore P. Carcinoma of the tongue: a case series analysis of clinical presentation, risk factors, staging and outcome. Oral Surg Oral Med Oral Pathol Oral Radio Endod. 2004;98:546–52. doi: 10.1016/j.tripleo.2003.12.041. [DOI] [PubMed] [Google Scholar]
- 4.Shiboski CH, Schmidt BL, Jordan RCK. Tongue and tonsil carcinoma: increasing trends in the US population ages 20–44 years. Cancer. 2005;103:1843–9. doi: 10.1002/cncr.20998. [DOI] [PubMed] [Google Scholar]
- 5.Sano D, Myers JN. Metastasis of squamous cell carcinoma of the oral tongue. Cancer Metastasis Rev. 2007;26:645–62. doi: 10.1007/s10555-007-9082-y. [DOI] [PubMed] [Google Scholar]
- 6.Zhen W, Karnell LH, Hoffman HT, Funk GF, Buatti JM, Menck HR. The national cancer data base report on squamous cell carcinoma of the base of the tongue. Head and Neck. 2004;26:660–674. doi: 10.1002/hed.20064. [DOI] [PubMed] [Google Scholar]
- 7.Macfarlane Gl, Boyle P, Evstifeeva TV, Robertson C, Scully C. Rising trends of oral cancer mortality among males worldwide: the return of an old public health problem. Cancer Causes Control. 1994;5:2259–265. doi: 10.1007/BF01830246. [DOI] [PubMed] [Google Scholar]
- 8.Ziober AF, Patel KR, Alawi F, Gimotty Ph, Weber RS, Feldman MM, Chalian AA, Weinstein GS, Hunt J, Ziober BL. Identification of a gene signature for rapid screening of oral squamous cell carcinoma. Clin Can Res. 2006;12:5960–71. doi: 10.1158/1078-0432.CCR-06-0535. [DOI] [PubMed] [Google Scholar]
- 9.Nagata M, Fujita H, Ida H, Hoshina H, Inoue T, Seki Y, Ohnishi M, Ohyama T, Shingaki S, Kaji M, et al. Identificaiton of potential biomarkers of lymph node metastasis in oral squamous cell carcinoma by cDNA microarray analysis. Int J Cancer. 2003;106:683–689. doi: 10.1002/ijc.11283. [DOI] [PubMed] [Google Scholar]
- 10.Shi Z, Stack MS. Urinary-type plasminogen activator (uPA) and its receptor (uPAR) in squamous cell carcinoma of the oral cavity. Biochem J. 2007;407:153–9. doi: 10.1042/BJ20071037. [DOI] [PubMed] [Google Scholar]
- 11.Shi Z, Stack MS. Molecules of cell adhesion and extracellular matrix proteolysis in oral squamous cell carcinoma. Histol Histopathol. 2010 doi: 10.14670/HH-25.917. in press. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Guo F, Wei H, Dong J, Wu J. Expression of urokinase-type plasminogen activator receptor is correlated with metastases of lingual squamous cell carcinoma. Br J Oral Maxillofacial Surg. 2006;44:515–519. doi: 10.1016/j.bjoms.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Curino A, Patel V, Nielsen BS, Iskander AJ, Ensley JF, Yoo GH, Holsiger FC, Myers JN, El-Naggar A, Kellman RM, Shillitoe EJ, Molinolo AA, Gutkind JS, Bugge TH. Detection of plasminogen activators in oral cancer by laser capture microdissection combined with zymography. Oral Oncol. 2004;40:1026–1032. doi: 10.1016/j.oraloncology.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Clayman G, Wang SW, Nicolson GL, el-Naggar A, Mazar A, Henkin J, Blasi F, Goepfert H, Boyd DD. Regulation of urokinase-type plasminogen activator expression in squamous cell carcinoma of the oral cavity. Int J Cancer. 1993;54:73–80. doi: 10.1002/ijc.2910540113. [DOI] [PubMed] [Google Scholar]
- 15.Nozaki S, Endo Y, Kawashiri S, Nakagawa K, Yamamoto E, Yonemura Y, Sasaki T. Immunohistochemical localization of a urokinase-type plasminogen activator system in squamous cell carcinoma of the oral cavity: association with mode of invasion and lymph node metastasis. Oral Oncol. 1998;34:58–62. doi: 10.1016/s1368-8375(97)00028-6. [DOI] [PubMed] [Google Scholar]
- 16.Lindberg P, Larsson A, Nielsen BS. Expression of plasminogen activator inhibitor-1, urokinase receptor and laminin gamma 2 chain is an early coordinated event in incipient oral squamous cell carcinoma. Int J Cancer. 2006;118:2948–2956. doi: 10.1002/ijc.21568. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda T, Sakata Y, Kitamura K, Morita M, Ishida T. Localization of plasminogen activators and their inhibitor in squamous cell carcinomas of the head nad neck. Head Neck. 1997;19:611–6. doi: 10.1002/(sici)1097-0347(199710)19:7<611::aid-hed8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 18.Hundsdorfer B, Zeilhofer HF, Bock KP, Dettmar P, Schmitt M, Kolk A, Pautke C, Horch HH. Tumor associated urokinase type plasminogen activator and its inhibitor PAI1 in normal and neoplastic tissues of patients with squamous cell cancer of the oral cavity – clinical relevance and prognostic value. J Craniomaxillofac Surg. 2005;33:191–6. doi: 10.1016/j.jcms.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh S, Brown R, Jones JCR, Ellerbroek SM, Stack MS. Urinary-type plasminogen activator (uPA) expression and uPA receptor localization are regulated by α3β1 integrin in oral keratinocytes. J Biol Chem. 2000;275:23869–76. doi: 10.1074/jbc.M000935200. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh S, Johnson JJ, Sen R, Mukhopadhyay S, Liu Y, Zhang F, Wei Y, Chapman HA, Stack MS. Functional relevance of urinary-type plasminogen activator receptor-α3β1 integrin association in proteinase regulatory pathways. J Biol Chem. 2006;281:13021–29. doi: 10.1074/jbc.M508526200. [DOI] [PubMed] [Google Scholar]
- 21.Fidler IJ. Rationale and methods for the use of nude mice to study the biology and therapy of human cancer metastasis. Cancer Metastasis Rev. 1986;5:29–49. doi: 10.1007/BF00049529. [DOI] [PubMed] [Google Scholar]
- 22.Morifuji M, Taniguchi S, Sakai H, Nakabeppu Y, Ohisi M. Differential expression of cytokeratin after orthotopic implantation of newly established human tongue cancer cell lines of defined metastatic ability. Am J Path. 2000;156:1317–26. doi: 10.1016/S0002-9440(10)65002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myers JN, Holsinger FC, Jasser SA, Bekele BN, Fidler IJ. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clinical Cancer Research. 2002;8:293–298. [PubMed] [Google Scholar]
- 24.Yazici YD, Kim S, Jasser SA, Wang Z, Carter KB, Bucana CD, Myers JN. Antivascular therapy of oral tongue squamous cell carcinoma with PTK787. The Laryngoscope. 2005;115:2249–55. doi: 10.1097/01.mlg.0000183861.53765.77. [DOI] [PubMed] [Google Scholar]
- 25.Al Moustafa Ae, Alaoui-Jamali MA, Batist G, Hernandez-Perez M, Serruya C, Alpert L, Black MJ, Sladek R, Foulkes WD. Identification of genes associated with head and neck carcinogenesis by cDNA microarray comparison between matched primary normal epithelial and squamous carcinoma cells. Oncogene. 2002;21:2634–40. doi: 10.1038/sj.onc.1205351. [DOI] [PubMed] [Google Scholar]
- 26.Lengyel E, Gum R, Stepp E, Juarez J, Wang H, Boyd D. Regulation of urokinase-type plasminogen activator expression by an ERK1-dependent signaling pathway in a squamous cell carcinoma cell line. J Cell Biochem. 1996;61:430–443. doi: 10.1002/(sici)1097-4644(19960601)61:3<430::aid-jcb10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 27.Barnes L, et al. World Health Organization classifications of tumors: Pathology and Genetics, Head and Neck Tumours. 2005;4:174–175. [Google Scholar]
- 28.Fu Y, et al. Head and Neck Pathology with Clinical Correlations. 2001;9:474. [Google Scholar]
- 29.Gnepp DR. Diagnostic Surgical Pathology of the Head and Neck. 2001;2:22–26. [Google Scholar]
- 30.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 31.Pettus JR, Johnson JJ, Shi Z, Davis JW, Koblinski J, Ghosh S, et al. Multiple kallikrein (KLK 5,7,8, and 10) expression in squamous cell carcinoma of the oral cavity. Histol Histopathol. 2009;24:197–207. doi: 10.14670/hh-24.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borgono CA, Iacovos PM, Eleftherios PD. Human tissue kellikreins: physiologic roles and applications in cancer. Mol Can Res. 2004;2:257–80. [PubMed] [Google Scholar]
- 33.Clements JA, Willemsen NM, Myers SA, Dong Y. The tissue kallikrein family of serine proteases: functional roles in human disease and potential as clinical biomarkers. Crit Rev Clin Lab Sci. 2004;41:265–312. doi: 10.1080/10408360490471931. [DOI] [PubMed] [Google Scholar]
- 34.Providence KM, Higgins SP, Mullen A, Battista A, Samarakoon R, Higgins CE, et al. SERPINE1 (PAI1) is deposited into keratinocyte migration trails and required for optimal monolayer wound repair. Arch Dermatol Res. 2008;300:303–10. doi: 10.1007/s00403-008-0845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Res. 2008;34:122–36. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Andreasen PA. PAI-1 – a potential therapeutic target in cancer. Curr Drug Targets. 2007;8:1030–41. doi: 10.2174/138945007781662346. [DOI] [PubMed] [Google Scholar]
- 37.Marinkovich MP. Tumor microenvironment: laminin 332 in squamous cell carcinoma. Nat Rev Cancer. 2007;7:370–80. doi: 10.1038/nrc2089. [DOI] [PubMed] [Google Scholar]
- 38.Tsuruta D, Kobayashi H, Imanishi H, Sugawara K, Ishii M, Jones JC. Laminin-332-integrin interaction: a target for cancer therapy? Curr Med Chem. 2008;15:1968–75. doi: 10.2174/092986708785132834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mognetti B, DiCarlo F, Berta GN. Animal models in oral cancer research. Oral Oncol. 2006;42:448–60. doi: 10.1016/j.oraloncology.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Bell RB, Kademani D, Homer L, Dierks EJ, Potter BE. Tongue cancer: is there a difference in survival compared with other subsites in the oral cavity? J Oral Maxillofac Surg. 2007;65:229–236. doi: 10.1016/j.joms.2005.11.094. [DOI] [PubMed] [Google Scholar]
- 41.Suhr ML, Dysvik B, Bruland O, Warnakulasuriya S, Amaratunga AN, Jonassen I, et al. Gene expression profile of oral squamous cell carcinomas from Sri Lankan betel quid users. Oncol Rep. 2007;18:1061–75. [PubMed] [Google Scholar]
- 42.Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nature Reviews Cancer. 2005;5:127–135. doi: 10.1038/nrc1549. [DOI] [PubMed] [Google Scholar]
- 43.Roepman P, Wessels KFA, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P, Tilanus MGJ, Koole R, Hordijk G-J, vad der Vliet PC, Reinders MJR, Slootweg PJ, Holstege FCP. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nature Genetics. 2005;37:182–6. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Su L, Pirani AA, Wu H, Zhang H, Shin DM, Gernert KM, Chen ZG. Understanding metastatic SCCHN cells from unique genotypes to phenotypes with the aid of an animal model and DNA microarray analysis. Clin Exp Metastasis. 2006;23:209–222. doi: 10.1007/s10585-006-9031-0. [DOI] [PubMed] [Google Scholar]
- 45.Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Curr Opin Cell Biol. 2000;12:613–20. doi: 10.1016/s0955-0674(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 46.Jo M, Thomas KS, Wu L, Gonias SL. Soluble urokinase-type plasminogen activator receptor inhibits cancer cell growth and invasion by direct urokinase-independent effects on cell signaling. J Biol Chem. 2003;278:46692–8. doi: 10.1074/jbc.M308808200. [DOI] [PubMed] [Google Scholar]
- 47.Blasi F, Carmeliet P. uPAR: a versatile signaling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–43. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 48.Zhang F, Tom CC, Kugler MC, Ching TT, Kreidberg JA, Wei Y, Chapman HA. Distinct ligand binding sites in integrin a3b1 regulate matrix adhesion and cell-cell contact. J Cell Biol. 2003;163:177–188. doi: 10.1083/jcb.200304065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kishimoto K, Sasaki A, Yoshihama Y, Mese H, Tsukamoto G, Matsumura T. Expression of vascular endothelial growth factor C predicts regional lymph node metastasis in early oral squamous cell carcinoma. Oral Oncology. 2003;39:391–6. doi: 10.1016/s1368-8375(02)00143-4. [DOI] [PubMed] [Google Scholar]
- 50.Tanigaki Y, Nagashima Y, Kitamura Y, Matsuda H, Mikami Y, Tsukuda M. The expression of vascular endothelial growth factor-A and –C and receptors 1 and 3: correlation with lymph node metastasis and prognosis in tongue squamous cell carcinoma; Int J Mol Med. 2004;14:389–95. [PubMed] [Google Scholar]
- 51.Shintani S, Li C, Ishikawa T, Mihara M, Nakashiro K, Hamakawa H. Expression of vascular endothelial growth factor A, BC and D in oral squamous cell carcinoma. Oral Oncology. 2004;40:13–20. doi: 10.1016/s1368-8375(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 52.Miyahara M, Tanuma J, Sugihara K, Semba I. Tumor lymphangiogenesis correlates with lymph node metastasis and clinicopathologic parameters in oral squamous cell carcinoma; Cancer. 2007;110:1287–94. doi: 10.1002/cncr.22900. [DOI] [PubMed] [Google Scholar]
- 53.Siriwardena B, Kudo Y, Ogawa I, Udagama k, Tilakaratne WM, Takata T. VEGF-C is associated with lymphatic status and invasion in oral cancer. J Clinical Path. 2007 doi: 10.1136/jcp.2007.047662. in press. [DOI] [PubMed] [Google Scholar]
- 54.Pampalakis G, Sotiorpoulou G. Tissue kallikrein proteolytic cascade pathways in normal physiology and cancer. Biochim Biophys Acta. 2007;1776:22–31. doi: 10.1016/j.bbcan.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Paliouras M, Borgono C, Diamandis EP. Human tissue kallikrins: the cancer biomarker family. Cancer lett. 2004;249:61–79. doi: 10.1016/j.canlet.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 56.Hundsdorfer B, Zeilhofer HF, Bock KP, Dettmar P, Schmitt M, Kolk A, et al. Tumor associated urokinase-type plasminogen activator and its inhibitor PAI-1 in normal and neoplastic tissues of patients with squamous cell cancer of the oral cavity – clinical relevance and prognostic value. j Craniomaxillofac Surg. 2005;33:191–6. doi: 10.1016/j.jcms.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 57.Vairaktaris E, Yapijakis C, Serefoglou Z, Vylliotis A, Ries J, Nkenke E, et al. Plasminogen activator inhibitor-1 polymorphism is associated with increased risk for oral cancer. Oral Oncol. 2006;42:888–92. doi: 10.1016/j.oraloncology.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 58.Lindberg P, Larsson A, Niesen BS. Expression of plasminogen activator inhibitor-1, urokinase receptor and laminin gamma-2 chain is an early coordinated event in incipient oral squamous cell carcinoma. Int J Cancer. 2006;118:2948–56. doi: 10.1002/ijc.21568. [DOI] [PubMed] [Google Scholar]
- 59.Driemel O, Dahse R, Hakim SG, Tsioutsias T, Pistner H, Reichert TE, Kosmehl H. Laminin-5 imunocytochemistry: a new tool for identifying dysplastic cells in oral brush biopsies. Cytopathology. 2997;18:329–30. doi: 10.1111/j.1365-2303.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 60.Chen C, Mendez E, Houck J, Fan W, Lohavanichbutr P, Doody D, et al. Gene expression profiling identifies genes predictive of oral squamous cell carcionma. Cancer Epidemiol Biomarkers Prev. 2008;17:2152–62. doi: 10.1158/1055-9965.EPI-07-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuratomi Y, Kumamoto M, Kidera K, Toh S, Masuda M, Nakashima T, et al. Diffuse expression of laminin gamma2 chain in disseminating and infiltrating cancer cells inducates a highly malignant state in advanced tongue cancer. Oral Oncol. 2006;42:73–6. doi: 10.1016/j.oraloncology.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 62.Zhang X, Liu Y, Gilcrease MZ, Yuan XH, Clayman GL, Adler-Storthz K, Chen Z. A lymph node metastatic mouse model reveals alterations of metastasis-related gene expression in metastatic human oral carcinoma sublines selected from a poorly metastatic parental cell line. Cancer. 2002;95:1663–72. doi: 10.1002/cncr.10837. [DOI] [PubMed] [Google Scholar]
- 63.Henson B, Li F, Coatney DD, Carey TE, Mitra RS, Kirkwood KL, D’Silva NJ. An orthotopic floor-of-mouth model for locoregional growth and spread of human squamous cell carcinoma. J Oral Pathol Med. 2007;36:363–70. doi: 10.1111/j.1600-0714.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 64.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. Article 3 http://www.bepress.com/sagmb/vol3/iss1/art3. [DOI] [PubMed]
- 65.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–20. [Google Scholar]
- 66.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Research. 2002;30(4):e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Characterization of SCC25-uPAR-KD and SCC25-uPAR+ cells. (A) Relative surface expression levels of uPAR were measured by flow cytometric analysis using primary antibody 3937, as described in Experimental Procedures. Mean fluorescence index values are shown. (vec) – SCC25 parental cells expressing empty vector; uPAR-KD – SCC25 cells with downregulated uPAR; uPAR+ - SCC25 cells that overexpress uPAR. (B) Invasive activity was determined by seeding cells in the top of a Boyden chamber (8 um pore size, coated with Matrigel). After 24 h, cells in the upper chamber were removed with a cotton swab and invading cells adherent to the bottom chamber were fixed and stained with Diff-quik prior to enumeration. Bars, mean +/− SEM. *p<.05. (C) Invasive activity was determined as in (B) above in the presence of function blocking antibodies directed against uPAR or a3 integrin (20 ug/ml), the MEK inhibitor PD98059 (10 uM) or DMSO (vehicle control). Bars, mean +/− SEM *p<.05.
Supplemental Figure 2. Analysis of tumor parameters. (A) Loss of differentiation of SCC25-uPAR+ cells, observed from analysis of H&E stained sections, was confirmed using qPCR analysis of keratin 19 (K19, black bar) and keratin 13 (K13, white bar) expression. Relative expression levels were normalized to housekeeping gene PGK. Each bar depicts the mean +/− SD of replicate values expressed as fold-difference in keratin mRNA level relative to SCC25-uPAR-KD cells (designated as 1). *p<.05 relative to corresponding gene in SCC25-uPAR+. (B) Quantitation of relative tumor area and level of inflammation as described in Experimental Procedures. Bars, mean +/− SEM. (open bar) – SCC25-vec, (grey bar) – SCC25-uPAR-KD, (black bar) – SCC25-uPAR+.
Supplemental Figure 3. Quantitation of blood vessel density and perineural invasion. (A) Blood vessel density and (B) perineural invasion was quantified from H&E stained sections by counting a minimum of 140 high powered fields from three biologic replicate experiments. Bars, mean +/− SEM. (black bar) – SCC25-uPAR+; (grey bar) – SCC25-uPAR-KD.