

Abstract
Combinations of antiangiogenic agents (AAs) with cytotoxic agents have shown significant promise and several such clinical trials are currently underway. We have designed, synthesized and evaluated two compounds that each inhibit vascular endothelial growth factor receptor-2 (VEGFR-2) and platelet derived growth factor receptor-beta (PDGFR-β) for antiangiogenic effects and also inhibit human thymidylate synthase (hTS) for cytotoxic effects in single agents. The synthesis of these compounds involved the nucleophilic displacement of the common intermediate 5-chloro-9H-pyrimido[4,5-b]indole-2,4-diamine with appropriate benzenethiols. The inhibitory potency of both these single agents against VEGFR-2, PDGFR-β and hTS is better than or close to standards. In a COLO-205 xenograft mouse model one of the analogs significantly decreased tumor growth (TGI = 76% at 35 mg/kg), liver metastases and tumor blood vessels compared to a standard drug and to control and thus demonstrated potent tumor growth inhibition, inhibition of metastasis and antiangiogenic effects in vivo. These compounds afford combination chemotherapeutic potential in single agents.
Antiangiogenic agents (AA) have established a new paradigm in cancer chemotherapy and have allowed significant progress towards the control and treatment of various cancers.1 Normal cell angiogenesis is initiated under conditions of injured or hypoxic tissues, occurs in wound healing, menstrual cycle and pregnancy and is promoted by vascular endothelial growth factor (VEGF) and similar growth factors. Except for these conditions angiogenesis is mostly absent in normal tissues.2 Heterogeneous solid growing tumors however are in a state of angiogenesis and continually overexpress growth factors.
Receptor tyrosine kinases (RTKs) are a subclass of cell surface growth factor receptors with intrinsic, ligand-controlled tyrosine kinase activity.3, 4 Epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR) and vascular endothelial growth factor receptor (VEGFR) families of receptors are all RTKs and despite their diverse biological roles, share similarities in structure and domain arrangement.
Clinically useful RTK inhibitors have resulted from small molecule ATP competitive inhibitors (Figure 1) that target the kinase domain of RTKs. While RTK inhibitors have afforded a new mechanism for the treatment of a variety of cancers it was quickly realized that single RTK inhibitors allowed the development of resistance by point mutations in the ATP binding site. In addition, the molecular pathways responsible for tumor growth, survival and metastasis are redundant and adaptable between individual patients and within tumors in the same patient. Thus treatment targeting a single RTK would be highly unlikely to provide long term tumor control in most patients. Proangiogenic growth factors are redundantly expressed by both tumor and stromal cells.5 Thus there is now a paradigm shift in the utility of RTK inhibitors for cancer treatment in that rather than single RTK inhibition it is now desirable to use multi-targeted RTK inhibitors to overcome possible resistance and thwart the escape mechanism of alternative pathways for tumor growth. Recent FDA approvals of just such multi-targeted RTK inhibitors sorafenib (VEGFR-2, PDGFR-β, FMS-like tyrosine kinase 3 (Flt-3), raf kinase and stem cell factor receptor (c-kit)) and sunitinib (VEGFR-1, 2 and 3, PDGFR-β, α, Flt-3, and colony stimulating factor 1 Receptor (CSF-1R)) attest to the importance of this new paradigm in cancer chemotherapy.3-12 In addition, vandetanib is a VEGFR-2 inhibitor that combines EGFR (rather than PDGFR-β) inhibitory activity, and lapatinib also combines EGFR and human epidermal growth factor receptor-2 (ErbB-2) inhibitory activity. All of these multi-targeting RTK inhibitors have some monotherapy potential but are mostly cytostatic (some cytotoxicity has been observed via apoptosis in a limited number of tumor types) thus for optimal benefit they must be combined with cytotoxic, conventional chemotherapeutic agents and/or radiation therapy.13 The rationale and mechanisms for the success of these combinations continues to provide discussion in the literature.13, 14 Hundreds of such ongoing clinical trials (clinicaltrials.gov) attest to the importance of the concept and the individual drugs both cytostatic and cytotoxic in the combinations.
Figure 1.
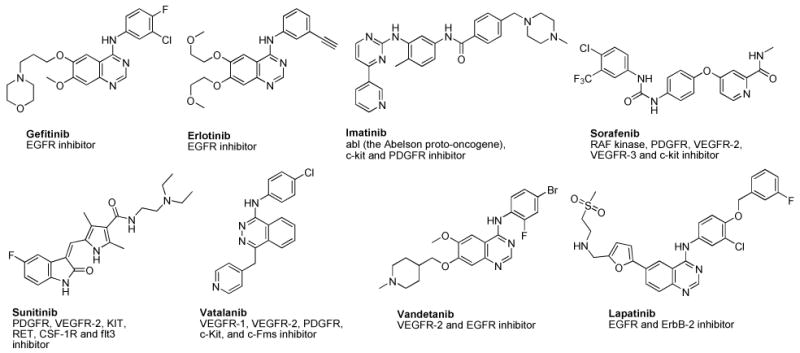
Single and multi-targeted RTK inhibitors.
Combination therapy with VEGFR-2 (endothelial cell inhibition) along with PDGFR-β inhibition (pericytes inhibition) increases the antiangiogenic effect even in the often intractable, late state of solid tumors.15, 16 Thus targeting both VEGFR-2 and PDGFR-β simultaneously is a desirable goal for AAs that have cytostatic and perhaps cytotoxic activity. The clinical success of sunitinib9, 11 and sorafenib11 that target both VEGFR and PDGFR attests to the viability of this combination in single agent multi RTK inhibitors. Preclinical and clinical evidence indicates that combinations of AAs with conventional cytotoxic agents and/or radiation therapy results in additive or even synergistic antitumor effects15-17 and that monotherapy is usually unsuccessful with antiangiogenic agents. Rapid vascular regrowth in tumors after the removal of AAs attests to their cytostatic mechanism.18, 19
Combination cancer chemotherapy is not a new idea. Recent studies indicate that the combination of antiangiogenic agents with cytotoxic agents is more effective in cancer treatment.20 However, what would be novel is if a single agent could be found that had both antiangiogenic activity by multi-targeting RTKs and also possessed cytotoxic activity to afford combination chemotherapy in a single agent. We21 synthesized such agents with VEGFR-2, PDGFR-β and dihydrofolate reductase (DHFR) inhibition with good results. However, the potency of the cytotoxic component in these agents was extremely low.21 Thus we elected to structurally engineer multi RTK inhibitory attributes along with the deliberate design of cytotoxicity in single agents as a second iteration of our previous report. Such single agents would circumvent the pharmacokinetic problems of multiple agents, avoid drug-drug interactions, could be used at lower doses to alleviate toxicity, be devoid of overlapping toxicities, and delay or prevent tumor cell resistance. Most importantly providing the cytotoxic agent, by structural design, in the same molecule allows the cytotoxicity to be mainfested as soon as the antiangiogenic effects are operable. A separately dosed cytotoxic agent may miss the timing window and hence preclude the intent of the combination. Such multi-targeted agents could exert their cytotoxic action as soon as or even during transient tumor vasculature normalization13, 22 due to the antiangiogenic effects. Thus such agents might not need to be as potent as conventional separately dosed cytotoxic agents. Dosing of such an antiangiogenic multi-targeted RTK inhibitor with a built-in cytotoxic mechanism would be tantamount to providing a combination of multi-targeted RTK inhibitors along with a metronomic dosing of a cytotoxic agent. Thus these single agents would be in keeping with the two important mechanisms that explain the rationale for the success of separate antiangiogenic and cytotoxic chemotherapeutic agents in combination for cancer chemotherapy.13, 22-25 Other advantages of such single agents are in the reduced cost and increased patient compliance which are sometimes as significant contributors to chemotherapy failure as resistance, toxicity and lack of efficacy.
The antiangiogenic component is usually targeted to tumor cells and is, under most circumstances, not targeted to normal cells. However, the cytotoxic component is targeted to the tumor cells but not exclusively. Thus the challenge in designing single agents with a cytotoxic component is that the cytotoxic component should be potent enough to kill tumor cells that have been compromised via the antiangiogenic effect but not potent enough to cause serious toxicity to normal cells not affected by the antiangiogenic effect. Clearly one of the problems with conventional cytotoxic chemotherapeutic agents is dose limiting toxicities. These single agents should avoid these toxicities since they do not need to be as potent as conventional chemotherapeutic agents.
We have designed and synthesized compounds 1 and 2 (Figure 2) that each target VEGFR-2 and PDGFR-β and provide antiangiogenic effects in vivo and also have human thymidylate synthase (hTS) inhibitory activity to afford the cytotoxic component in vivo in single agents. The choice of VEGFR-226 and PDGFR-β27 inhibition for antiangiogenic activity was obvious since these are the principal direct and indirect mediators of angiogenesis. The choice of inhibition of human dihydrofolate reductase (hDHFR) and/or hTS as the cytotoxic component was based in part on our long standing interest in inhibitors of folate metabolizing enzymes and on structural, architectural and molecular modeling considerations in the design of the molecules. In addition, the successful clinical and preclinical combinations of capecitibine,28, 29 5-FU30 (TS inhibitors) and pemetrexed (PMX)31, 32 (TS and DHFR inhibitor) (Figure 3) with AAs in combination therapy with and without radiation was also an important factor in selecting TS and/or DHFR as the possible cytotoxic targets in the design of 1 and 2.
Figure 2.
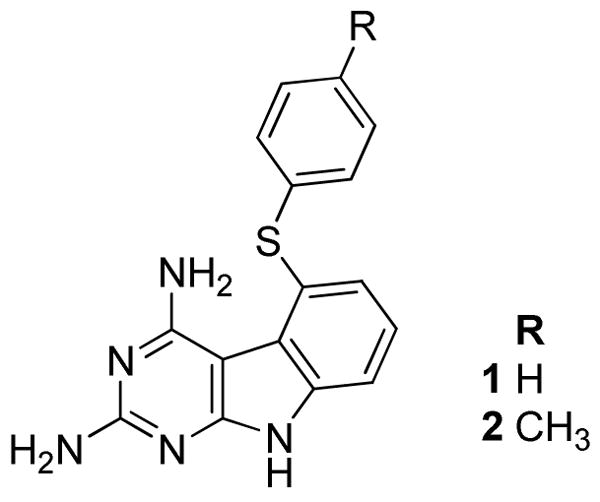
Target compounds 1 and 2.
Figure 3.
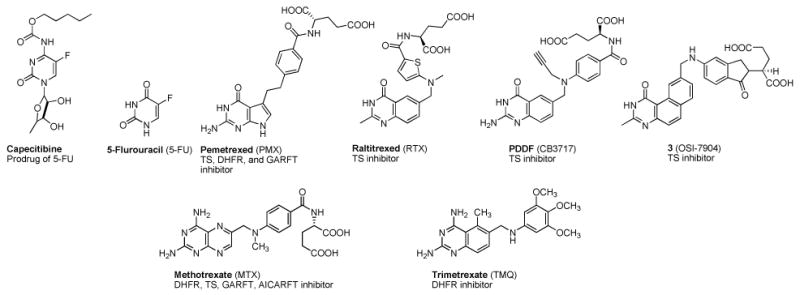
Dihydrofolate reductase and thymidylate synthase inhibitors
DHFR carries out the reduction of dihydrofolate to tetrahydrofolate and maintains the pool of reduced folate cofactors that function in one carbon transfers in a variety of metabolic transformations crucial for cell survival.33 Thus inhibitors of DHFR such as methotrexate (MTX) and PMX (Figure 3) have found utility as antitumor agents. TS carries out the sole de novo biosynthesis of TMP from dUMP. It utilizes 5,10-methylenetetrahydrofolate as a cofactor to transfer the methyl group to dUMP.34 Because of its pivotal role in DNA synthesis and cell growth it is a viable target for several clinically used cancer chemotherapeutic agents.35 The fluoropyrimidine 5-FU and its derivatives, in particular, capecitabine (Figure 3) have found extensive utility in ovarian, breast, colon and several other cancers alone and in combinations and is a mainstay in cancer chemotherapy.36 Folate based TS inhibitor antimetabolites (Figure 3) that are clinically used alone or in combination in a variety of cancers include PMX37 and in Europe raltitrexed (RTX) a derivative of PDDF. In addition 3 (Figure 3), also a TS inhibitor, and its derivatives are in various stages of clinical development alone and in combinations against a variety of cancers.38
Tricyclic scaffolds reported in the literature with appropriate 4-anilino substitutions have shown excellent RTK inhibition.39, 40 An example of such an analog is presented in Figure 4A.39 In a general RTK model it is shown41-44 that the 2-NH2, the N3 and the 4-anilino nitrogen of the pyrimidine ring of RTK inhibitors H-bond with the Hinge region as depicted in Figure 4A. In addition, the phenyl ring of the 4-anilino moiety lies in the Hydrophobic region 1, and the tricyclic scaffold mimics the purine ring of ATP.39, 40 We reasoned that transposing the phenyl ring from the 4-position (Figure 4A) to the 5-position of the tricyclic scaffold as shown in Figure 4B maintains access to Hydrophobic region 1 and allows the H-bonds with the Hinge region. Such compounds with phenyl substitutions in the 5-position should maintain RTK inhibitory activity. However, moving the phenyl ring from the 4- to the 5-position unveils a 2,4-diaminopyrimidine motif on the tricyclic scaffold (Figure 4B) that is highly conducive for DHFR and/or TS inhibition.45, 46 MTX and trimetrexate (TMQ) (Figure 3) are well known 2,4-diamino fused pyrimidines with hDHFR inhibition and for MTX, hTS inhibitory activity as well.47 Usually DHFR and TS inhibitors have a 2,4-diamino and a 2-amino-4-oxo substitution on the pyrimidine ring respectively. There are however reports where analogs with a 2,4-diamino substitution have TS and/or DHFR inhibitory activity45, 46 while a 2-amino-4-oxo substitution provides DHFR and/or TS inhibitory activity.48, 49 Thus we designed 2,4-diamino-5-thio phenyl substituted pyrimido[4,5-b]indoles as potential RTK inhibitors with DHFR and/or TS inhibitory attributes.
Figure 4.
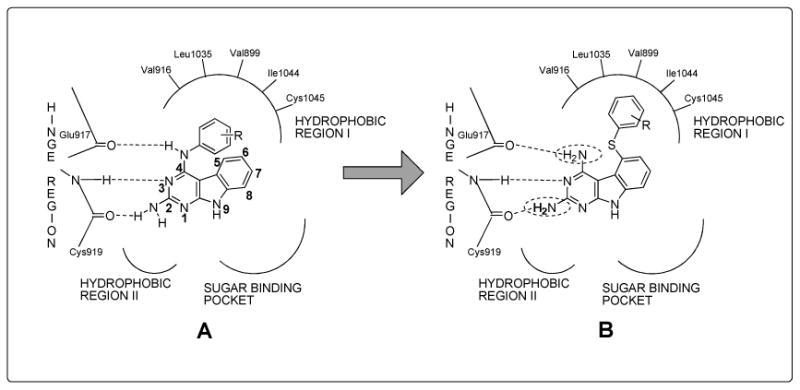
Rationale for the synthesis of target molecules.
Molecular modeling with hDHFR confirmed that the third ring of the tricyclic scaffold could afford additional interaction with Ile60 of hDHFR50 over the previously synthesized bicyclic analogs.21 Modeling (Figure 5) using SYBYL 7.351 and superimposition of one of the energy minimized conformations of 1 with the pyrimidine ring of 1 on to the X-ray crystal structure of the pyrimidine ring of the pyrido[2,3-d]pyrimidine inhibitor (not shown) (1PDB)50 in hDHFR, shows that the 2,4-diamino motif is maintained for hDHFR inhibitory activity as is the S-Ph which mimics the phenyl ring of MTX52, 53 and TMQ.50 The 2NH2 and N1 make a salt bridge with Glu30 in hDHFR and the S-Ph ring makes hydrophobic contact with Phe31 and Val115 much like TMQ and MTX and the third ring of the tricyclic scaffold contacts Ile60. The conformation of the phenyl ring of 1 in Figure 5 is not the usual conformation of this ring in other reported hDHFR inhibitors. With this exception all other interactions of the scaffold of 1 were similar to the pteridine of MTX and the quinazoline of TMQ. Thus we anticipated DHFR inhibitory activity for 1.
Figure 5.
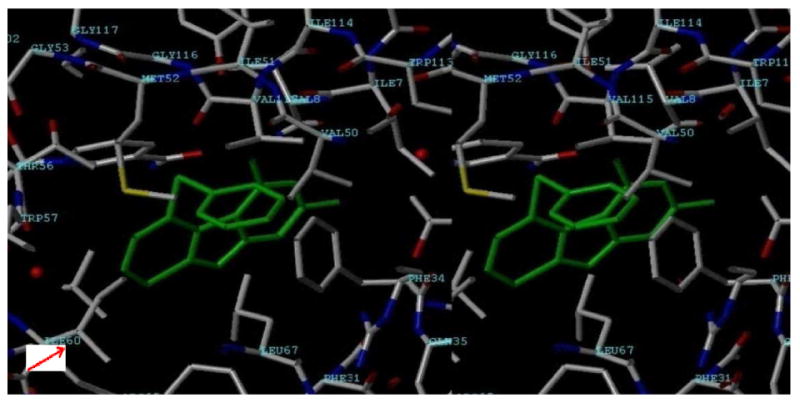
Stereoview (SYBYL7.3) Pyrimidine ring of 1 superimposed on to the pyrimidine ring of the DHFR inhibitor in the X-ray crystal structure of hDHFR (1PDB)50
For TS inhibitory activity, modeling 1 by superimposing the pyrimidine ring of the energy minimized 1 (yellow) onto the pyrimidine ring of the X-ray crystal structure of PMX (green) in hTS (PDB:1JU6),54 Figure 6 (SYBYL 7.3), showed that the superimposition is perfect for the two scaffolds. In the molecular modeling, the hydrophobic interactions of the tricyclic C-ring of 1 with Trp109 (bottom right) are somewhat better than the bicyclic B-ring of PMX. In addition, the S-phenyl ring of 1 and its close proximity to the benzoyl ring of PMX (green) is evident as is the hydrophobic interactions with Ile108, Leu221, Phe225 (top center) (and Met311 not shown). Also evident is the stacking interaction of 1 and dUMP (directly below 1). Thus on basis of molecular modeling we anticipated compound 1 to have DHFR and/or TS inhibitory activity.
Figure 6.
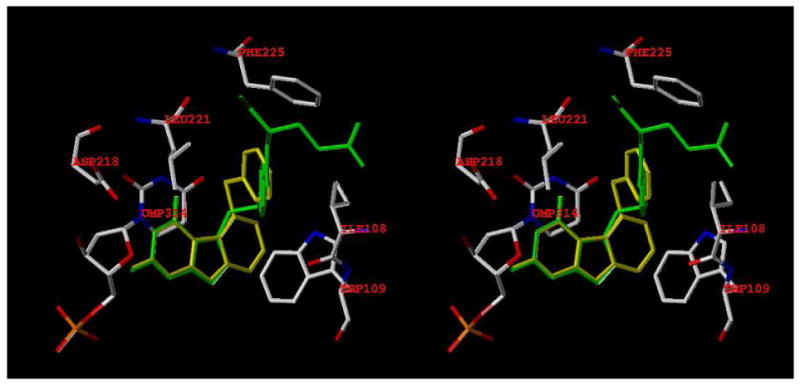
Stereoview (SYBYL 7.3). 1 superimposed on the X-ray crystal structure of pemetrexed (green) in hTS. (PDB ID: 1JU6).54
In Figure 7, energy minimized 1 was superimposed (SYBYL 7.3) onto a furo[2,3-d]pyrimidine55 inhibitor (not shown) of VEGFR-2 in the X-ray crystal structure of VEGFR-2 (PDB: 1YWN)55 that indicated the Hinge region binding of the 4NH2 group of 1 with Glu915 (C = O); N3 with Cys917 (N-H); 2NH2 with Cys917 (C = O). The 5-thiophenyl ring lies in Hydrophobic region 1 and interacts with Val897, Leu1033 and Cys1043. Thus molecular modeling supports the inhibition of VEGFR-2 by 1.
Figure 7.
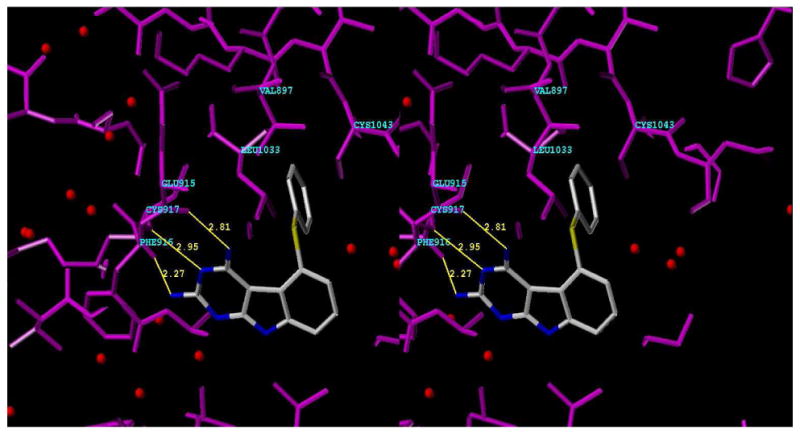
Stereoview (SYBYL7.3) of 1 superimposed on to furo[2,3-d]pyrimidine X-ray crystal structure (not shown) in VEGFR2. Hinge region 4NH2- Glu915 (C=O); N3-Cys917 (N-H); 2NH2-Cys917 (C=O); Hydrophobic region 1 5-thiophenyl-(Leu1033, Cys1043) (PDB ID: 1YWN).55
There is currently no crystal structure of PDGFR-β bound to a ligand. Using the structure of colony-stimulating factor-1 receptor (cFMS) (PDB: 1PKG, chain A)56 as the template a homology model of inactive PDGFR-β was generated using the Molecular Operating Environment (MOE 2007.09)57 suite and methods indicated in the literature.58-60 A conformational database was built for 1 and minimized and docked into the homology modeled active site shown in Figure 8. The docked poses were then ranked. The 2- and 4- NH2 groups of 1 form hydrogen bonds with the backbone residues of the Hinge region (Tyr683, Cys684). Additionally, the 5-S-Ph is involved in a cation-pi interaction (10-15 kcal/mol stabilization) with the protonated Arg604. Figure 8 shows only one of the low energy binding poses of 1 with PDGFR-β and provides a working model for binding to PDGFR-β.
Figure 8.
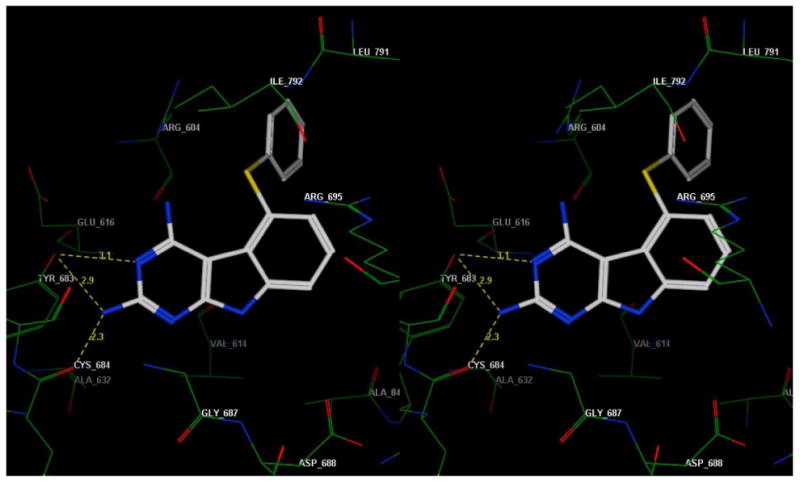
Stereoview of a docked pose of 1 in the putative PDGFR-β active site model.
Thus we anticipated that the synthesis and evaluation of 1 and 2 would afford VEGFR-2 and/or PDGFR-β inhibition and that these compounds would also possess DHFR and/or TS inhibitory activity thus providing combination chemotherapy potential in single molecules.
Chemistry
The synthesis of the target compounds is outlined in Scheme 1. Condensation of 1,2-dichloro-3-nitro-benzene 4 with the potassium salt of ethyl cyanoacetate anion provided adduct 2-(2-chloro-6-nitrophenyl)-2-cyanoacetate 5. Zinc dust reduction of 5, utilizing literature conditions39 furnished ethyl 2-amino-4-chloro-1H-indole-3-carboxylate 6. Cyclocondensation of 6 with carbamimidic chloride hydrochloride61 afforded 2-amino-5-chloro-3H-pyrimido[4,5-b]indol-4(9H)-one 7. Protection of the 2-amino group of 7 using 2,2-dimethyl propanoic anhydride provided 8. The 4-chloro derivative 9 was prepared by treating 8 with phosphorus oxychloride at reflux. Displacement of the 4-chloro group with ammonia, and concomitant deprotection of the 2-amino group of 9 was achieved by means of a sealed vessel reaction; thus the 2, 4-diamino compound 10 was obtained as the common intermediate. Treatment of 10 with the appropriate substituted benzenethiols in a microwave apparatus (from Biotage) provided target compounds 1 and 2.
Scheme 1a.
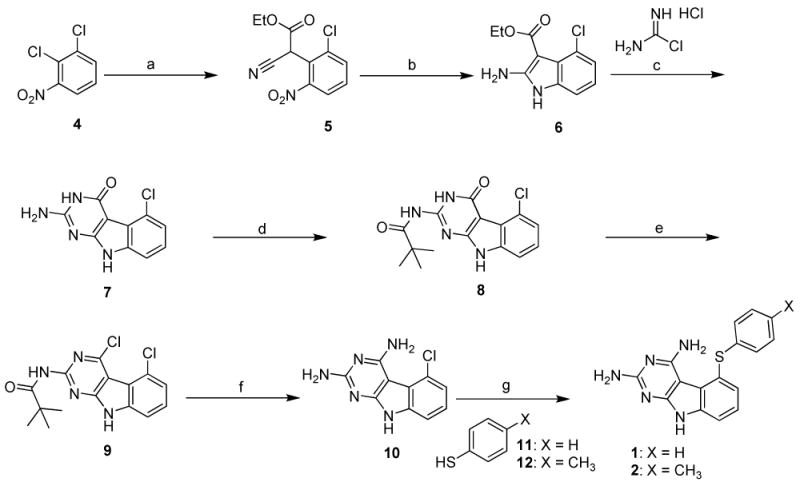
a Reagents and conditions: (a) EtO2CCH2CN, t-BuOK, THF, reflux, 48 h; (b) Zn, HOAc, 50-55°C, 2 hrs 30 mins; (c) Methyl sulfone, 110°C, 30 mins; (d) 2,2-dimethylpropanoic anhydride, DMAP, NEt3, DMF, 60°C, 48 h; (e) POCl3, reflux, 4 hrs; (f) NH3/CH3OH, sealed vessel, 130°C, 48 h; (g) K2CO3, NMP, microwave, 250°C, 30 mins.
Biological Evaluation and Discussion
Discrepancies in IC50 values obtained from isolated RTK inhibitory assays when compared to IC50 values obtained against whole tumor cell assays are sometimes quite large (as much as 1000-fold). There are several reasons for this and the conditions (e.g. ATP concentrations for ATP competitive agents) of the assay dictate the IC50 values. Variations in assay conditions afford different IC50 values. Thus we have elected to evaluate the RTK inhibitory activity of our compounds using human tumor cells known to express high levels of the appropriate RTK.
We believe that tumor cell inhibitory assays are the most meaningful assays of the activity of the analogs evaluated and allow the most appropriate extrapolation regarding candidate selection and chances of success in in vivo evaluations. In addition to this, we recognize that the IC50 of standard compounds also vary under different assay conditions, thus we use a standard (control) compound in each of our assays. These standard compounds were evaluated side by side with our analogs and afford direct comparison of IC50s of our analogs with the standard compounds.
Compounds 1 and 2 were evaluated as inhibitors of EGFR, VEGFR-1, VEGFR-2, PDGFR-β, phosphatidylinositol 3-kinase (PI3K), Flt-3 and for antiproliferative activity against the A431 tumor cell line that over expresses EGFR. For the RTK and PI3K assays, cells overexpresssing each kinase were exposed to compound followed by the ligand for each RTK as described in the detailed methods section. This was followed by a “cytoblot,” or in-cell ELISA, developed by our laboratory for the evaluation of kinase activity42. The compounds were compared with standards N-(3-bromophenyl)-6,7-dimethoxyquinazolin-4-amine (13) for EGFR (Figure 9); semaxinib for VEGFR-2; N-(4-chloro-2-fluorophenyl)-6,7-dimethoxyquinazolin-4-amine (14) for VEGFR-1; (Z)-3-((4-dimethylamino)benzylidene)indolin-2-one (DMBI) for PDGFR-β; cisplatin (CDDP) for A431; 2-morpholino-8-phenyl-4H-chromen-4-one (15) for P13K, and 2-(3,4-dimethoxybenzamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (16) for Flt-3 (Figure 9). The results are listed in Table 1 (below). The compounds are active against VEGFR-2 and within 2- to 4-fold of the standard semaxinib with 1 more potent than 2. Against PDGFR-β, 1 is somewhat more potent than the standard DMBI and 2 is about 11-fold less potent. Against the growth of A431 tumor cells in culture 2 is similar to cisplatin while 1 is much less potent. Since neither compound is a potent EGFR inhibitor compared to standard 13, the marginal inhibitory activity against A431 tumor cells of 1 and 2 was not surprising. The involvement of Flt-311 inhibition for both sorafenib and sunitinib in addition to other RTKs prompted the modeling of 1 in the X-ray crystal structure of Flt-3.62 This afforded an excellent docked conformation and hence we initiated the whole cell assay for Flt-3 kinase for 1 and 2 (Table 1). Compounds 1 and 2 were about 13-fold less active against Flt-3 compared with standard 16. Due to the importance of PI3K in tumor growth63 we also evaluated 1 and 2 in the PI3K assay using 15 as the standard (Table 1), compounds 1 and 2 are 8-fold and 11-fold less active respectively than the standard 15 against PI3K.
Figure 9.
Standard drugs and control agents.
Table 1. IC50 values (μM) of kinase inhibition, A431 cytotoxicity assay and EC50 (μM) against P13K.
| Compd # | EGFR Kinase Inhibition | VEGFR-2 (Flk-1) Kinase Inhibition | VEGFR-1 (Flt-1) kinase Inhibition | PDGFR-β Kinase Inhibition | A431 Cytotoxicity | PI3K Activity | Flt-3 Activity |
|---|---|---|---|---|---|---|---|
| 1 | 15.07 ± 3.1 | 22.6 ± 4.5 | 118.1 ± 19.4 | 2.8 ± 0.42 | 49.2 ± 4.7 | 21.6 | 41.2 ± 5.7 |
| 2 | 10.41 ± 1.2 | 56.3 ± 7.1 | 160.1 ± 28.9 | 40.3 ± 5.1 | 14.1 ± 2.0 | 12.2 | 39.6 ± 4.1 |
| 13 | 0.23 ± 0.05 | ||||||
| Semaxinib | 12.9 ± 2.9 | ||||||
| 14 | 14.1 ± 2.8 | ||||||
| DMBI | 3.75 ± 0.31 | ||||||
| Cisplatin | 10.6 ± 3.5 | ||||||
| 15 | 1.5 | ||||||
| 16 | 2.9 ± 0.031 |
In-cell kinase activity was assessed by a “cytoblot” developed in our laboratory using A431 cells which overexpress EGFR, SF539 cells for PDGFRβ, U251 cells for VEGFR2, A498 cells for VEGFR1, MV 3:11 cells for Flt-3, and HeLa cells for PI3K as described in the detailed methods section.
The compounds were also evaluated against isolated DHFR and TS for the cytotoxic component. The results are listed in Table 2 and are compared with standards for DHFR (MTX, trimethoprim (TMP)) and TS (PMX, RTX). The compounds were potent inhibitors of hTS at submicromolar levels that were 54-fold (1) and 76-fold (2) more active than clinically used PMX as its monoglutamate, and comparable to RTX. The better hTS inhibitory activity of both 1 and 2 compared to PMX could be due to a better C-ring interaction with Trp109.
Table 2. Inhibitory Concentrations (IC50, μM) of DHFR and TS.
| Compd | TS Inhibitory Activitya | DHFR Inhibitory Activitya | ||||
|---|---|---|---|---|---|---|
| IC50 Values (μM) of Thymidylate Synthase Inhibition | IC50 Values (μM) of Dihydrofolate Reductase Inhibition | |||||
| Humanc | E. colic | Toxoplasma gondiid | Humane | E. colif | Toxoplasma gondiid | |
| 1 | 0.54 | > 27 | 0.11 | > 33 (17)b | > 33 (35) | 33 |
| 2 | 0.39 | > 26 | > 26 | > 31 (7) | > 31 (27) | > 31 (22) |
| Pemetrexed g | 29.0 | 15 | 14 | |||
| Raltitrexed h | 0.29 | 2.3 | 0.48 | |||
| MTX | 0.022 | 0.0066 | 0.019 | |||
| Trimethoprim | 680 | 0.02 | 2.9 | |||
The percent inhibition was determined at a minimum of four inhibitor concentrations within 20% of the 50% point. The standard deviations for determination of 50% points were within ± 10% of the value given.
Numbers in parentheses indicate the percent inhibition at the stated concentration.
Kindly provided by Dr. Frank Maley, New York State Department of Health.
Kindly provided by Dr. Karen Anderson, Yale University, New Haven, CT.
Kindly provided by Dr. Andre Rosowsky, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA.
Kindly provided by Dr. R. L. Blakley, St. Jude Children's Hospital, Memphis, TN.
Kindly provided by Dr. Chuan Shih, Eli Lilly and Co.
Kindly provided by Dr. Ann Jackman, CRC Centre for Cancer Therapeutics, Surrey SM2 5NG, England.
However 1 and 2 were not appreciably potent against hDHFR as well as other DHFR. Why was 1 poorly active against hDHFR? Molecular modeling had suggested that superimposition of 1 onto DHFR inhibitors in X-ray crystal structures (Fig. 5) should afford inhibition of hDHFR. The modeling provides the 5-S-phenyl of 1 in a binding mode that is at 90° to other hDHFR inhibitor (such as MTX52, 53 and TMQ50) side chain phenyl rings. On the basis of molecular modeling, other binding modes of 1 afford clashes of the C-ring with side chains and backbone of hDHFR. This could perhaps explain, in part, the poor activity against hDHFR.
Thus 1 and 2 were good to moderate inhibitors of VEGFR-2 and PDGFR-β comparable or better than the standards (except 2 for PDGFR-β) for RTK inhibition and potential antiangiogenic activity and also had hTS inhibitory activity (IC50) in the submicromolar range to provide the cytotoxic effects needed for combination chemotherapy potential in single agents. The hTS IC50 values were not in the single digit nanomolar range, and hence should not cause severe toxicity to normal cells not compromised by the antiangiogenic effects of 1 and 2. The usual next step is to determine the inhibitory activity against tumor cells in culture. The NCI preclinical 60 tumor panel was the obvious choice, however since angiogenesis is not part of these tumors in culture, only the cytotoxic effects were expected to play a role in the inhibition of these cells in culture. Compound 1 was evaluated against the growth of the NCI 60 tumor cell lines and the results were as anticipated, 1 inhibited the tumors at GI50 values of 1 μM - 10 μM. This was about 10-fold less than the isolated hTS inhibitory activity (Table 2) and indicates a relatively low potency completely in keeping with the absence of the antiangiogenic component and perhaps an indication of low toxicity to normal cells.
Pharmacodynamic (PD) data in MDA-MB-435 cells
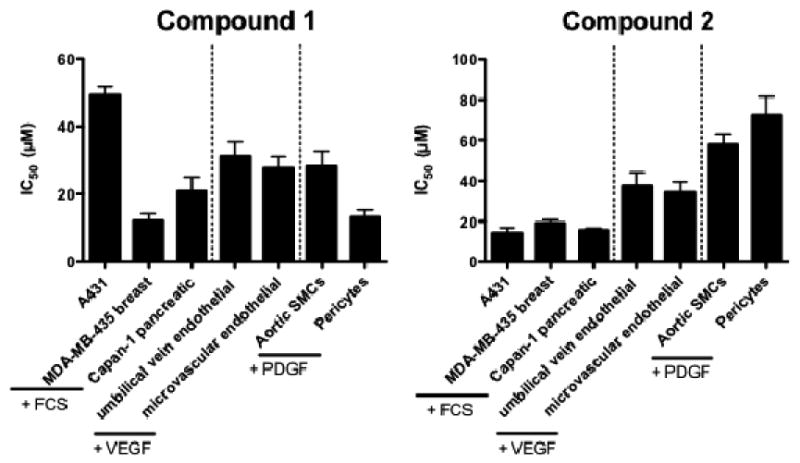
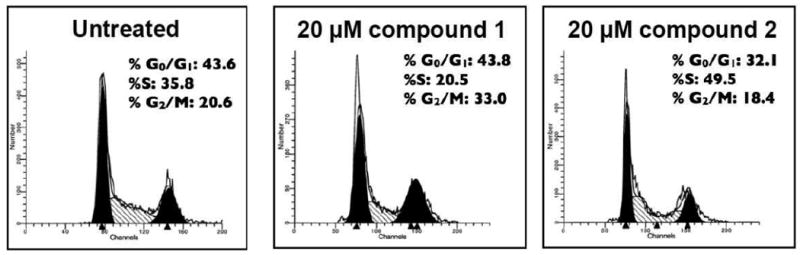
Compounds 1 and 2 were evaluated for cell viability (IC50 of cell kill) in several cell types (Fig. 10). In addition MDA MB 435 metastatic dermal breast cancer cells were exposed to 20 μM of 1 and 2 for cell cycle analysis (Fig. 11), and cell signaling analysis (Fig. 12) were carried out. First, cell viability was evaluated on cells serum-starved overnight before the addition of 1% FCS to the cancer cells, 200 ng/ml hrVEGF-165 to the endothelial cells, and 100 ng/ml hrPDGF-BB to the SMC variants together with drug for 24 h followed by 24 h incubation in serum-containing media and a cell viability assay carried out as described in the Experimental. It was found that 1 had varied effects (Fig. 10) on three cancer cell lines (MDA-MB-435, A431, Capan-1); and fairly consistent effects on two endothelial cell types (HUVEC, HMEC-1). Human retinal pericytes (Huperi-3),64 (not human aortic SMCs), were very sensitive to 1 cell kill (Fig. 10, left panel). Compound 2 had a different profile of sensitivity than did 1 with tumor cells being the most sensitive, endothelial cells falling someplace in the middle, and SMC variants being the least sensitive to 2 (Fig. 10, right panel). For cell cycle analysis it was found that after 24 h exposure, 1 resulted in a buildup in G2/M (Fig. 11, right panel) while 2 resulted in an increase in S phase (Fig. 11, left panel). Next, a cell signaling experiment was performed on the MDA-MB-435 breast cells exposed for either 1 or 4 h to 20 μM 1 or 2. The same cells exposed to 30 J/m2 UV-B for 4 h were used as a positive control. The first downstream target of RTKs was activated phospho-p42/44 MAPkinase (ERK 1/2), a MAP kinase member involved in cell proliferation in many cancers. It was found both 1 and 2 reduced levels of activated ERK 1/2, but not to the same extent as UV-B exposure (Fig. 12; top gel). In contrast, exposure to neither agent resulted in increased levels of activated phospho-Akt, a cell survival pathway also downstream of RTKs with levels of (non-phospho) Akt used as a loading control (Fig. 12, middle two gels). Finally, neither 20 μM 1 or 20 μM 2 resulted in increased levels of cleaved caspase 3, the terminal step in apoptosis (Fig. 12; bottom most gel).
Figure 10.
Cell viability assay after 48 hr treatment.
Figure 11.
Cell cycle analysis
Figure 12.
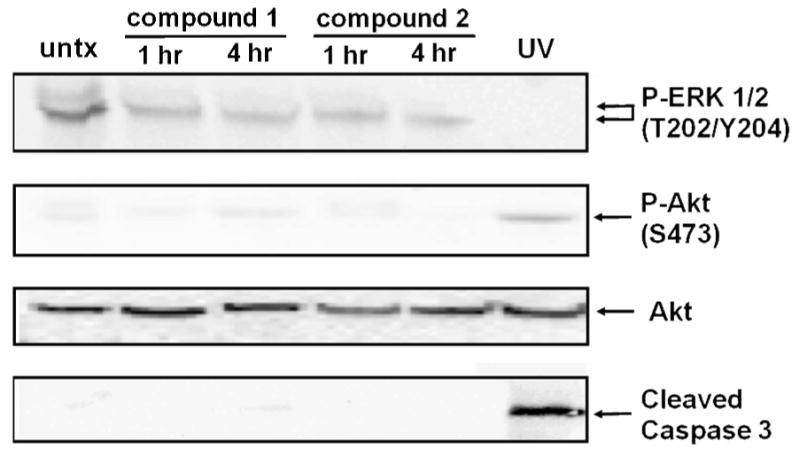
Cell signaling
MDA-MB-435 chorio-allantoic membrane (CAM) xenograft
We have developed an intermediate systemic in vivo xenograft system using the chicken embryo CAM. This method allows for in vivo testing of more compounds in a given time and cost than the mouse tumor xenograft studies. For the CAM xenograft model, MDA-MB-435 cells (250,000) were implanted under the vascularized CAM of 10 DI chicken embryos and then the embryos were treated systemically with 25 mg/kg 1 on days one and two; and CAMs were fixed, excised, and imaged with representative images shown from 5-6 CAMs (Figure 13). It was found that 1 reduced both the size and the vascularity of resulting tumors, at least to the same extent as the standard VEGFR2 kinase inhibitor semaxanib at 10 mg/kg (Fig. 13). These results indicate the antitumor and antiangiogenic activity of 1. Taken together the preliminary in vitro and the in vivo results (see below) suggest that compound 1 has profound antiangiogenic activity both in vitro and in vivo. Further, these data indicate that 1 may act more on the vasculature to decrease RTK growth factor signaling perhaps decreasing cell proliferation. The data from compound 2 are more complex, it appears that his compound acts both on the tumor and cells of the vasculature to decrease RTK and MAPK signaling. The increase in S phase content observed in response to 2 could indicate that cells enter into DNA synthesis but cannot exit into G2, a phenomenon consistent with TS inhibition.
Figure 13.
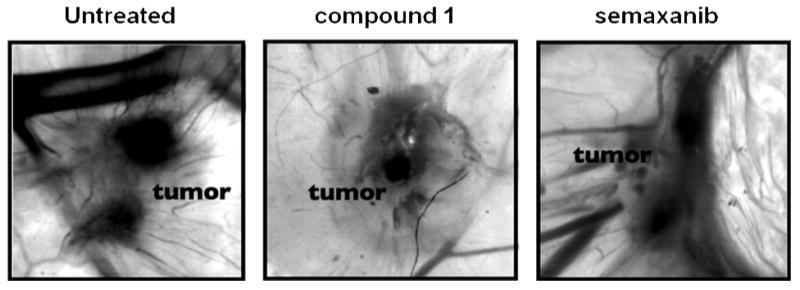
MDA-MB-435 CAM 72 hr xenograft
The logical next step was to demonstrate antitumor activity of 1 via an in vivo animal model. Compound 1 was the first choice over 2 mainly because of its better PDGFR-β inhibitory activity. To determine the antitumor effect of 1 in vivo, athymic mice were first implanted with COLO-205 metastatic colon cancer cells, shown to over express PDGFR-β.65 At day 9 after implantation, treatment was initiated with the PDGFR-β inhibitor DMBI66 at the MTD of 15 mg/kg three times weekly and with 1 at two doses, 25 mg/kg and 35 mg/kg three times weekly. Tumor growth was measured using calipers. It was found that DMBI and 1 resulted in significant decreases in primary tumor growth rate (Fig. 14A, and 14B), and that 1 resulted in a further significant decrease in tumor growth rate as compared to the control compound, DMBI. At the end of the experiment, tumors were taken and stained for blood vessels. It was found that 1 at 35 mg/kg three times weekly resulted in a significant decrease in primary tumor vascularity as compared to control treated animals (Fig. 14C) suggesting an antiangiogenic effect in vivo. Finally, livers were stained for the presence of metastases and it was found that 1 at both 25 and 35 mg/kg, but not DMBI, resulted in significantly reduced numbers of COLO-205 metastases to the liver (Fig 14D). TGI for 1 is 76% at 35 mg/kg in this study. No MTD was determined for 1; doses up from 75 mg/kg were well tolerated without any visual ADRs or loss in weight. Thus in vivo compound 1 has considerable antitumor, antiangiogenic activity and antimetastatic activity. Further in vitro and in vivo studies are currently underway to afford further evidence of the triple mechanism(s) of action of 1 and 2 with a triple combination of a VEGFR2, a PDGFR-β and a hTS inhibitor as standard as well as a combination of two of these standards together to determine the mechanism of action, in vivo, of 1 and 2 responsible for the in vivo antitumor, antiangiogenic and antimetastatic activities of 1.
Figure 14. Tumor growth (A), primary tumor growth rate (B), primary tumor growth, (C) primary tumor vascularity, (D) liver metastasis of COLO-205 tumor xenografts after various treatments.
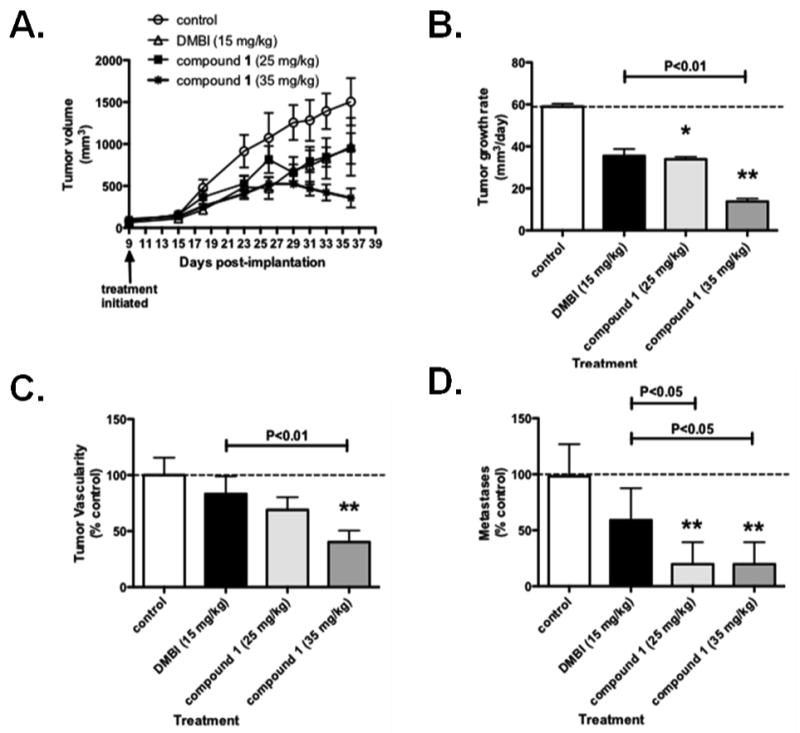
COLO-205 cells were implanted into male athymic mice as in the research design and methods section. Compounds were administered at the listed doses three times weekly and tumor growth measured using calipers. At the experiment end, tumors were stained for vascularity and livers stained for metastases as in research design and methods section. Data represent the average +/- SD of 7-11 animals. * P>0.05, ** P<0.01, *** P<0.001 by one way ANOVA and Neuman-Keuls post test. The average tumor growth rate for control treated animals (B) is 58.9 mm3/day, average number of tumor vessels in control-treated animals, (C) are 11.2/field and average metastases in control treated animals, (D) are 0.65/lobe.
We designed, synthesized and discovered two compounds, 1 and 2 that inhibit VEGFR-2 and PDGFR-β in whole cells at IC50 values comparable to standard compounds. In addition, 1 and 2 also inhibit isolated hTS better than the monoglutamate of clinically used PMX. Thus remarkably 1 and 2 possess both multi RTK inhibitory activity (VEGFR-2 and PDGFR-β) for antiangiogenic effects as well as hTS inhibitory activity (cytotoxic effects) comparable or better than clinically used single agents. Compound 1 in vivo in a COLO-205 metastatic colon cancer xenograft mouse model, at both 25 mg/kg and 35 mg/kg showed significantly decreased tumor growth, significantly decreased liver metastasis as well as significantly decreased primary tumor blood vessels (angiogenesis) remarkably without any toxicity, compared to untreated control and a standard PDGFR-β inhibitor DMBI. The combination chemotherapeutic attributes of 1 and 2 of VEGFR-2, PDGFR-β as well as hTS inhibitory activity makes them unique and distinct from all other known agents in clinical or investigational use. The relevance of this combination of activities in a single agent is that it mimics the clinically relevant combinations of RTK inhibitors with TS inhibitors. In addition, the in vivo antitumor, antiangiogenic and antimetastatic activity of 1 supports the optimization and development of this agent and design of analogs as potential clinically useful antitumor agents, that affords combination chemotherapy potential in single agents. Such single agents are designed to be used as monotherapy but could also be used with other antitumor agents or radiation as part of the therapy for cancers.
Experimental Section
Analytical samples were dried in vacuo (0.2 mm Hg) in a CHEM-DRY drying apparatus over P2O5 at 80 °C. Melting points were determined on a MEL-TEMP II melting point apparatus with FLUKE 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) were recorded on a Bruker WH-300 (300 MHz) spectrometer. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad singlet. Thin-layer chromatography (TLC) was performed on Whatman Sil G/UV254 silica gel plates with a fluorescent indicator, and the spots were visualized under 254 and 366 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230-400 mesh silica gel (Fisher, Somerville, NJ) column. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Element compositions are within ±0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples of antifolates could not be prevented in spite of 24-48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. Elemental analysis was used to determine the purity of the final compounds 1 and 2. All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and were used as received. Purity of the final compounds 1 and 2 were > 95% and were determined by elemental (C, H, N, S) analysis.
Ethyl 2-amino-4-chloro-1H-indole-3-carboxylate (6)
To an ice-cold solution of ethyl cyanoacetate (10.9 mL, 102.4 mmol) in anhydrous THF (170 mL) under N2 was added potassium tert-butoxide (12.07 g, 107.5 mmol). The formed white suspension was stirred for 15 min then treated with 1,2-dichloro-3-nitrobenzene 4 (9.83 g, 51.2 mmol). The suspension was heated at reflux for 48 h. The resulting reddish brown solution was poured into H2O, and the aqueous mixture was acidified to pH 2 with concentrated HCl. The mixture was extracted with ether (3×) and then the combined organic phases were dried using sodium sulfate and concentrated to give ethyl (2-chloro-2-nitrophenyl)(cyano)acetate 5 as a dark oil. The oil was purified by column chromatography eluting with 10:1 hexane:EtOAc. TLC Rf 0.23 (hexane/EtOAc, 3:1). 1H NMR (DMSO-d6) δ 1.17-1.22 (t, 3 H, CH3), 4.18-4.25 (q, 2 H, CH2), 6.36 (bs, 2 H, 2-NH2, exch), 7.75-7.81 (t, J = 8.1 Hz, 1 H, C4-CH), 8.04-8.06 (dd, J = 6.9, 1.2 Hz, 1 H, Ar), 8.13-8.16 (dd, J = 6.9, 1.2 Hz, 1 H, Ar). The material was used directly for the next step.
A solution of impure 5 (18 g, 67 mmol) in glacial AcOH (185 mL) was treated with a single charge of Zn dust (12.1 g, 185 mmol). The mixture was heated at 55 °C for 45 min, then treated with more Zn dust (6 g). After heating for another 105 min, the mixture was filtered through a pad of celite. The pad was washed well with AcOH and the filtrate was concentrated to a residue that was distributed between CHCl3 and H2O. The organic phase was washed with 5% aq NaHCO3 and concentrated to a residue that was purified by column chromatography, eluting sequentially with 0%, 5%, and 10% EtOAc in CHCl3. The fractions containing the pure product (TLC) were pooled and evaporated to give a pink solid. The overall yield from 4 to 6 was 63% (7.7 g of 6 was obtained). TLC Rf 0.18 (hexane/CHCl3, 1:1); mp 140-142 °C; 1H NMR (DMSO-d6) δ 1.26-1.30 (t, 3 H, CH3), 4.15-4.23 (q, 2 H, CH2), 6.82-6.87 (t, J = 7.8 Hz, 1 H, C6-CH), 6.92-6.96 (dd, J = 7.8 Hz, 1 H, Ar), 6.96 (bs, 2 H, 2-NH2, exch), 7.05-7.08 (dd, J = 7.8 Hz, 1 H, Ar), 11.35 (bs, 1H, 9-NH, exch). Anal. (C11H11ClN2O2) C, H, N
2-Amino-5-chloro-3, 9-dihydro-4H-pyrimido[4,5-b]indol-4-one (7)
A mixture of ethyl 2-amino-4-chloro-1H-indole-3-carboxylate (6) (200 mg, 0.837 mmol), carbamimidic chloride hydrochloride61 (106.22 mg, 1.37 mmol) and methyl sulfone (1 g) was stirred and heated at 110-120 °C for 30 minutes. About 10 mL water was added to quench the reaction. Ammonia water was added to neutralize the reaction mixture. The precipitated solid was filtered dissolved in chloroform and methanol, dried (Na2SO4) and the solvent evaporated, and recrystallized from chloroform and methanol (1:1). in overall yield of 78%. TLC Rf 0.33 (CHCl3/MeOH, 1:1); mp >250 °C; 1H NMR (DMSO-d6) δ 6.58 (bs, 2H, 2-NH2, exch), 7.04-7.06 (t, J = 3.3 Hz, 1 H, C7-CH), 7.17-7.20 (m, 2 H, Ar), 10.42 (s, 1H, 3-NH, exch), 11.65 (s, 1H, 9-NH, exch). HRMS calcd for C10H7ClN4O 234.0309, found 234.0308.
N-(5-chloro-4-oxo-4,9-dihydro-3H-pyrimido[4,5-b]indol-2-yl)-2,2-dimethyl propanamide (8)
To a round bottomed flask were added 7 (300 mg, 1.27 mmol), 2,2-dimethylpropanoic anhydride (713.32mg, 3.83 mmol), dimethylaminopyridine (7 mg, 0.06mmol) and triethylamine (514.05mg, 5.08mmol), along with 30 mL DMF. The mixture was stirred at 60 °C for 48 h. The DMF was removed using an oil pump to afford a residue which was purified by column chromatography, eluting sequentially with 0%, 1%, and 5% CH3OH in CHCl3. Fractions containing the product 8 (TLC) were pooled and evaporated to give solid compound. The yield was 40 % (163 mg). TLC Rf 0.45 (CHCl3/MeOH, 10:1); mp 185.8-190.1 °C; 1H NMR (DMSO-d6) δ 1.27 (s, 9 H, C(CH3)3), 7.18-7,21 (t, J = 2.4 Hz, 1 H, C7-CH), 7.24-7.26 (dd, J = 7.5 Hz, 1 H, Ar), 7.37-7.40 (dd, J = 6.3 Hz, 1 H, Ar), 11.15 (s, 1H, 3-NH, exch), 11.93 (s, 1H, 2-NH, exch), 12.11 (s, 1H, 9-NH, exch). HRMS calcd for C15H15ClN4O2 [M+Na]+ 341.0757, found 341.0781.
N-(4,5-dichloro-9H-pyrimido[4,5-b]indol-2-yl)-2,2-dimethyl propanamide (9)
To round bottomed flask was added 8 (2 g, 6.274 mmol) and dissolved in 30 mL of POCl3. The reaction mixture was refluxed at 110-120 °C for 4 hours. After evaporation of the POCl3, ice-cold water was added. The reaction mixture was neutralized with NH3.H2O, and extracted with CHCl3. The organic phase was dried with Na2SO4. The crude mixture was then purified by column chromatography, eluting sequentially with 0%, 1% and 5% methanol in chloroform. Fractions containing the product 9 (TLC) were pooled and evaporated to give a solid. The yield was 70% (1.48 g). TLC Rf 0.86 (CHCl3/MeOH, 5:1); mp 245.6-246.1 °C; 1H NMR (DMSO-d6) δ 1.25 (s, 9H, C(CH3)3), 7.36-7.39 (t, J = 4.5 Hz, 1 H, C7–CH), 7.49-7.51 (m, 2 H, Ar), 10.30 (s, 1H, 9-NH, exch), 12.96 (s, 1H, 2-NH, exch). HRMS calcd for C15H14Cl2N4O2 [M+Na]+ 359.0428, found 359.0442.
5-Chloro-9H-pyrimido[4,5-b]indole-2,4 diamine propanamide (10)
Compound 9 (200 mg, 0.6 mmol) was added to 5 mL of methanol saturated with ammonia. The solution was stirred at 130 °C for 2 days in a sealed vessel. The methanol was evaporated to give a solid that was purified by column chromatography, eluting sequentially with 0% and 1% methanol in chloroform. Fractions containing the product 10 (TLC) were pooled and evaporated to give a solid. The yield was 39 % (54 mg). TLC Rf 0.43 (CHCl3/MeOH, 5:1); mp 245.2-246.3 °C; 1H NMR (DMSO-d6) δ 6.15 (bs, 2H, 4-NH2, exch), 6.85 (bs, 2H, 2-NH2, exch), 7.10-7.15 (t, J = 7.2 Hz, 1 H, C7-CH), 7.15-7.18 (dd, J = 9 Hz, 1 H, Ar), 7.22-7.24 (dd, J = 6.9 Hz, 1 H, Ar), 11.54 (bs, 1 H, 9-NH, exch). Anal. (C10H8ClN5) C, H, N, Cl.
General procedure for the synthesis of 5-(substitutedphenylthio)-9H-pyrimido[4,5-b]indole-2,4-diamines 1 and 2
Compound 10 (50 mg, 0.2 mmol), the appropriate thiol (0.9 mmol), potassium carbonate (120 mg, 0.9 mmol) were added to a 2-5 mL biotage® microwave vial. 3 mL NMP was added as solvent and the tube was sealed. The reaction was run in a microwave for 30 minutes at 250 °C. After cooling to room temperature, the reaction mixture was transferred on top of a silica gel column and eluted with 0% and 4% methanol in chloroform. Fractions containing the product (TLC) were pooled and evaporated to afford the product 1 and 2.
5-(phenylthio)-9H-pyrimido[4,5-b]indole-2,4-diamine (1)
Using the general procedure described above, the reaction of 10 with benzene thiol 11 afforded 1 as an off-white solid: yield 97% (64 mg). TLC Rf 0.23 (CHCl3/MeOH 10:1); mp 251 °C; 1H NMR (DMSO-d6) δ 6.04 (bs, 2 H, 4-NH2, exch), 7.03-7.04 (d, J = 4.5 Hz, 2 H, Ar), 7.13-7.16 (t, J = 4.2 Hz, 1 H, C7-CH), 7.21-7.27 (m, 2 H, Ar), 7.24 (bs, 2 H, 2-NH2, exch), 7.32-7.33 (dd, J = 4.2 Hz, 1 H, Ar), 7.40-7.42 (dd, J = 4.5 Hz, 1 H, Ar), 11.49 (bs, 1H, 9-NH, exch). Anal. (C16H13N5S) C, H, N, S.
5-[(4-methylphenyl)thio]-9H-pyrimido[4,5-b]indole-2,4-diamine (2)
Using the general procedure described above, the reaction of 10 with 4-methyl benzene thiol 12 afforded 1 as a off-white solid: yield 87% (60 mg). TLC Rf 0.54 (CHCl3/MeOH 5:1); m.p. >250 °C; 1H NMR (DMSO-d6) δ 2.19 (s, 3H, CH3), 6.02 (bs, 2H, 4-NH2, exch); 7.19 (bs, 2H, 2-NH2, exch), 6.95-6.97 (d, J = 6 Hz, 2 H, Ar), 7.05-7.07 (d, J = 6 Hz, 2 H, Ar), 7.18-7.22 (t, J = 6 Hz, 1 H, C7-CH), 7.29-7.31 (dd, J = 5.7 Hz, 1 H, Ar), 7.38-7.40 (dd, J = 5.7 Hz, 1 H, Ar), 11.46 (bs, 1H, 9-NH, exch). Anal. Calculated (C17H15N5S. 0.4 H2O) C, H, N, S.
Molecular Modeling
There is currently no known crystal structure of PDGFR-β bound to a ligand. A homology model was hence built for evaluating the binding of 1 in PDGFR-β. The amino acid sequence of the PDGFR-β kinase domain was obtained from the SWISS-PROT database (ID: PGFRB_HUMAN [P09619]). As has been reported earlier in the literature67, a DISOPRED2.068 analysis of the amino acid sequence was performed to predict the ordered and disordered regions. The results from this analysis predicted amino acids 700 – 792 were disordered. A homology model was then built using MOE 2007.09 using the structure of c-KIT kinase complex (PDB: 1PKG, chain A) as the template. A BLASTP58 search indicated that chain A of the c-KIT kinase complex shows high sequence similarity with PDGFR-β (E-value: 1e-58). Sequence alignment was performed using MOE_Align using the ‘actual secondary structure’ option in MOE. Using the ‘actual secondary structure’ option in MOE was necessary to correctly identify the disordered kinase insertion domain (amino acids 700-792). The final homology model returned by the program was subjected to further energy minimization using Amber99 as the forcefield and a 0.5 RMS gradient. A Ramachandran plot of the model showed the presence of six outlying residues (Tyr562, Glu563, Asp590, Ser623, Ser642, Glu911). Since these residues were not in the proximity of the ATP binding site, the model was used without further refinement. Docking studies were performed using the docking suite of MOE 2007.09. After addition of hydrogen atoms, the protein was then “prepared” using the LigX function in MOE.
LigX is a graphical interface and collection of procedures for conducting interactive ligand modification and energy minimization in the active site of a flexible receptor. In LigX calculations, the receptor atoms far from the ligand are constrained and not allowed to move while receptor atoms in the active site of the protein are allowed to move but are subject to tether restraints that discourage gross movement. The procedure was performed with the default settings.
Ligands were built using the molecule builder function in MOE and were energy minimized to its local minima using the MMF94X forcefield to a constant of 0.05 kcal/mol. Ligands were docked into the active site of the prepared protein using the docking suite as implemented in MOE. The docking was restricted to the active site pocket residues using Alpha triangle placement method. Refinement of the docked poses was carried out using the Forcefield refinement scheme and scored using Affinity dG scoring system. Around 30 poses were returned for each compound at the end of each docking run. The docked poses were manually examined in the binding pocket to ensure quality of docking and to confirm absence of steric clashes with the amino acid residues of the binding pocket.
Detailed Methods
Specific assays
CYQUANT cell proliferation marker assay
As a measure of cell proliferation, the CYQUANT cell counting/proliferation assay was used as previously described.69 We have found that this assay is comparable in sensitivity to a clonogenic (colony-forming) assay70 and lacks many of the problems associated with metabolic-based assays. For the CYQUANT assay, cells were serum-starved overnight, 1% FCS was then added along with drug to the media of cancer cells (MDA-MB-435, Capan-1, J82, A431, U251), 200 ng/ml hrVEGF-165 was added with drug to the media of endothelial cell (HUVEC, HMEC-1), and 100 ng/ml hrPDGF-BB was added to the media of SMCs (HASMCs, HuPeri-3) for 24 hr then replaced with fresh (drug-free) completed media for an additional 24 hr. The cells were then lysed and the CYQUANT dye, which intercalates into the DNA of cells, was added and after 5 min the fluorescence of each well was measured using a plate reader. A positive control used for cytotoxicity was cisplatin. Data are graphed as a percent of cells receiving no compound and IC50 values were estimated from 2-3 separate experiments (n=6-15) using sigmoidal-dose response analysis with Prism 3.0 (GraphPad software, San Diego).
Phosphotyrosine (PY) cell-based ELISA
Because of the need for screening many compounds in multiple replicates, a higher throughput 96-well phosphotyrosine (PY) ELISA was developed.71, 72 Briefly, cells at 60-75% confluence were placed in serum-free medium for 18 hr to reduce the background of phosphorylation. Cells used for these experiments have been shown to overexpress particular RTKs; specifically A431 for EGFR, SF539 for PDGFRβ, U251 for VEGFR2, A498 for VEGFR1, and MV 3:11 cells for Flt-3 (but not VEGFR1/2). Cells were then pre-treated for 60 minutes with 10, 3.33, 1.11, 0.37 and 0.12 μM compound followed by an optimized dose and time of purified growth factor (EGF, PDGF-β, VEGF). The reaction was stopped and cells permeabilized by quickly removing the media from the cells and adding ice-cold Tris-buffered saline (TBS) containing 0.05% triton X-100, protease inhibitor cocktail and tyrosine phosphatase inhibitor cocktail (both from Sigma Chemical). The TBS solution was then removed and cells fixed to the plate by heat and further incubation in 70% ethanol. Cells were further exposed to block (TBS with 1% BSA), washed, and then a horseradish peroxidase (HRP)-conjugated phosphotyrosine antibody was added. The antibody was removed, cells are washed again, and exposed to an enhanced luminol ELISA substrate and light emission was measured using a plate reader. The known RTK-specific kinase inhibitors (PD153035, SU5416, CB67645, DMBI and compound 16) discussed above were used as positive controls for kinase inhibition. Data are graphed as a percent of cells receiving growth factor alone and EC50 values estimated from 2-3 separate experiments (n=8-24) using sigmoidal dose-response relations in Prism 3.0 software (GraphPad). In every case, the activity of a positive control inhibitor did not deviate more than 10% from the EC50 values historically collected in this assay.21, 42, 73
PI3 kinase assay
The PI3 kinase assay is a cytoblot for the phosphorylated (active) form of the p85 subunit of PI3K. HeLa cells have been shown to possess large PI3K activity without any stimulus,74 so for these studies no inducer is necessary. HeLa cells grown in log phase for one day after passaging in serum-containing media were exposed as above to various concentrations of unknown compounds or the known PI3K inhibitor, LY 294002 for one hour at 37 °C. As above, cells were permeabilized with Triton, fixed by heat and ethanol, blocked, and a phospho-(Tyr) p85 PI3K binding motif antibody (Cell Signaling) was added at a 1:100 dilution overnight at 4 °C. The antibody was then removed and an anti-rabbit HRP conjugated secondary antibody was then added (Pierce Chemical) for 30 min at RT, the antibody was removed, cells were washed again, and exposed to an enhanced luminol ELISA substrate and light emission measured using a chemilluminescent plate reader.
Dihydrofolate Reductase (DHFR) Assay75
All enzymes were assayed spectrophotometrically in a solution containing 50 μM dihydrofolate, 80 μM NADPH, 0.05 Tris-HCl, 0.001 M 2-mercaptoethanol, and 0.001 M EDTA at pH 7.4 and 30 °C. The reaction was initiated with an amount of enzymes yielding a change in optical density at 340 nm of 0.015/min.
Thymidylate Synthase (TS) Assay
TS was assayed spectrophotometrically at 30 °C and pH 7.4 in a mixture containing 0.1 M 2-mercaptoethanol, 0.0003 M (6R,S)-tetrahydrofolate, 0.012 M formaldehyde, 0.02 M MgCl2, 0.001 M dUMP, 0.04 M Tris-HCl, and 0.000 75 M NaEDTA. This was the assay described by Wahba and Friedkin76 except that the dUMP concentration was increased 25-fold according to the method of Davisson et al.77 The reaction was initiated by the addition of an amount of enzyme yielding a change in absorbance at 340 nm of 0.016/min in the absence of inhibitor.
In Vitro Pharmacodynamic studies
Cell Cycle Analysis
After exposure to drug, cells were fixed in 50% ethanol overnight, permeabilized and labeled with PBS containing 0.1% triton X-100, 100 μg/ml Rnase A, and 25 μg/ml propidium iodide for 45 min to 1 hr at 37°C before being subjected to flow cytometry as previously described.70 Cell cycle analysis of mean fluorescence data were performed using MODFIT software to generate percent S phase, G1/G0 phase and G2/M phase (Verity Software, Topsham, ME). Aphidicolin (250 ng/ml), an agent that inhibits DNA polymerase, was used as a control for G1 arrest78 and 50 ng/ml nocodazole, a reversible mitotic spindle poison, was used as a control for G2/M arrest (data not shown).79
Cell signaling analysis
Cells were treated with compounds at around the IC50 dose for 1 and 4 hrs and whole cell lysates were prepared using M-PER reagent (Pierce Chemical, Rockford, IL) with 300 mM NaCl to lyse the nucleus. Western blots against phosphorylated (activated) (ERK1/2 (Cell Signaling CAT#4370)) and against phospho (Cell Signaling CAT# 4060) and non-phosphorylated Akt (Cell Signaling CAT# 2920), two major signaling pathways downstream of RTKs, were done per manufacturer's instructions. Additionaly, cleaved caspase 3 (Cell Signaling CAT# 9661) was assessed as a marker of apoptosis.
Chorioallantoic membrane (CAM) xenograft assay
The procedure used was a modification of a previously published procedure80 Fertile Leghorn chicken eggs (CBT Farms, Chestertown, MD) were incubated until 10 DI, eggs candled for viability, then small window was made in the superior shell to expose the chorion and CAM. The CAM was then candled to visualize the vasculature and a small mark was made on the chorion in a location away from major vessels. Human MDA-MB-435 cancer cells (American Type Culture Collection, Manassas, VA), 250,000 in 50 μl inert extracellular matrix (Humatrix, Care-Tech Laboratories, St Louis, MO) were implanted just under the CAM using a 1 ml syringe with a 25G needle. Neomycin-polymyxin ointment was then applied to injection site and the shell hole covered with Micropore surgical tape (3M, St. Paul, MN). Four and 48 hr after tumor implantation, the tape was partially removed and drug or solvent (DMSO in SWFI) was placed onto the chorion using a micropipettor, yielding a systemic dose to the embryo. In an initial dose-finding experiment, either semaxanib or compound 1 were given at doses of 2.5, 5, 10, 15, 25, and 35 mg/kg embryo body weight81 and at 3 days after injection embryo lethality was evaluated. It was found that 10 mg/kg gemcitabine and 25 mg/kg compound 1 were the highest doses resulting in no lethality or overt toxicity in embryos, thus these doses were used in subsequent experiments. Three days after tumor implantation, CAMs were fixed in situ by pipetting on a solution of 0.1% triton X-100 in 4% paraformaldehyde for 1 minute. The solution was removed and CAM/chorion were excised in an area surrounding the tumor implantation site. The CAM/chorion were placed into 4% paraformaldehyde, allowed to fix an addition 2 minutes, and the chorion separated from the CAM. CAMs were removed to 6-well plates containing 4% paraformaldehyde. CAMs were spread gently to remove wrinkles and images captured at 6.25× magnification using a brightfield dissecting microcope (Wild M400 photomakroscop, Bern, Switzerland) connected to a cooled camera (QImaging, Burnaby, BC, Canada). Images were then converted to grayscale and the number of vessels surrounding the tumor manually counted and the size of the tumor determined using NIH Image J densitometry software.
Animal numbers
Consulting with the Biostatistics Program at OUHSC and using StatMate software (www.graphpad.com) and a power analysis, we have found that a sample size of 10 animals is optimal for 80% power, a significance level of 0.05 in a (two-tailed) unpaired t-test, and an expected SD of 24 (from previous experiments). Four animals per treatment group are used for short-term toxicity studies and five animals per group are used for the longer term toxicity studies.
Whole animal toxicity assay
To first determine the maximal tolerated dose (MTD) of a drug, male NCr nu/nu mice at 8 weeks of age without tumors were injected with 5, 10, 12.5, 15, 17.5 and 20 mg/kg compound 1 or the PDGFRβ kinase inhibitor DMBI three times weekly, on Monday AM, Wednesday noon and Friday PM. Weights were taken and animals observed for acute distress during the first 96 hr after injection and in a 3 and 6 week period. Significance in weights were calculated after each weighing using one-way ANOVA and Neuman-Keuls post-test with the null hypothesis rejected if P<0.05. Animals with significant (>7%) weight loss, were humanely euthanized, inspected for any overt toxicity and organs (heart, lung, kidney, liver, colon) removed for inspection by a veterinarian pathologist at OUHSC.
COLO-205 human metastatic colon mouse xenograft
It has been shown that the natural killer (NK) T cell activity in athymic mice affects the metastasis of human tumor xenografts.82, 83 Thus for these experiments, NIH-III nude mice, deficient in NK cells, were used. One million COLO-205 (liver colonizing; ATCC) human metastatic colon cancer cells were injected SQ in the lateral flank of athymic NIH-II male mice, 8 weeks in age. Animals are monitored every other day for the presence of tumors. At the time in which most tumors are measurable by calipers (day 9 after implantation), animals with tumors were randomly sorted into treatment groups. DMSO stocks (30 mM) of drugs are further dissolved into sterile water for injection and the optimal dose (from toxicity studies) injected intraperitoneally (IP). The length (long side), width (short side) and depth of the tumors are measured using digital Vernier Calipers each Monday, Wednesday, and Friday. Tumor volume was calculated using the formula length × width × depth. Tumor growth rate was then calculated using a linear regression analysis algorithm with the software GraphPad Prism 4.0.c. At the experiment's end, animals were humanly euthanized tumors and liver excised, fixed in 20% neutral buffered formalin for 8-10 h, embedded into paraffin, and hematoxylin-eosin (H&E) stain of three separate tissue sections completed to span the tumor/liver. Together with the OUHSC Department of Pathology core, metastases per liver lobe were counted using the H&E stained sections. The metastases can be seen as purple clusters of disorganized cells on the highly organized largely pink liver. Together with the OUHSC Department of Pathology core, blood vessels per unit area on primary tumors were also counted in 5 fields at 100× magnification and averaged. Tumor growth rate per day, tumor vascularity and liver metastases were calculated and significance determined from the growth curves using two-way ANOVA with a repeated measures post-test and the null hypothesis rejected if P<0.05.
Supplementary Material
Acknowledgments
This work was supported, in part, by a grant from the National Institutes of Health, National Cancer Institute (Grant CA136944 (A.G.)).
Abbreviations
- AA
Antiangiogenic agents
- VEGF
vascular endothelial growth factor
- RTK
receptor tyrosine kinases
- EGFR
epidermal growth factor receptor
- PDGFR
platelet derived growth factor receptor
- abl
abelson proto-oncogene
- c-kit
stem cell factor receptor
- CSF-1R
colony stimulating factor 1 Receptor
- Flt-3
FMS-like tyrosine kinase 3
- ErbB-2
human epidermal growth factor receptor 2
- hTS
human thymidylate synthase
- hDHFR
human dihydrofolate reductase
- GARFT
glycinamide-ribonucleotide formyl transferase
- AICARFT
aminoimidazole-4-carboxamide-ribonucleotide formyl transferase
- 5-FU
5-fluorouracil
- PMX
pemetrexed
- RTX
raltitrexed
- MTX
methotrexate
- TMQ
trimetrexate
- PDB
protein data bank
- DMSO2
methyl sulfone
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- POCl3
phosphorus oxychloride
- NMP
N-methyl-2-pyrrolidone
- PI3K
phosphatidylinositol 3-kinase
- CAM
chorioallantoic membrane
Footnotes
Supporting Information Available: Results from elemental analysis and high resolution mass spectrometry. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Davis DW, Herbst RS, Abbruzzese JL. Antiangiogenic Cancer Therapy. CRC Press, Taylor and Francis Group; Boca Raton, Fl: 2008. p. 841. [Google Scholar]
- 2.Folkman J. Angiogenesis. Ann Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 3.Fabbro D, McCormick F, editors. Protein Tyrosine Kinases: From Inhibitors to Useful Drugs. 2006. p. 290. [Google Scholar]
- 4.Gschwind A, Fischer OM, Ullrich A. Timeline: The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 6.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 7.Gorre Mercedes E, Sawyers Charles L. Molecular mechanisms of resistance to STI571 in chronic myeloid leukemia. Curr Opin Hematol. 2002;9:303–307. doi: 10.1097/00062752-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 9.Oestman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15:275–286. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Pearson M, C GE, Fabbro D. Protein Tyrosine Kinases as Targets for Cancer and Other Indications in Protein Tyrosine Kinases. In: Fabbro D, McCormick F, editors. Inhibitors to Useful Drugs. Human Press; Totowa, NJ: 2005. pp. 1–29. [Google Scholar]
- 11.Stein MN, Flaherty KT. CCR drug updates: Sorafenib and sunitinib in renal cell carcinoma. Clin Cancer Res. 2007;13:3765–3770. doi: 10.1158/1078-0432.CCR-06-2844. [DOI] [PubMed] [Google Scholar]
- 12.Tabernero J. The Role of VEGF and EGFR Inhibition: Implications for Combining Anti-VEGF and Anti-EGFR Agents. Mol Cancer Res. 2007;5:203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 14.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 15.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 17.Gasparini G, Longo R, Fanelli M, Teicher BA. Combination of antiangiogenic therapy with other anticancer therapies: Results, challenges, and open questions. J Clin Oncol. 2005;23:1295–1311. doi: 10.1200/JCO.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 18.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Mancuso MR, Davis R, Norberg SM, O'Brien S, Sennino B, Nakahara T, Yao VJ, Inai T, Brooks P, Freimark B, Shalinsky DR, Hu-Lowe DD, McDonald DM. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma J, Waxman DJ. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther. 2008;7:3670–3684. doi: 10.1158/1535-7163.MCT-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gangjee A, Zeng Y, Ihnat M, Warnke LA, Green DW, Kisliuk RL, Lin FT. Novel 5-substituted, 2,4-diaminofuro[2,3-d]pyrimidines as multireceptor tyrosine kinase and dihydrofolate reductase inhibitors with antiangiogenic and antitumor activity. Bioorg Med Chem. 2005;13:5475–5491. doi: 10.1016/j.bmc.2005.04.087. [DOI] [PubMed] [Google Scholar]
- 22.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: Targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O'Reilly MS, Folkman J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- 24.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 25.Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24. doi: 10.1172/JCI8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho QT, Kuo CJ. Vascular endothelial growth factor: Biology and therapeutic applications. Int J Biochem Cell Biol. 2007;39:1349–1357. doi: 10.1016/j.biocel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindahl P, J BR, Levéen P, Betsholtz C. Pericyte Loss and Microaneurysm Formation in PDGF-B-Deficient Mice. Science. 1997;11:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]; Sjöblom TPK, Östman A, Heldin CH. Platelet-derived Growth Factor. Normal Function, Role in Disease and Application of PDGF Antagonists, in Protein Tyrosine Kinases. In: Fabbra D, McCormick F, editors. Inhibitors to Useful Drugs. Humana Press; Totowa, NJ: 2006. pp. 161–186. [Google Scholar]
- 28.Clinical Trials - Combination of Capecitabine with Kinase Inhibitors: Capecitabine (Xeloda) and Lapatinib (Tykerb) as First-line Therapy in HER2/Neu-positive Breast Cancer. http://www.clinicaltrials.gov/ct2/show/NCT00496366?term=capecitabine&rank=4; http://www.clinicaltrials.gov/ct2/show/NCT00496366?term=capecitabine&rank=4; A study of Sunitinib in Combination with Capecitabine Compared with Capecitabine in Patients with Breast Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00435409?term=capecitabine&rank=5; Study of Sunitinib in Combination with Cisplatin/Capecitabine or Oxaliplatin/Capecitabine in Patients with Advanced Gastric Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00555620?term=capecitabine&rank=6; Bevacizumab, Erlotinib and Capecitabine for Locally Advanced Rectal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00543842?term=capecitabine&rank=15; Capecitabine/Erlotinib Followed of Gemcitabine Versus Gemcitabinie/Erlotnib Followed of Capecitabine, http://www.clinicaltrials.gov/ct2/show/NCT00440167; A Phase I Trial of Capecitabine in Combination with Gemcitabine and Erlotinib for Advanced Pancreatic Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00480584; Gemcitabine/Vinorelbine Versus Gemcitabine/Cisplatin Versus Gemcitabine/Capecitabine in Metastatic Breast Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00480597; A Phase II Trial of Gemcitabine, Capecitabine and Bevacizumab in Metastatic Renal Cell Carcinoma, http://www.clinicaltrials.gov/ct2/show/NCT0000523640; Brain Mets - Capecitabine and WBRT, http://www.clinicaltrials.gov/ct2/show/NCT00570908?term=capecitabine&rank=19; Combination Study of Capecitabine and Erlotinib Concurrent with Radiotherapy for Non-operable Advanced Pancreatic Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00565487?term=capecitabine&rank=30; Phase I Vandetanib Plau Capecitabine, Oxaliplatin and Bevacizumab for Metastatic Colorectal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00532909?term=capecitabine&rank=44; Phase I Study of Lapatinib in combination with Oxaliplatin and Capecitabine in Sugjects with Advanced Colorectal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00536809?term=capecitabine&rank=74; Enzastaurin in combination of Capecitabine to Treat Breat Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00437294?term=capecitabine&rank=92; Imatinib Mesylate, Gemcitabine and Capecitabine in Treating Patients with Advanced Solid Tumors, http://www.clinicaltrials.gov/ct2/show/NCT00483366?term=capecitabine&rank=93; Lapatinib + Capecitabine Treatment for Advanced Metastatic Breast Cancer in Women from China, http://www.clinicaltrials.gov/ct2/show/NCT00508274?term=capecitabine&rank=101; A clinical Trial Comparing Efficacy and Safety of Sunitinib and Capecitabine, http://www.clinicaltrials.gov/ct2/show/NCT00373113?term=capecitabine&rank=150; Study for Patients with Untreated Gastric Cancer Who will Receive Capecitabine and Lapatinib, http://www.clinicaltrials.gov/ct2/show/NCT0000570908?term=capecitabine&rank=152; EAP (Expanded Access Protocol) of Lapatinib combinied with Capecitabine in Metastatic Breast Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00338247?term=capecitabine&rank=167; Phase I/II XP + Sorafenib in AGC, http://www.clinicaltrials.gov/ct2/show/NCT00565370?term=capecitabine&rank=250. A Brain Metastases in ErbB2-Positive Breast Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00437073?term=capecitabine&rank=267; Lapatinib in Combination with Capecitabine in Japanese Patients with Metastatic Breast Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00477464?term=capecitabine&rank=309; Clinical Trial on the Mixture of Gemcitabine, Capecitabine and sorafenib (Bay 43-9006) in the Treatment of Patients with Renal Cell Carcinoma (RCC) (SOGUG-02-06), http://www.clinicaltrials.gov/ct2/show/NCT00496301?term=capecitabine&rank=311; Erlotinib, Combination Chemotherapy and Radiation Therapy in Treating Patients with Stage I or Stage II Pancreatic Cancer that can be Removed by Surgery, http://www.clinicaltrials.gov/ct2/show/NCT00313560?term=capecitabine&rank=352.
- 29.Tripathy D. Capecitabine in combination with novel targeted agents in the management of metastatic breast cancer: underlying rationale and results of clinical trials. Oncologist. 2007;12:375–389. doi: 10.1634/theoncologist.12-4-375. [DOI] [PubMed] [Google Scholar]
- 30.Clinical Trials Combination of 5-FU with Kinase Inhibitors: Bevacizumab, Erlotinib and 5-Fluorouracil with External Beam Radiation Therapy in Locally Advanced Rectal Cancer,. http://www.clinicaltrials.gov/ct2/show/NCT00307736; Study of erlotinib and Chemotherapy for Unresectable or Metastatic Cancer of the Esophagus and Gastric Cardia, http://www.clinicaltrials.gov/ct2/show/NCT00591123; Sunitinib, Irinotecan, Fluorouracil and Leucovorini in Treating Patients with Advanced Stomach Cancer or Gastroesophageal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00524186; A Phase I study of Sunitinib in combination with Cisplatin and 5-Fluorouracil in Patients with Advanced Gastric Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00555672; Oxaliplatin, Fluorouracil, Erlotinib and Radiation Therapy before Surgery and Erlotinib After Sursery in Treating Patients with Locally Advanced Cancer of the Esophagus or Gastroesophageal Junction, http://www.clinicaltrials.gov/ct2/show/NCT00499564; Cisplatin, Fluorouracil, Gefitinib and Radiation Therapy in Treating Patients with Locally Advanced Head and Neck Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00352105; Study of FOLFIRI Chemotherapy with or without Sunitinib in Patients with Metastatic Colorectal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00457691; Docetaxel and Lapatinib with or Without combination chemotherapy or Docetaxel and Trastuzumab with Combination Chemotherapy in Treating Women with Locally Advanced, Inflammatory or Resectable Breast Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00450892; Chemotherapy, Radiation Therapy and Immunotherapy Prior to Surgery in Operable Esophageal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00393068; Combination Chemotherapy and Radiation therapy with or Without Lapatinib in Treating Patients with Locally Advanced Cancer of the Larynx or Hypopharynx That Can Be Removed by Surgery, http://www.clinicaltrials.gov/ct2/show/NCT00498953; Lapatinib +/-Trastuzumab in addition to Standard Neoadjuvant Breast Cancer Therapy, http://www.clinicaltrials.gov/ct2/show/NCT00524303; Chemotherapy & Erlotinib in Treating Patients with Esophageal or Gastroesophageal Cancer That Cannot be Removed by Surgery, http://www.clinicaltrials.gov/ct2/show/NCT0000539617; A Phase II Study of 2 Doses of ZD6474 (Vandetanib) in combination with FOLFOX vs FOLFOX Alone for the Treatment of Colorectal Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00500292.
- 31.Clinical Trials - Combination of Pemetrexed with Kinase Inhibitors: A Study to Find the Best Dose of SU011248 When Given with Pemetrexed, Pemetrexed and Cisplatin or Pemetrexed and Carboplatin in Patients with Advanced Solid Tumors,. http://www.clinicaltrials.gov/ct2/show/NCT00528619?term=pemetrexed&rank=1; Efficacy Study Comparing ZD6474 in Combination with Pemetrexed and Pemetrexed Alone in 2nd Line NSCLC Patients (ZEAL), http://clinicaltrials.gov/ct2/show/NCT00418886?term=pemetrexed&rank=2; Docetaxel or Pemetrexed with or Without Cetuximab in Patients with Recurrent or Progressive Non-small Cell Lung Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00095199?term=pemetrexed&rank=3; A Phase II Trial of Lapatinib (TYKERB) + Pemetrexed (ALIMTA) Versus Pemetrexed in Advanced Non-small Cell Lung Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00528281?term=pemetrexed&rank=7; Study of Pemetrexed Versus Pemetrexed + Erlotinib as Treatment of Non-small Cell Lung Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00447057?term=pemetrexed&rank=14; Cisplatin, Imatinib Mesylate, and Pemetrexed in Malignant Mesothelioma Patients, http://www.clinicaltrials.gov/ct2/show/NCT00402766?term=pemetrexed&rank=17; ZD6474 (Vandetanib) + Alimta Combo Study, http://www.clinicaltrials.gov/ct2/show/NCT00506051?term=pemetrexed&rank=22; Study of Enzastaurin Versus Placebo with Pemetrexed for Patients with Advanced or Metastatic Lung Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00530621?term=pemetrexed&rank=27; Pemetrexed Disodium with or Without Sorafenib as Second-line Therapy in Treating Patients with Stage IIIB or Stave IV Non-small Cell Lung Cancer, http://www.clinicaltrials.gov/ct2/show/NCT00454194?term=pemetrexed&rank=31; AZD6244 Versus Pemetrexed (Alimta®) in Patients with Non-small Cell Lung Cancer, Who Have Failed One or Two Prior Chemotherapy Regimen, http://clinicaltrials.gov/ct2/show/NCT00372788?term=pemetrexec&rank=32; Randomized Phase III Trial of Pemetrexed vs Erlotinib in Pretreated Patients with NSCLC, http://clinicaltrials.gov/ct2/show/NCT00440414?term=pemetrexed&rank=34; Vatalanib and Pemetrexed Disodium in Treating Patients with Advanced Solid Tumors, http://clinicaltrials.gov/ct2/show/NCT00390000?term=pemetrexed&rank=37; Sorafenib Combined with Cisplatin and Etoposide or Carboplatin and Pemetrexed in Treating patients with Metastatic Solid Tumors, http://clinicaltrials.gov/ct2/show/NCT00573690?term=pemetrexed&rank=92; Study in Patients with Advanced Non-small Cell Lung Cancer Treated with Pemetrexed and Carboplatin Plus or Minus Sorafenib (PECASO), http://clinicaltrials.gov/ct2/show/NCT00473486?term=pemetrexed&rank=98; Chemotherapy for patients with Non-small Cell Lung Cancer Who Are Non-smokers, http://clinicaltrials.gov/ct2/show/NCT00409006?term=pemetrexed&rank=152; Phase I/II Trial of Bevacizumab, Pemetrexed and Erlotinib in Elderly Patients with Non-small Cell Lung Cancer, http://clinicaltrials.gov/ct2/show/NCT00351039?term=pemetrexed&rank=202; A Study to Evaluate Bevacizumab and Chemotherapy or Tarceva in Treating Recurrent or Refractory NSCLC (Non-small Cell Lung Cancer), http://clinicaltrials.gov/ct2/show/NCT00095225?term=pemetrexed&rank=203; A Study of Tarceva (Erlotinib) and Standard of Care Chemotherapy in Patients with Advanced, Recurrent or Metastatic Non-small Cell Lung Cancer (NSCLC), http://clinicaltrials.gov/ct2/show/NCT00556322?term=pemetrexed&rank=206.
- 32.Huber PE, Bischof M, Jenne J, Heiland S, Peschke P, Saffrich R, Groene HJ, Debus J, Lipson KE, Abdollahi A. Trimodal cancer treatment: Beneficial effects of combined antiangiogenesis, radiation, and chemotherapy. Cancer Res. 2005;65:3643–3655. doi: 10.1158/0008-5472.CAN-04-1668. [DOI] [PubMed] [Google Scholar]
- 33.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 34.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 35.Lehman NL. Future potential of thymidylate synthase inhibitors in cancer therapy. Expert Opin Invest Drugs. 2002;11:1775–1787. doi: 10.1517/13543784.11.12.1775. [DOI] [PubMed] [Google Scholar]
- 36.Pizzorno G, D RB, Cheng YC. In: Pyrimidine and Purine Antimetabolites in Cancer Medicine. Holland JF, Frei E III, editors. B. C. Decker, Inc.; Hamilton, London: 2003. pp. 739–744. [Google Scholar]
- 37.Rollins KD, Lindley C. Pemetrexed: A multitargeted antifolate. Clin Ther. 2005;27:1343–1382. doi: 10.1016/j.clinthera.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 38.Clamp AR, Schoeffski P, Valle JW, Wilson RH, Marreaud S, Govaerts AS, Debois M, Lacombe D, Twelves C, Chick J, Jayson GC. A phase I and pharmacokinetic study of OSI-7904L, a liposomal thymidylate synthase inhibitor in combination with oxaliplatin in patients with advanced colorectal cancer. Cancer Chemother Pharmacol. 2008;61:579–585. doi: 10.1007/s00280-007-0509-5. [DOI] [PubMed] [Google Scholar]
- 39.Showalter HDH, Bridges AJ, Zhou H, Sercel AD, McMichael A, Fry DW. Tyrosine Kinase Inhibitors. 16. 6,5,6-Tricyclic Benzothieno[3,2-d]pyrimidines and Pyrimido[5,4-b]- and -[4,5-b]indoles as Potent Inhibitors of the Epidermal Growth Factor Receptor Tyrosine Kinase. J Med Chem. 1999;42:5464–5474. doi: 10.1021/jm9903949. [DOI] [PubMed] [Google Scholar]
- 40.Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T, Lydon N. 4-(Phenylamino)pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase. J Med Chem. 1996;39:2285–2292. doi: 10.1021/jm960118j. [DOI] [PubMed] [Google Scholar]
- 41.Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Therapeut. 1999;82:195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- 42.Gangjee A, Yang J, Ihnat MA, Kamat S. Antiangiogenic and antitumor agents. Design, synthesis, and evaluation of novel 2-amino-4-(3-bromoanilino)-6-benzylsubstituted pyrrolo[2,3-d]pyrimidines as inhibitors of receptor tyrosine kinases. Bioorg Med Chem. 2003;11:5155–5170. doi: 10.1016/j.bmc.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 43.Laufer SA, Domeyer DM, Scior TRF, Albrecht W, Hauser DRJ. Synthesis and Biological Testing of Purine Derivatives as Potential ATP-Competitive Kinase Inhibitors. J Med Chem. 2005;48:710–722. doi: 10.1021/jm0408767. [DOI] [PubMed] [Google Scholar]
- 44.Abu Thaher B, Koch P, Schattel V, Laufer S. Role of the hydrogen bonding heteroatom-Lys53 interaction between the p38α mitogen-activated protein (MAP) kinase and pyridinyl-substituted 5-membered heterocyclic ring inhibitors. J Med Chem. 2009;52:2613–2617. doi: 10.1021/jm801467h. [DOI] [PubMed] [Google Scholar]
- 45.Gangjee A, Lin X, Kisliuk RL, McGuire JJ. Synthesis of N-{4-[(2,4-Diamino-5-methyl-4,7-dihydro-3H- pyrrolo[2,3-d]pyrimidin-6-yl)thio]benzoyl}-L-glutamic Acid and N-{4-[(2-Amino-4-oxo-5-methyl-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin- 6-yl)thio]benzoyl}-L-glutamic Acid as Dual Inhibitors of Dihydrofolate Reductase and Thymidylate Synthase and as Potential Antitumor Agents. J Med Chem. 2005;48:7215–7222. doi: 10.1021/jm058234m. [DOI] [PubMed] [Google Scholar]
- 46.Rosowsky A, Forsch RA, Freisheim JH, Danenberg PV, Moran RG, Wick MM. Methotrexate analogs. 29. Effect of γ-aminobutyric acid spacers between the pteroyl and glutamate moieties on enzyme binding and cell growth inhibition. J Med Chem. 1986;29:1872–6. doi: 10.1021/jm00160a014. [DOI] [PubMed] [Google Scholar]
- 47.Gangjee A, Jain HD, Kurup S. Recent advances in classical and non-classical antifolates as antitumor and antiopportunistic infection agents: part I. Anti-Cancer Agents Med Chem. 2007;7:524–542. doi: 10.2174/187152007781668724. [DOI] [PubMed] [Google Scholar]
- 48.Gangjee A, Qiu Y, Li W, Kisliuk RL. Potent Dual Thymidylate Synthase and Dihydrofolate Reductase Inhibitors: Classical and Nonclassical 2-Amino-4-oxo-5-arylthio-substituted-6-methylthieno[2,3-d]pyrimidine Antifolates. J Med Chem. 2008;51:5789–5797. doi: 10.1021/jm8006933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gangjee A, Li W, Yang J, Kisliuk RL. Design, Synthesis, and Biological Evaluation of Classical and Nonclassical 2-Amino-4-oxo-5-substituted-6-methylpyrrolo[3,2-d]pyrimidines as Dual Thymidylate Synthase and Dihydrofolate Reductase Inhibitors. J Med Chem. 2008;51:68–76. doi: 10.1021/jm701052u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cody V, Luft Joseph R, Pangborn W, Gangjee A. Analysis of three crystal structure determinations of a 5-methyl-6-N-methylanilino pyridopyrimidine antifolate complex with human dihydrofolate reductase. Acta Crystallogr D Biol Crystallogr. 2003;59:1603–1609. doi: 10.1107/s0907444903014963. [DOI] [PubMed] [Google Scholar]
- 51.Tripos Inc., 1699 South Hanley Road, St. Louis, MO 63144.
- 52.Davies JF, II, Delcamp TJ, Prendergast NJ, Ashford VA, Freisheim JH, Kraut J. Crystal structures of recombinant human dihydrofolate reductase complexed with folate and 5-deazafolate. Biochemistry. 1990;29:9467–9479. doi: 10.1021/bi00492a021. [DOI] [PubMed] [Google Scholar]
- 53.Oefner C, D'Arcy A, Winkler FK. Crystal structure of human dihydrofolate reductase complexed with folate. Eur J Biochem. 1988;174:377–385. doi: 10.1111/j.1432-1033.1988.tb14108.x. [DOI] [PubMed] [Google Scholar]
- 54.Sayre PH, Finer-Moore JS, Fritz TA, Biermann D, Gates SB, MacKellar WC, Patel VF, Stroud RM. Multi-targeted Antifolates Aimed at Avoiding Drug Resistance Form Covalent Closed Inhibitory Complexes with Human and Escherichia coli Thymidylate Synthases. J Mol Biol. 2001;313:813–829. doi: 10.1006/jmbi.2001.5074. [DOI] [PubMed] [Google Scholar]
- 55.Miyazaki Y, Matsunaga S, Tang J, Maeda Y, Nakano M, Philippe RJ, Shibahara M, Liu W, Sato H, Wang L, Nolte RT. Novel 4-aminofuro[2,3-d]pyrimidines as Tie-2 and VEGFR2 dual inhibitors. Bioorg Med Chem Lett. 2005;15:2203–2207. doi: 10.1016/j.bmcl.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 56.Mol CD, Lim KB, Sridhar V, Zou H, Chien EYT, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-Kit Product Complex Reveals the Basis for Kinase Transactivation. J Biol Chem. 2003;278:31461–31464. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- 57.Molecular Operating environment (MOE 2007.09) C. C. G., Inc.; 1255 University Street, Suite 1600, Montreal, Quebec, Canada, H3B 3X3: [Google Scholar]
- 58.Altschul SF, Koonin EV. Iterated profile searches with PSI-BLAST - a tool for discovery in protein databases. Trends Biochem Sci. 1998;23:444–447. doi: 10.1016/s0968-0004(98)01298-5. [DOI] [PubMed] [Google Scholar]
- 59.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahboobi S, Uecker A, Sellmer A, Cenac C, Hoecher H, Pongratz H, Eichhorn E, Hufsky H, Truempler A, Sicker M, Heidel F, Fischer T, Stocking C, Elz S, Boehmer FD, Dove S. Novel Bis(1H-indol-2-yl)methanones as Potent Inhibitors of FLT3 and Platelet-Derived Growth Factor Receptor Tyrosine Kinase. J Med Chem. 2006;49:3101–3115. doi: 10.1021/jm058033i. [DOI] [PubMed] [Google Scholar]
- 61.Henderson EA, Bavetsias V, Theti DS, Wilson SC, Clauss R, Jackman AL. Targeting the a-folate receptor with cyclopenta[g]quinazoline-based inhibitors of thymidylate synthase. Bioorg Med Chem. 2006;14:5020–5042. doi: 10.1016/j.bmc.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 62.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–178. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 63.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 64.Ihnat MA, Kaltreider RC, Thorpe JE, Green DE, Kamat CD, Leeper M, Shanner AC, Warnke LA, Piconi L, Ceriello A. Attenuated superoxide dismutase induction in retinal cells in response to intermittent high versus continuous high glucose. Am J Biochem Biotechnol. 2007;3:16–23. [Google Scholar]
- 65.Schroeder MC, Hamby JM, Connolly CJ, Grohar PJ, Winters RT, Barvian MR, Moore CW, Boushelle SL, Crean SM, Kraker AJ, Driscoll DL, Vincent PW, Elliott WL, Lu GH, Batley BL, Dahring TK, Major TC, Panek RL, Doherty AM, Showalter HD. Soluble 2-substituted aminopyrido[2,3-d]pyrimidin-7-yl ureas. Structure-activity relationships against selected tyrosine kinases and exploration of in vitro and in vivo anticancer activity. J Med Chem. 2001;44:1915–1926. doi: 10.1021/jm0004291. [DOI] [PubMed] [Google Scholar]
- 66.Zaman GJR, Vink PMF, van den Doelen AA, Veeneman GH, Theunissen HJM. Tyrosine kinase activity of purified recombinant cytoplasmic domain of platelet-derived growth factor β-receptor (β -PDGFR) and discovery of a novel inhibitor of receptor tyrosine kinases. Biochem Pharmacol. 1999;57:57–64. doi: 10.1016/s0006-2952(98)00271-8. [DOI] [PubMed] [Google Scholar]
- 67.Mori Y, Hirokawa T, Aoki K, Satomi H, Takeda S, Aburada M, Miyamoto Ki. Structure activity relationships of quinoxalin-2-one derivatives as platelet-derived growth factor- β receptor (PDGF β R) inhibitors, derived from molecular modeling. Chem Pharm Bull. 2008;56:682–687. doi: 10.1248/cpb.56.682. [DOI] [PubMed] [Google Scholar]
- 68.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 69.Wilson SM, Barsoum MJ, Wilson BW, Pappone PA. Purine nucleotides modulate proliferation of brown fat preadipocytes. Cell Proliferat. 1999;32:131–140. doi: 10.1046/j.1365-2184.1999.32230131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maurer BJ, Ihnat MA, Morgan C, Pullman J, O'Brien C, Johnson SW, Rasey JS, Cornwell MM. Growth of human tumor cells in macroporous microcarriers results in p53-independent, decreased cisplatin sensitivity relative to monolayers. Mol Pharmacol. 1999;55:938–947. [PubMed] [Google Scholar]
- 71.Fong TA, Shawver LK, Sun L, Tang C, App H, Powell TJ, Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth KP, McMahon G. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- 72.Stockwell BR, Haggarty SJ, Schreiber SL. High-throughput screening of small molecules in miniaturized mammalian cell-based assays involving post-translational modifications. Chem Biol. 1999;6:71–83. doi: 10.1016/S1074-5521(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 73.Gangjee A, Namjoshi OA, Yu J, Ihnat MA, Thorpe JE, Warnke LA. Design, synthesis and biological evaluation of substituted pyrrolo[2,3-d]pyrimidines as multiple receptor tyrosine kinase inhibitors and antiangiogenic agents. Bioorg Med Chem. 2008;16:5514–5528. doi: 10.1016/j.bmc.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- 75.Kisliuk RL, Strumpf D, Gaumont Y, Leary RP, Plante L. Diastereoisomers of 5,10-methylene-5,6,7,8-tetrahydropteroyl-D-glutamic acid. J Med Chem. 1977;20:1531–1533. doi: 10.1021/jm00221a038. [DOI] [PubMed] [Google Scholar]
- 76.Wahba AJ, Friedkin M. Enzymic synthesis of thymidylate. I. Early steps in the purification of thymidylate synthetase of Escherichia coli. J Biol Chem. 1962;237:3794–3801. [PubMed] [Google Scholar]
- 77.Davisson VJ, Sirawaraporn W, Santi DV. Expression of human thymidylate synthase in Escherichia coli. J Biol Chem. 1989;264:9145–9148. [PubMed] [Google Scholar]
- 78.Tsuda H, Sakaguchi M, Kawakita M, Nakazawa S, Mori T, Takatsuki K. Alteration of cell cycle progression in human leukemia cell line (KOPM-28) induced by 12-o-tetradecanoylphorbol-13-acetate. Int J Cell Cloning. 1988;6:209–220. doi: 10.1002/stem.5530060306. [DOI] [PubMed] [Google Scholar]
- 79.Iliakis G, Nusse M. Arrest of irradiated G1, S, or G2 cells at mitosis using nocodazole promotes repair of potentially lethal damage. Radiat Res. 1984;99:346–351. [PubMed] [Google Scholar]
- 80.Lucarelli E, Sangiorgi L, Benassi S, Donati D, Gobbi GA, Picci P, Vacca A, Ribatti D. Angiogenesis in lipoma: An experimental study in the chick embryo chorioallantoic membrane. Int J Mol Med. 1999;4:593–596. doi: 10.3892/ijmm.4.6.593. [DOI] [PubMed] [Google Scholar]
- 81.Romanoff AL. Biochemistry of the Avian Embryo; A Quantitative Analysis of Prenatal Development. 1967. p. 398. [Google Scholar]
- 82.Hanna N, Fidler IJ. Role of natural killer cells in the destruction of circulating tumor emboli. J Natl Cancer Inst. 1980;65:801–809. doi: 10.1093/jnci/65.4.801. [DOI] [PubMed] [Google Scholar]
- 83.Hanna N, Davis TW, Fidler IJ. Environmental and genetic factors determine the level of NK activity of nude mice and affect their suitability as models for experimental metastasis. Int J Cancer. 1982;30:371–376. doi: 10.1002/ijc.2910300319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.