

Abstract
Immunohistochemistry for mismatch repair proteins has shown utility in the identification of Lynch syndrome, but majority of tumours with loss MLH1 expression are due to sporadic hypermethylation of the MLH1 promoter. These tumours can also show epigenetic silencing of other genes, such as p16. The aim of our study is to evaluate the utility of p16 immunohistochemistry in the prediction of MLH1 germline mutations.
METHODS:
p16 immunohistochemistry was appropriately evaluated in 79 colorectal cancers with loss of MLH1 expression. Methylation of MLH1 and p16 were quantitatively studied using real time PCR assay Methylight. BRAF V600E mutation in tumour tissue was also investigated. Genetic testing for germline mutation of MLH1 was made on 52 patients.
RESULTS:
Loss of p16 expression was seen in 21 out of 79 samples (26,6%). There was found statistically significant association between p16 expression and p16 methylation (p<0.001), MLH1 methylation (p<0.001) and BRAF mutation (p<0.005). All tumours with loss of p16 expression showed hypermethylation of p16 (21/21), 95.2% (20/21) showed MLH1 methylation and 71.4% (15/21) were mutated for BRAF V600E Mutational analysis showed pathogenic germline mutations in 8 of the patients, harbouring 10 tumours. All 10 of these tumours showed normal staining of p16 in the immunochemical analysis.
CONCLUSIONS:
p16 immunohistochemistry is a good surrogate marker for p16 and MLH1 epigenetic silencing due to hypermethylation, and is useful as screening tool in the selection of patients for genetic testing in Lynch syndrome.
Keywords: colorectal cancer, Lynch syndrome, p16, immunohistochemistry, diagnosis
BACKGROUND
Lynch syndrome is an autosomal dominant disorder that accounts for approximately 3-4% of all colorectal cancers (CRC) (1). Lynch syndrome is caused by germline mutations in the DNA mismatch repair (MMR) genes, mainly MLH1, MSH2, MSH6 and PMS2 (1). Defects in this pathway lead to changes in the length of nucleotide repeat sequences, a phenomenon called microsatellite instability (MSI), which constitutes the molecular hallmark of HNPCC (2). These tumours can also be identified by immunohistochemical loss of MMR proteins (3;4). The presence of MSI may be observed in up to 10-15% of sporadic CRC. In these cases, MMR impairment is caused by epigenetic silencing of MLH1, due to MLH1 promoter methylation (5).
Since molecular characterization of Lynch syndrome was established, the identification of gene carriers has become a critical issue. Identification of patients with Lynch syndrome has important clinical implications because surveillance for CRC and other cancers in this population is able to reduce cancer mortality and is cost-effective (6). A previous study from our group established that fulfilment of revised Bethesda criteria (7), followed by either MSI testing or MMR proteins immunohistochemistry is a sensible approach to pre-select patients for genetic testing (4). Patients having tumours with loss of expression of MSH2 or MSH6 are suspected carriers of germline mutations of any of these genes, but patients whose tumours show loss of MLH1 may either have hereditary or sporadic disease. The majority of sporadic tumours with loss of MLH1 expression belong to a group of colorectal cancers that are hypermethylated at multiple genetic loci. These CRC have been described as displaying the CpG Island Methylator Phenotype (CIMP) (8;9), and a panel of markers has been proposed for its diagnosis (10). One of the loci frequently methylated in these CIMP tumours is CDKN2A (p16). Presumably, some of the tumours with loss of MLH1 caused by epigenetic silencing through aberrant methylation, should also have a silenced p16, and, therefore, immunohistochemical loss of staining of this protein. The aim of our study is to evaluate the value of p16 immunohistochemistry in the prediction of MLH1 germline mutations in patients with tumours that show loss of MLH1.
METHODS
Subjects
Immunohistochemical analysis of MLH1 was performed in 2401 CRC tumours. Tumour tissue was collected from a series of 2,246 non-selected surgical CRC specimens from the EPICOLON study (n=1.281) (11) and from the Pathology Department of the Hospital General Universitario of Alicante, collected between the years 1999-2007 (n=965). The remaining 155 tumours were collected from patients of the Genetic Counselling in Cancer Department of the Hospital General Universitario of Elche. Demographic, clinical, and tumor-related characteristics of probands, as well as a detailed family history were obtained using a pre-established questionnaire, as described elsewhere (4). Loss of MLH1 expression was found in 124 tumors (5.2%), from 120 patients. All these tumours showed normal expression of MSH2 and MSH6. In 32 cases there was not enough tissue to perform immunohistochemical or molecular studies and they were excluded from this study. Finally, the study was performed in 92 tumours from 88 patients that showed loss of MLH1 immunohistochemical expression. Eighty-three tumours were non selected population-based CRC specimens and 9 were from the Genetic Counselling Unit. Figure 1 shows a flow chart of the molecular analysis performed on the samples.
Figure 1.
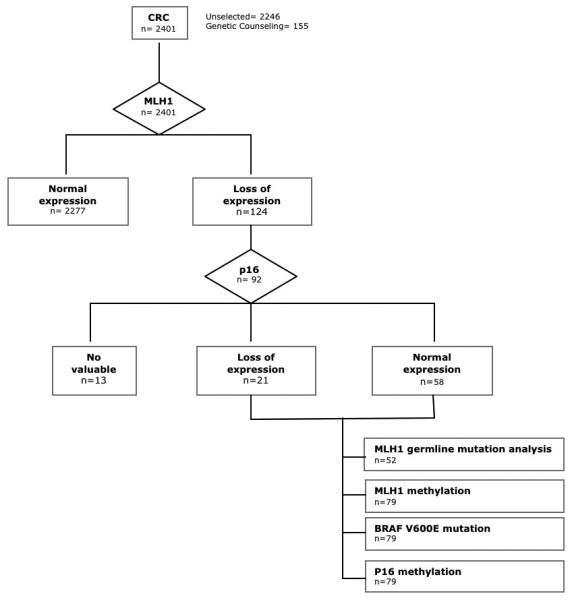
Flow diagram for the immunohistochemical and molecular analysis performed in tumors.
Immunohistochemistry
Immunohistochemical analysis of MLH1, MSH2, MSH6 and PMS2 was performed in blocks of formalin-fixed paraffin-embedded tumour tissue as previously described (4;12).
Immunohistochemical analysis of p16 expression was performed on tissue microarray (TMA). One of the requirements for inclusion in the study was that enough tumour tissue was present within the block of wax-embedded tissue to facilitate subsequent TMA construction. The representative tumour regions were identified and marked on the H&E stained slides and subsequently identified on the corresponding tissue blocks. Tissue cylinders of diameter of 1 mm were punched out from the marked areas of each block and incorporated into a recipient paraffin block using a precision instrument—the tissue arrayer (Beecher Instruments, Durviz). A total of 6 TMAs were constructed for the study. TMAs contained between 30 and 50 cores of 1 mm needle size. For inclusion in the study, at least two evaluable cores of tumour tissue were required per case. Four-micrometer-thick sections were cut from TMAs. The slides were put on a TechMate 500 immunostainer and incubated for 30 minutes at room temperature with the mouse monoclonal antibody JC2, which recognizes the first ankyrin repeat of p16 (provided by Dr. Jim Koh, Duke University, Durham, NC, USA) (13). The antibody was detected by the Envision+ technique (Dako). Processed immunohistochemical slides were evaluated by two pathologists. A tumour was considered to have normal expression for p16 when unequivocal nuclear staining was seen in some neoplastic epithelial cells, with or without cytoplasmatic staining. Cases with loss of expression included those cases with lack of expression in tumour cells in presence of internal positive control (stromal cells or blood vessels). Samples were considered not scored when no staining of internal control was seen.
MLH1 and CDKN2A methylation analysis
Genomic DNA was extracted from tumour paraffin-embedded tissue blocks. Two tissue cylinders of 1 mm of diameter were punched out with the tissue arrayer from the previously selected tumour areas. QiaAmp DNA Mini kit for DNA extraction was used according to the manufacturer's protocol after removal of paraffin by xylene.
The MLH1 and CDKN2A (p16) methylation analysis was performed by real time PCR assay Methylight as previously described (Applied Biosystems, Foster City, CA, USA) (14). Bisulphite conversion was made with the EZ DNA methylation-Gold kit as described by the manufacturer (Zymo Research, Orange, CA, USA). Quantitative PCR was performed by ABI 7500 (Applied Biosystems, Foster City, CA, USA). Primers and a probe, designed to detect bisulphite converted fully methylated MLH1 and p16 DNAs, have been described and used previously (10;15-17). The PCR reactions were performed according to the protocols (16;18).
In order to calculate the percentage of methylated reference (PMR) we established the dichotomization threshold at PMR= 4, to obtain a bimodal distribution in the MLH1 and CDKN2A methylation loci. Methylation positive (PMR > 4) MLH1 and CDKN2A samples could be distinguished from negative (PMR ≤ 4) ones.
BRAF V600E Mutation
V600E BRAF mutation was detected using specific TaqMan probes by real time PCR (ABI PRISM 7500, Applied Biosystems, Foster City, CA, USA) and the allelic discrimination software (Applied Biosystems) as previously described by Benlloch et al. (19)
MLH1 germline genetic testing
Germline genetic alteration studies were performed on genomic DNA isolated from peripheral blood leukocytes or from non-tumour colon tissue as previously described (4). Point mutation analysis of MLH1 gene was done by polymerase chain reaction (PCR) amplification and direct sequencing of the entire coding region and the exon-intron boundaries. PCR primers and conditions have been described elsewhere (20-22). Large genomic rearrangements (insertions and/or deletions) in MLH1 loci were screened by multiplex ligation-dependent probe amplification (MLPA) according to the manufacturer protocols (Salsa MLPA Kit P003 and P008; MRC-Holland, Amsterdam, The Netherlands).
Data management and analysis
Data were collected and entered into the computer using MICROSOFT ACCESS software for storage and initial analysis. Further analysis was done using SPSS software (SPSS 15.0). For continuous variables relevant measures of central tendency (means for normally distributed data and medians and interquartile ranges for skewed data) were used to explore data. The Chi2 test was used for comparison of qualitative variables. A Student's t test was used for comparison of normally distributed continuous variables and a Mann-Whitney U test was used for unpaired comparison of non-normally distributed continuous variables. A p value of less than 0.05 was considered significant.
RESULTS
p16 immunohistochemistry was performed in 92 tumours with loss of MLH1 expression (Figure 2) from 88 patients. In 13 of the tumours, p16 immunohistochemistry could not be confidently assessed and was classified as not scored, due to absence of clear staining in stromal cells, which served as internal positive controls. Loss of p16 expression was seen in 21 out of 79 samples (26,6%) (Fig.2). Characteristics of tumours according to p16 expression status can be seen in table 1. There was a statistically significant association between p16 expression and p16 methylation (p<0.001), MLH1 methylation (p<0.001) and BRAF mutation (<0.005). All tumours with loss of p16 expression showed hypermethylation of p16 (21/21), 95.2% (20/21) showed MLH1 methylation and 71.4% (15/21) were mutated for BRAF V600E (table 1). However, 20 out of 41 (50%) of the tumours with p16 methylation retained p16 expression (Table 1). Tumours with loss of p16 expression showed more frequently poor differentiation. p16 immunohistochemistry was also performed in 60 sporadic tumours with normal expression of MLH1 and microsatellite stability, loss of p16 expression was seen in only 5 of these tumours (3%).
Figure 2.




p16 immunohistochemistry. A-C. Positive cases with nuclear and cytoplasmatic staining of tumour cells. B-D. Negative cases with stromal staining as a internal control
Table 1.
Characteristics of tumors regarding p16 expression
| P16 normal expression (n=58) |
P16 loss of expression (n=21) |
TOTAL (n=79) |
Odds ratio | p | ||||
|---|---|---|---|---|---|---|---|---|
| me | P25-P75 | Me | P25-P75 | me | P25-P75 | |||
| Age at diagnosis | 73 | 52-80 | 77 | 69-79 | 75 | 50-80 | N.S. | |
| n | % | N | % | n | % | |||
| Age at diagnosis | ||||||||
| <50 years | 14 | 24.1 | 2 | 9.5 | 16 | 20.3 | 3.0 (0.6-14.6) | N.S. |
| >50 years | 44 | 75.9 | 19 | 90.5 | 63 | 79.7 | ||
| Sex | ||||||||
| Male | 27 | 46.6 | 8 | 38.1 | 35 | 44.3 | 1.4 (0.5-3.9) | N.S. |
| Female | 31 | 53.4 | 13 | 61.9 | 44 | 55.7 | ||
| Revised Bethesda guidelines | ||||||||
| Fulfilling | 31 | 53.4 | 5 | 23.8 | 36 | 45.6 | 3.7 (1.2-11.4) | <0.05 |
| Not fulfilling | 27 | 46.6 | 16 | 76.2 | 43 | 54.4 | ||
| Amsterdam II criteria | ||||||||
| Fulfil | 11 | 19.0 | 1 | 4.8 | 12 | 15.2 | 4.7 (0.6-38.7) | N.S. |
| No fulfil | 47 | 81.0 | 20 | 95.2 | 67 | 84.8 | ||
| Tumour location | ||||||||
| Right-sided | 47 | 81.0 | 15 | 71.4 | 62 | 78.5 | 1.8 (0.6-5.8) | N.S. |
| Left-sided | 11 | 19.0 | 6 | 28.6 | 17 | 21.5 | ||
| Grade | ||||||||
| Poorly differenciated | 37 | 63.8 | 9 | 42.9 | 46 | 58.2 | 2.3 (0.9-6.5) | N.S. |
| Other | 21 | 36.2 | 12 | 57.1 | 33 | 41.8 | ||
| Methylation p16 | ||||||||
| Not methylated | 38 | 65.5 | 0 | 0 | 38 | 48.1 | - | <0.001 |
| Methylated | 20 | 34.5 | 21 | 100 | 41 | 51.9 | ||
| BRAF V600E mutation | ||||||||
| Not mutated | 41 | 70.7 | 6 | 28.6 | 47 | 59.5 | 5.6 (1.9-17.1) | <0.005 |
| Mutated | 17 | 29.3 | 15 | 71.4 | 32 | 40.5 | ||
| Methylation MLH1 | ||||||||
| Not methylated | 28 | 48.3 | 1 | 4.8 | 29 | 36.7 | 18.7 (2.3-148.4) | <0.001 |
| Methylated | 30 | 51.7 | 20 | 95.2 | 50 | 63.3 | ||
Me: median;
- Odds ratio of p16 cannot be calculated.
Mutational analysis of MLH1 was performed in 52 out of 88 patients whose tumours showed loss of MLH1 staining. Fifty-four CRC from these 52 patients were analyzed. Thirty of these patients fulfilled some of the revised Bethesda criteria and 11 fulfilled Amsterdam II criteria. Mutational analysis showed pathogenic mutations in 8 of the patients, harbouring 10 tumours. All 10 of the tumours analyzed from the 8 patients with germline pathogenic mutations in MLH1 showed normal staining of p16. All patients with germline mutations met Bethesda criteria. Moreover, all tumours from patients with germline mutations showed non-mutated BRAF and non-methylated MLH1 (table 2).
Table 2.
Characteristics of tumours in patients with germline mutation.
| Germline mutation | Yes (tumours n=10) (patients n=8) |
No (tumours n=44) (patients n=44) |
|||
|---|---|---|---|---|---|
| n | % | n | % | P | |
| IHC p16 | |||||
| Normal expression | 10 | 100 | 30 | 69.8 | <0.05 |
| Loss of expression | 0 | 0 | 14 | 30.2 | |
| Methylation p16 | |||||
| Not methylated | 8 | 80 | 20 | 46.5 | 0.056 |
| Methylated | 2 | 20 | 24 | 53.5 | |
| Methylation MLH1 | |||||
| Not methylated | 10 | 100 | 9 | 20.9 | <0.001 |
| Methylated | 0 | 0 | 35 | 79.1 | |
| BRAF V600E mutation | |||||
| Not mutated | 10 | 100 | 25 | 55.8 | <0.01 |
| Mutated | 0 | 0 | 19 | 44.2 | |
| Bethesda guidelines | |||||
| Fulfilling | 10 | 100 | 21 | 46.5 | <0.005 |
| No fulfilling | 0 | 0 | 23 | 53.5 | |
| Ámsterdam II criteria | |||||
| Fulfilling | 7 | 70 | 4 | 9.3 | <0.001 |
| No fulfilling | 3 | 30 | 40 | 90.7 | |
Me: median
Table 3 shows values of sensitivity, specificity, positive and negative predictive value, and positive Likelihood ratio for Bethesda criteria, BRAF mutation, MLH1 methylation, p16 immunohistochemistry. Different combinations of these variables for the prediction of germline MLH1 mutation can also be seen in table 3. Values for p16 immunohistochemistry and BRAF mutation are similar, and combination of these techniques improves separately obtained results.
Table 3.
Values of different strategies for detecting germline mutation in tumours with MLH1 loss of expression.
| GERMLINE MUTATION | ||||||
|---|---|---|---|---|---|---|
| Strategies without clinical information | Sen | Spe | PPV | NPV | +LR | NNT |
| p16 IHC | 100 | 30.2 | 25 | 100 | 1.4 | 3.1 |
| BRAF mutation | 100 | 44.2 | 29.4 | 100 | 1.8 | 2.3 |
| p16 IHC + BRAF mutation | 100 | 51.2 | 32.3 | 100 | 2.0 | 1.9 |
| MLH1 methylation | 100 | 79.1 | 52.6 | 100 | 4.8 | 1.4 |
| BRAF mutation + MLH1 methylation | 100 | 79.1 | 52.6 | 100 | 4.8 | 1.3 |
| p16 IHC + MLH1 methylation | 100 | 79.1 | 52.6 | 100 | 4.8 | 1.2 |
| p16 IHC + BRAF mutation + MLH1 methylation | 100 | 79.1 | 52.6 | 100 | 4.8 | 1.2 |
| Strategies needing clinical information | ||||||
| BETHESDA | 100 | 53.5 | 33.3 | 100 | 2.2 | 1.9 |
| p16 IHC + BETHESDA | 100 | 62,8 | 38,5 | 100 | 2.7 | 1.6 |
| BRAF mutation + BETHESDA | 100 | 67.4 | 41.7 | 100 | 3.1 | 1.5 |
| p16 IHC + BRAF mutation + BETHESDA | 100 | 69.8 | 43.5 | 100 | 3.3 | 1.4 |
| BRAF mutation + BETHESDA + MLH1 methylation | 100 | 83.7 | 58.8 | 100 | 6.1 | 1.2 |
| BETHESDA + MLH1 methylation | 100 | 83.7 | 58.8 | 100 | 6.1 | 1.2 |
| p16 IHC + BRAF mutation + BETHESDA + MLH1 methylation | 100 | 83.7 | 58.8 | 100 | 6.1 | 1.2 |
Sen: Sensitivity, Spe: Specificity, PPV: Positive Predictive Value, NPV: Negative Predictive Value, +LR: Positive Likelihood Ratio, NNT: Number Needed to Test for detecting one germline mutation
DISCUSSION
Selection of patients for genetic testing in Lynch syndrome is frequently difficult in clinical practice. The use of Amsterdam criteria is capable to detect Lynch Syndrome with a high specificity but with very low sensitivity. When clinical presentation and family history are most compelling, the yield of mutational testing is often no better than 50%, and even in the best case-scenario, when Amsterdam criteria are met and a tumour shows high MSI and loss of MMR protein expression, the likelihood of germline mutation detection is approximately 70-80% (23). Other strategies, such as the revised Bethesda criteria (7), improve the sensitivity but with a high lack of specificity. With this approach, a high number of patients are sent for genetic testing based only on clinical criteria, with the subsequent expending of resources and the consequent generation of anxiety to patients and their families. Several approaches have been used for refining the selection of patients for genetic testing. The observation that patients with Lynch syndrome show a characteristic phenotype with microsatellite instability prompted to use these markers as a first pre-screening modality. Then, the demonstration of the role of the immunohistochemistry and its equivalence to microsatellite instability analysis in the diagnostic algorithm of Lynch syndrome (4) improved the availability of these tools and its generalization in clinical practice, due to the possibility of performing immunohistochemistry in any pathology department. Moreover, patients with tumours showing MSH2 or MSH6 lack of expression should be directly sent for genetic testing, because it is a strong indicator for mutation in these genes (23). However, this clinical-molecular strategy have had some detractors, because a number of patients with Lynch syndrome might not fulfil revised Bethesda criteria (24). Sometimes family history is difficult or even impossible to obtain. Furthermore, recent studies show that, even among patients with a known high risk for Lynch syndrome, there is a marked underutilization of MSI analysis (25). For these reasons, some authors advocate for routine molecular screening of patients with CRC for Lynch syndrome using immunohistochemistry (24). Another fact that can support routine immunohistochemical study of CRC is the recognized better prognosis of mismatch repair deficient tumours (26), and the different response to 5-fluorouracil based chemotherapy that these tumours have (27-29). In our study, we included only patients with MLH1 loss of expression, and compared molecular only with clinical-molecular approaches for diagnosis, showing that combinations of only molecular tests are at least as good as strategies that include clinical data (Table 3). Our results show that p16 immunohistochemistry can improve the results of this strategy, avoiding germline genetic testing in approximately a third of patients with loss of MLH1 expression.
Instruments for the refinement of the selection of patients with loss of MLH1 for genetic testing have been proposed. Mutation V600E in the oncogene BRAF has been suggested as characteristic of sporadic colorectal tumours with MSI and this mutation is not detected in tumours from patients with germline mutations in MLH1 or MSH2 genes (30;31). Several studies have demonstrated that detection of BRAF V600E mutation could simplify the selection of CRC patients for genetic testing for Lynch syndrome (32;33). However, the use of BRAF mutational analysis in clinical practice has been limited, probably due to the need of molecular biology resources for its implementation. The main strength of p16 immunohistochemistry for clinical use in selection of suspected Lynch syndrome patients for genetic testing is its feasibility, in contraposition to other methylation markers such as V600E BRAF mutation or MLH1 methylation (34), that are time consuming and not available for the majority of clinical centresAberrant promoter hypermethylation associated with transcriptional silencing of multiple tumour suppressor genes has been proposed as a mechanistic component in the evolution of multiple cancers (35). Tumours with a critical degree of aberrant methylation have the CpG island methylator phenotype (CIMP). CIMP tumours show promoter hypermethylation in multiple genes, including p16, p14, MGMT and MLH1 among others. Loss of the INK4a/ARF/INK4b locus is among the most frequent cytogenetic events in human cancer. The products of this locus p15INK4a, p16INK4b and ARF play widespread and independent roles in tumour suppression (36). Specific somatic loss of p16, through point mutation or small deletion, has been reported in human cancer (37), but epigenetic silencing through aberrant promoter methylation is the most common mechanism of inactivation (36;38). p16 loss of expression provokes increase in proliferation and vascularisation in colon cancer cells (13;39).
Limitations of our study are the small number of patients with MLH1 germline mutations that we included. However, the excellent sensitivity of p16 expression for MLH1 methylation, with virtually all the cases with loss of p16 expression being methylated, makes p16 immunohistochemistry a robust marker for this event. Most samples include abundant stromal components that stain for p16, providing an internal positive control to verify adequate tissue preservation and technical success of the staining. Another limitation is the existence of cases with p16 hypermethylation that showed normal p16 staining. This fact may be caused, at least in part, by the target region analyzed for the p16 methylation. The Methylight system (primers and probe) used here has been described elsewhere, being useful to characterize the CpG island methylator phenotype (18). The amplicon sequence analyzed is located at exon 1α. Using in vitro models, Gonzalgo et al (1998) observed that p16 expression could occur in the presence of a relatively heavily methylated coding domain (exon 1α, named as region D). Methylation of certain regions upstream of the p16 exon 1α may be more critical for transcription activity (particularly region C) (40). Exonic CpG islands are more susceptible to de novo methylation than promoter islands. The cancer-specific promoter methylation might be a result of spreading from exonic foci and selection of cells whose growth is deregulated by the gene inactivation (41).
In conclusion, our results suggest that the immunohistochemical study of p16 could improve the selection of patients for genetic testing of germline mutations in MLH1. Patients with CRC and MLH1 loss of expression, whose tumours also show loss of p16 expression can be reasonably excluded for genetic testing, because this loss of expression indicates, with high possibility, aberrant hypermethylation and epigenetic silencing of both p16 and MLH1 genes.
STATEMENT OF TRANSLATIONAL RELEVANCE.
The main contribution of this manuscript is the utility of p16 immunohistochemistry in the identification of patients with colorectal cancer and high level of suspicion of Lynch syndrome. Patients with tumours showing loss of MLH1 expression can be hereditary or sporadic. In this study we demonstrate that p16 immunohistochemistry is a good surrogate marker for both p16 and MLH1 hypermethylation. Patients whose tumours have loss of both MLH1 and p16 expression have hypermethylated colorectal cancer and, therefore, their tumours are sporadic. These patients can be confidently excluded for genetic testing of MLH1. P16 immunohistochemistry is easy to perform and available for every pathology department, taking advantage over other more exigent techniques such as BRAF mutation.
Footnotes
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Gut and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence (http://gut.bmj.com/ifora/licence.pdf).
Reference List
- 1.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–38. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 2.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–61. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 3.Jover R, Paya A, Alenda C, Poveda MJ, Peiro G, Aranda FI, Perez-Mateo M. Defective mismatch-repair colorectal cancer: clinicopathologic characteristics and usefulness of immunohistochemical analysis for diagnosis. Am J Clin Pathol. 2004;122:389–94. doi: 10.1309/V9PG-K2Y2-60VF-VULR. [DOI] [PubMed] [Google Scholar]
- 4.Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, Xicola RM, Rodriguez-Moranta F, Paya A, Jover R, Bessa X. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293:1986–94. doi: 10.1001/jama.293.16.1986. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler JM, Bodmer WF, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000;47:148–53. doi: 10.1136/gut.47.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 1995;108:1405–11. doi: 10.1016/0016-5085(95)90688-6. [DOI] [PubMed] [Google Scholar]
- 7.Umar A, Boland CR, Terdiman JP, Syngal S, de la CA, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, O'Connor T, Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–87. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 9.Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–45. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 10.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 11.Pinol V, Andreu M, Castells A, Paya A, Bessa X, Rodrigo J. Frequency of hereditary non-polyposis colorectal cancer and other colorectal cancer familial forms in Spain: a multicentre, prospective, nationwide study. Eur J Gastroenterol Hepatol. 2004;16:39–45. doi: 10.1097/00042737-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Xicola RM, Llor X, Pons E, Castells A, Alenda C, Pinol V, Andreu M, Castellvi-Bel S, Paya A, Jover R, Bessa X, Giros A, Duque JM, Nicolas-Perez D, Garcia AM, Rigau J, Gassull MA. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007;99:244–52. doi: 10.1093/jnci/djk033. [DOI] [PubMed] [Google Scholar]
- 13.Dai CY, Furth EE, Mick R, Koh J, Takayama T, Niitsu Y, Enders GH. p16(INK4a) expression begins early in human colon neoplasia and correlates inversely with markers of cell proliferation. Gastroenterology. 2000;119:929–42. doi: 10.1053/gast.2000.17952. [DOI] [PubMed] [Google Scholar]
- 14.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiegl H, Gattringer C, Widschwendter A, Schneitter A, Ramoni A, Sarlay D, Gaugg I, Goebel G, Muller HM, Mueller-Holzner E, Marth C, Widschwendter M. Methylated DNA collected by tampons--a new tool to detect endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:882–8. [PubMed] [Google Scholar]
- 16.Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–17. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Widschwendter M, Siegmund KD, Muller HM, Fiegl H, Marth C, Muller-Holzner E, Jones PA, Laird PW. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004;64:3807–13. doi: 10.1158/0008-5472.CAN-03-3852. [DOI] [PubMed] [Google Scholar]
- 18.Ogino S, Cantor M, Kawasaki T, Brahmandam M, Kirkner GJ, Weisenberger DJ, Campan M, Laird PW, Loda M, Fuchs CS. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–6. doi: 10.1136/gut.2005.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benlloch S, Paya A, Alenda C, Bessa X, Andreu M, Jover R, Castells A, Llor X, Aranda FI, Massuti B. Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology. J Mol Diagn. 2006;8:540–3. doi: 10.2353/jmoldx.2006.060070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, Penzone P, Lombardi J, Dunn P, Cohn DE, Copeland L, Eaton L, Fowler J, Lewandowski G, Vaccarello L, Bell J, Reid G, de la CA. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 21.Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, Wahlberg S, Fox EA, Peel D, Ziogas A, Garber JE, Syngal S, Anton-Culver H, Li FP. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–74. [PubMed] [Google Scholar]
- 22.Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, Kolodner RD, Fox E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002;62:3485–92. [PubMed] [Google Scholar]
- 23.Lynch HT, Boland CR, Rodriguez-Bigas MA, Amos C, Lynch JF, Lynch PM. Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J Clin Oncol. 2007;25:3534–42. doi: 10.1200/JCO.2006.10.3119. [DOI] [PubMed] [Google Scholar]
- 24.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, Comeras I, de la CA. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–60. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 25.van Lier MG, de Wilt J, Wagemakers J, Dinjens WN, Damhuis R, Kuipers EJ, van Leerdam ME. Poor compliance with MSI-analysis in patients with colorectal cancer at high risk for Lynch syndrome. Gastroenterology. 2008;134 doi: 10.1080/00365520802706008. Ref Type: Abstract. [DOI] [PubMed] [Google Scholar]
- 26.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–18. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 27.Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, Boland CR. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126:394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 28.Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Pinol V, Xicola RM, Bujanda L, Rene JM, Clofent J, Bessa X, Morillas JD, Nicolas-Perez D, Paya A, Alenda C. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55:848–55. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, Truta B, Sleisenger MH, Kim YS. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:191–5. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–12. [PubMed] [Google Scholar]
- 32.Bessa X, Balleste B, Andreu M, Castells A, Bellosillo B, Balaguer F, Castellvi-Bel S, Paya A, Jover R, Alenda C, Tito L, Martinez-Villacampa M, Vilella A, Xicola RM, Pons E, Llor X. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol. 2008;6:206–14. doi: 10.1016/j.cgh.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, Westra J, Frebourg T, Espin E, Armengol M, Hamelin R, Yamamoto H, Hofstra RM, Seruca R, Lindblom A, Peltomaki P, Thibodeau SN, Aaltonen LA, Schwartz S., Jr BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41:664–8. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bettstetter M, Dechant S, Ruemmele P, Grabowski M, Keller G, Holinski-Feder E, Hartmann A, Hofstaedter F, Dietmaier W. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res. 2007;13:3221–8. doi: 10.1158/1078-0432.CCR-06-3064. [DOI] [PubMed] [Google Scholar]
- 35.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 36.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, Stratton MR. COSMIC 2005. Br J Cancer. 2006;94:318–22. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–9. [PubMed] [Google Scholar]
- 39.Gibson SL, Boquoi A, Chen T, Sharpless NE, Brensinger C, Enders GH. p16(Ink4a) inhibits histologic progression and angiogenic signaling in min colon tumors. Cancer Biol Ther. 2005;4:1389–94. doi: 10.4161/cbt.4.12.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalgo ML, Hayashida T, Bender CM, Pao MM, Tsai YC, Gonzales FA, Nguyen HD, Nguyen TT, Jones PA. The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines. Cancer Res. 1998;58:1245–52. [PubMed] [Google Scholar]
- 41.Nguyen C, Liang G, Nguyen TT, Tsao-Wei D, Groshen S, Lubbert M, Zhou JH, Benedict WF, Jones PA. Susceptibility of nonpromoter CpG islands to de novo methylation in normal and neoplastic cells. J Natl Cancer Inst. 2001;93:1465–72. doi: 10.1093/jnci/93.19.1465. [DOI] [PubMed] [Google Scholar]