

Abstract
The plasma membrane of eukaryotic cells is coated with carbohydrates. By virtue of their extracellular position and recognizable chemical features, cell surface glycans mediate many receptor-ligand interactions. Recently, mammalian extracellular hydrolytic enzymes have been shown to modify the structure of cell surface glycans and consequently, alter their binding properties. These cell surface glycan remodeling events can cause rapid changes in critical signal transduction phenomena. This review highlights recent studies on the roles of eukaryotic extracellular sialidases, sulfatases, and a deacetylase in regulation of intracellular signaling. We also describe possible therapies that target extracellular glycan remodeling processes and discuss the potential for new discoveries in this area.
The glycocalyx stands between extracellular signals and intracellular responses. But the glycoproteins, glycolipids and proteoglycans that comprise the glycocalyx are not simply a barrier; rather, they serve essential roles in the transduction of signals from the outside to the inside of cells. Indeed, most cell surface receptors are glycosylated and many specifically recognize glycans that are attached to their ligands. Cell surface and extracellular glycans have been demonstrated to play critical roles in the control and modulation of a variety of signal transduction pathways (1–4). For example, in embryonic development, signaling through the Notch receptor requires that the receptor be modified with O-fucose glycans (5). If these glycans are absent, gestational death occurs. In the adult nervous system, the myelin-associated glycoprotein (MAG) binds to glycoproteins and glycolipids on axons, forming signaling complexes that inhibit axonal outgrowth (6). And in the immune system, specific cell surface glycans are essential components of the signal transduction pathways that lead to processes such as B-cell receptor activation and T-cell apoptosis (7, 8). Glycosylation also regulates intracellular signal transduction events: for example, the addition of O-linked N-acetylglucosamine (O-GlcNAc) to histone lysine methyl transferase MLL5 activates this enzyme, causing it to methylate histone H3 and thereby leading to cell lineage determination (9).
As more signaling roles for glycans are identified, an essential task is to understand the mechanisms that regulate the glycosylation state of a cell or molecule (Figure 1). Most glycans are assembled in the endoplasmic reticulum (ER) and Golgi through the coordinate action of many membrane-associated glycosyltransferases and other glycan-modifying enzymes, such as sulfotransferases and epimerases. ER- and Golgi-resident glycosyltransferases require nucleotide-sugar donors, such as uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), guanidine diphosphate-fucose (GDP-fucose), and cytidine monophosphate-sialic acids (CMP-sialic acids), that are synthesized in the cytoplasm or nucleus. Once assembled in the secretory pathway, glycoconjugates are displayed on the cell surface or secreted into the extracellular matrix (ECM). After a period of time, cell surface glycoconjugates can be internalized and delivered to the lysosome where glycosidases and sulfatases catalyze their degradation. Newly synthesized glycoconjugates take their place on the plasma membrane. However, emerging evidence indicates that at least some hydrolytic enzymes actually function on the cell surface, where they remodel glycoconjugates by removing terminal sugars or other modifying groups.
Figure 1. Methods of regulating cell surface glycans.

The diversity and abundance of cell surface glycans can be controlled at different points. Control can come at the level of metabolic flux through the pathways that connect monosaccharide building blocks with one another and with nucleotide-sugars. Activated nucleotide–sugars are substrates for glycosyltransferases. The relative abundance of these substrates influences which glycans are produced. Regulation also occurs at the level of glycosyltransferase expression. Golgi-resident glycosyltransferases catalyze the addition of specific sugars to acceptor molecules. Expression levels of these enzymes regulate which glycans are produced. Glycan display can also be regulated by internalization and degradation. Removal of glycoproteins or glycolipids from the cell surface is carried out by endocytosis, which transports them to the lysosome for recycling. Finally, glycan remodeling is an under-recognized mechanism for regulating glycan display. Glycans displayed on the cell surface can be modified through removal of sugars or functional groups by extracellular hydrolytic enzymes, including sialidases, sulfatases, and deacetylases.
Regulation of glycosylation can occur at any step in glycoprotein biosynthesis or catabolism. For example, the degree of N-glycan branching is exquisitely sensitive to changes in intracellular UDP-GlcNAc concentrations and leads to switch-like changes in responses to extracellular growth factors (10). Expression levels of glycosyltransferases also have demonstrated regulatory roles: expression of the Golgi-resident glycosyltransferase known as Fringe determines whether GlcNAc is added to Notch glycans and differentially modulates the receptor’s ability to respond to different classes of ligands (11). A cell’s glycosylation state is also regulated by the removal of specific glycoconjugates by internalization and subsequent degradation. But the recent discoveries of extracellular glycosidases and sulfatases suggest that cell surface glycosylation might be much more dynamic than previously appreciated. Extracellularly-oriented enzymes have the potential to catalyze rapid remodeling of cell surface glycans, actively inducing changes in signaling events. Here we review recent reports of hydrolytic enzymes that can expeditiously remodel cell surface glycans and describe the effects that these changes have on signaling pathways.
Glycan remodeling enzymes
The size of the human glycome remains unknown, although a recent estimate suggests that there are at least 7000 glycan determinants that could potentially be recognized by glycan-binding proteins (12). Sialic acids and sulfate are key features of many known glycan recognition motifs (13). Enzymes that remove or modify these groups (Figure 2) have the capability to have a significant impact on recognition and subsequent signaling events.
Figure 2. Cell surface remodeling reactions catalyzed by hydrolytic enzymes.
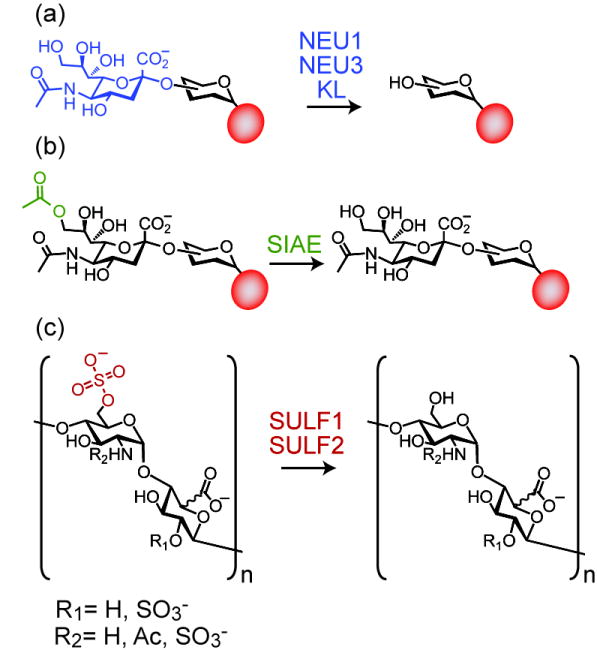
Extracellular or plasma membrane-associated enzymes hydrolytically cleave sugars and modifying groups from cell surface glycans. Some examples include a) sialidases, which hydrolyze the terminal sialic acid of a glycan chain; b) sialic acid esterase, which removes an acetyl group from the C-9 position of sialic acid; and c) sulfatases, which hydrolyze sulfate from the C-6 position of glucosamine residues located in heparan sulfate chains. In a) and b), the red circle represents a glycoprotein or glycolipid scaffold.
Sialic acid refers to a group of nine-carbon α-keto acids that are found at the non-reducing termini of many glycolipids and glycoproteins (14, 15). N-Acetyl neuraminic acid (NeuAc) is the most common sialic acid in humans. Cell surface glycoconjugates as well as circulating glycoproteins, such as antibodies and hormones, are often modified with sialylated glycans. Sialidases, also known as neuraminidases, are enzymes that hydrolytically cleave sialic acids from glycoconjugates. The most well-known enzyme in this family is the influenza neuraminidase, first identified in the 1950s (16). Influenza neuraminidase removes sialic acid from host receptors, allowing virions to be released from infected cells. Sialidases have been found in a variety of other viral, bacterial, and protozoal pathogens. In 1993, the first mammalian sialidase gene (now known as Neu2) was cloned (17) and the human ortholog, NEU2, was reported six years later (18). The NEU gene family now comprises four members (NEU1-NEU4), whose protein products have differing subcellular localization patterns. NEU1 is found primarily in lysosomes (19), where it participates in the degradation of free oligosaccharides as well as glycoproteins (20), but has also been observed at the plasma membrane (21, 22). NEU2 (18), also known as the soluble sialidase, is a cytosolic enzyme that accepts a variety of substrates, including oligosaccharides, glycoproteins, and gangliosides (sialic acid-containing glycolipids) (23). NEU3 (24) is associated with the plasma membrane and prefers ganglioside substrates (25). Some evidence points toward selective localization of NEU3 in endosomal structures or caveolae (26, 27). NEU4 is the most recently discovered member of the NEU family (28, 29). NEU4 localization has been reported in lysosomes, mitochondria, and other intracellular membranes and it has been observed to accept oligosaccharide, glycoprotein, and ganglioside substrates (29–31). In addition to the NEU family members, recent work indicates that the extracellular domains of the anti-aging hormone KL (also known as Klotho) have sialidase activity (32). This observation was quite surprising since KL shows no discernable sequence similarity with other mammalian sialidases and it suggests that additional mammalian sialidases could remain unidentified.
Sialic acids are subject to a variety of post-glycosylational modifications, including acetylation, sulfation, and phosphorylation (15). Acetylation is commonly found at the C-7 and C-9 positions. 9-O-acetylation, in particular, displays developmental and cell-type regulation. In eukaryotes, sialic acid acetylation is believed to occur in the late compartments of Golgi, but the responsible enzymes have thus far eluded identification (33, 34). Deactylation, on the other hand, can be catalyzed by sialic acid acetyl esterase (SIAE), which is expressed as two splice variants (35). These variants were originally believed to have cytosolic and lysosomal localization patterns, but recent work demonstrates very little lysosomal presence for the latter form. Rather, this protein is secreted and can be found associated with the plasma membrane where it is poised to act on cell surface glycoconjugates (36).
Glycoconjugates can also be modified by sulfation. Sulfates are found attached to glycoproteins, glycolipids, and glycosaminoglycans including heparin, heparan sulfate (HS), chondoitin sulfate, and dermatan sulfate. Heparan sulfate proteoglycans (HSPGs), in which HS is attached to a protein, are abundant cell surface molecules and important components of the extracellular matrix. Their sulfation patterns direct receptor binding of many key growth factors including WNTs, vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), hepatocyte growth factor (HGF), and heparin binding epidermal growth factor (HB-EGF) (2, 37). Heparan sulfate consists of a repeating glucosamine-glucuronic acid backbone of about 10 to 100 residues that has been modified by epimerization, sulfation and/or acetylation. A wide diversity of structures are theoretically possible, forming a potential ‘sulfation code” for recognition and signaling (38).
Until recently, HSPG sulfation patterns were believed to be determined exclusively by the Golgi-resident enzymes that synthesize these molecules. But in 2001, the first extracellular sulfatase, quail SULF1, was discovered. Two human extracellular sulfatases, SULF1 and SULF2, have now been identified (39). SULF1 and SULF2 are endosulfatases that hydrolytically remove 6-O-sulfates from glucosamine residues found in heparan sulfate polymers. The SULFs share sequence similarity with lysosomal exosulfatases, but exhibit an extracellular localization and maximal activity at near neutral pH, suggesting that they have evolved to function outside the cell, rather than in the lysosome. These enzymes contain four domains: an N-terminal signal sequence that targets them for secretion, a catalytic domain, a lysine- and arginine-rich hydrophilic domain and a C-terminal domain. The hydrophilic domain is important for membrane association, HS binding, and enzymatic activity (40).
Due to their extracellular location, the sialidases, deacetylase, and sulfatases discussed here have the potential to regulate cell-cell communication by a variety of mechanisms (Figure 3). Glycan remodeling enzymes can be displayed on the cell surface where they can modify substrates displayed on the same cell (autocrine signaling) or on adjacent cells (juxtacrine signaling). Indeed, in cell culture experiments, NEU3 has been observed to alter the glycosylation pattern of adjacent cells, suggesting a novel mechanism for cell-cell communication (41). Cell surface remodeling enzymes might also act on secreted proteins and thereby set up a gradient of signaling molecules. Alternatively, glycan remodeling enzymes themselves can be secreted (paracrine signaling) and then go on to modify the source cell, neighboring cells, or soluble glycoconjugates, each of which could lead to new signaling events. In the next sections, we discuss some of the known cases where signaling results from extracellular remodeling of glycans.
Figure 3. Modes of action of glycan remodeling enzymes.
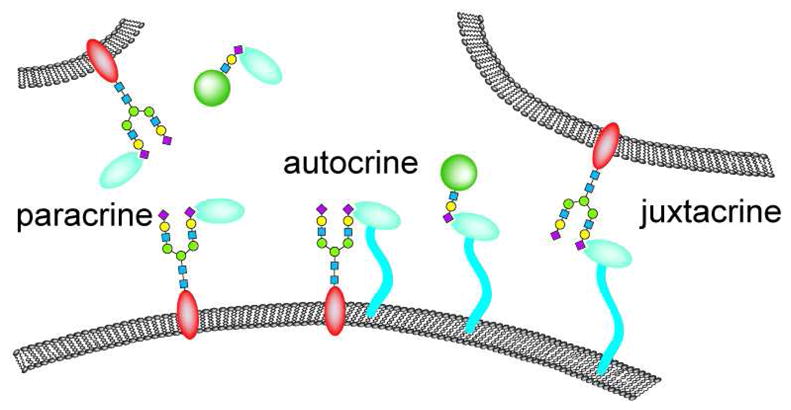
Cell surface hydrolytic enzymes can act on glycans positioned on the same cell (autocrine signaling), adjacent cells (juxtacrine signaling) or soluble glycoproteins (shown in green). Secreted enzymes can act on glycans positioned on distant cells (paracrine signaling) or soluble glycoproteins.
Desulfation signals in embryonic development
SULF1 was first identified on the basis of its activation during somite patterning in quail embryogenesis (42). Shortly after this discovery, two human sulfatases (SULF1 and SULF2) were cloned (39). From these and subsequent studies (43), a role for SULF1 in activation of Wnt signaling in muscle progenitor cells has emerged, although some details still remain unclear (Figure 4, panel a). Extracellular WNT ligands activate the intracellular Wnt signaling pathway by binding cell surface receptors of the Frizzled family. HSPGs inhibit Wnt signaling by binding WNT ligands and trapping them in the extracellular matrix. SULF1’s removal of sulfates from HSPG liberates WNTs, allowing them to bind Frizzled receptors and thereby intiate Wnt signaling. Some evidence suggests that HSPGs remain part of the WNT-Frizzled signaling complexes, so, at least in some cases, SULF1 may reduce the affinity of WNT-HSPG interactions without eliminating them.
Figure 4. Wnt and FGF2 signaling are regulated by extracellular sulfatase activities.
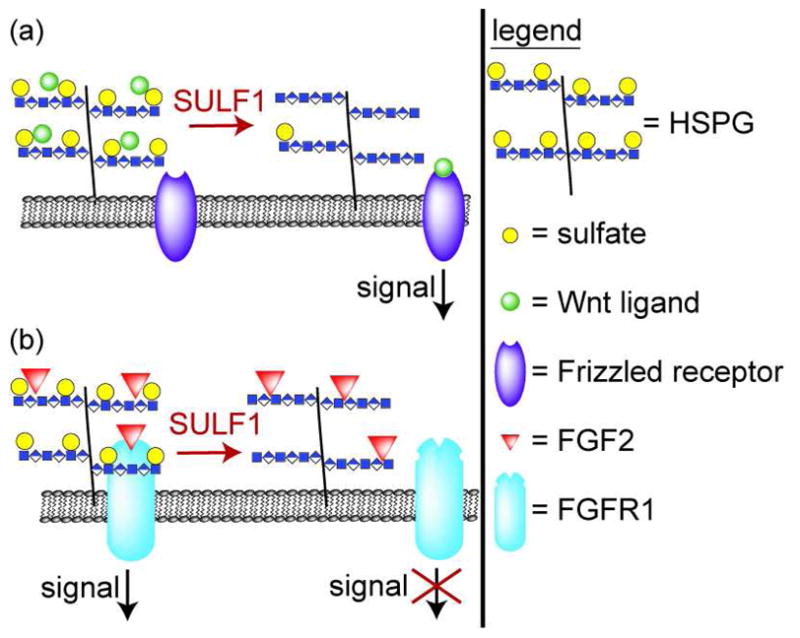
a) WNT ligands are sequestered by HSPGs. Desulfation of HSPGs by SULF1 liberates the WNTs to interact with Frizzled receptors, activating intracellular signaling pathways. b) FGF2 signaling requires a complex formed from FGF2 ligand, FGFR1, and HSPGs. Desulfation of HSPGs by SULF1 disrupts this complex and thereby reduces FGF2 signaling. SULF2 also affects FGF2 signaling, but direction of the effect and the mechanistic details remain unclear.
In addition to its role in the promotion of Wnt signaling, SULF1 also negatively regulates FGF2 signaling (Figure 4, panel b) (44). FGF2 signaling requires the formation of a ternary complex, in which HS mediates the interaction between FGF2 ligand and FGF receptor 1 (FGFR1). SULF1-mediated desulfation of HS reduces FGF2 signaling by interfering with formation of FGF2-HS-FGFR1 complexes. Wang et al. showed that SULF1’s effects on FGF2 signaling can prevent mesoderm induction in Xenopus embryos and angiogenesis in chick embryos (44).
Because quail SULF1 has distinct embryonic expression patterns in a number of cell lineages (42), it likely plays a role in specification of multiple developmental events. Reinforcing this idea is information obtained from mice with targeted disruptions in the sulfatase genes (45, 46). Mice lacking either SULF1 or SULF2 displayed only mild phenotypic changes, including a slight decrease in body weight for Sulf2−/− animals. On the other hand, the sulfatase double-knockout mice displayed renal hypoplasia, skeletal defects and a high incidence of neonatal mortality, indicating that SULF1 and SULF2 play redundant, yet essential roles in development. Sulf1−/− Sulf2−/− cells are hyper-responsive to fibroblast growth factors, consistent with the sulfatases’ postulated roles as negative modulators of FGF signaling (45, 47).
Some of the defects observed in Sulf1−/− Sulf2−/− mice could also be due to sulfatases’ effects on bone morphogenetic protein (BMP) signaling. BMPs are growth factors that are essential to many development processes including dorsal-ventral axis formation, skeletal morphogeneisis, neural tissue induction, and lung development (48). BMP function is regulated by extracellular antagonists. A HS-binding protein called Noggin is one of these antagonists. When Noggin is associated with the cell surface, it can bind BMPs and antagonize their signaling (49). In quail, SULF1 activity releases Noggin from the cell surface, freeing BMPs and activating them for signaling (50). More recent work in mice suggests that BMPs can also interact directly with HS (51). When HS is undersulfated, the BMPs may be liberated for signaling. In both of these studies, SULF1 seemed to function as a positive regulator of BMP signaling. However, the effects of SULF1 on Xenopus development suggest that it acts as an inhibitor, at least in the case of BMP4 signaling (52). Given this disparate evidence, more work will be needed to deconvolute the detailed mechanisms by which sulfatases affect BMP signaling.
In addition to their direct enzymatic effects on HS sulfation, sulfatases can also affect HS structures through a dynamic feedback mechanism. Recent work demonstrated that loss of sulfatase activity leads to changes in the expression patterns of the HS biosynthetic enzymes. For example, in mouse cells, loss of Sulf2 expression, which is expected to cause an increase in net 6-O-sulfation of HS, leads to a decrease in expression of a 2-O-sulfotransferase, Hs2st1 (53). These results suggest that HS can initiate signaling cascades that regulate its own production.
Glycan remodeling in cancer
The observation that SULFs can inhibit growth factor responses precipitated interest in their possible roles in cancer, where enhanced growth factor signaling is known to lead to increased proliferation and inhibition of apoptosis. mRNA expression profiling in cancer cell lines and in primary patient samples revealed that SULF1 is downregulated at early stages in ovarian and liver cancers (54–56). A number of studies have investigated the effects of altered SULF1 expression on signaling in cancer. Forced SULF1 expression in ovarian cancer cell lines causes decreased HSPG sulfation and results in decreased signaling through both HB-EGF and FGF2. SULF1-expressing cells also exhibit slowed proliferation and regained responsiveness to apoptotic stimuli, consistent with a role for SULF1 downregulation in cancer survival signaling (54). Similar results have been observed in vivo: induction of SULF1 expression in tumor xenografts leads to decreases in proliferation and angiogenesis (57, 58). The reduction in angiogenesis was attributed to effects on FGF signaling due to an observable decrease in the ability of HS to promote formation of a ternary FGF2/HS/FGFR1c complex.
While SULF1 may function as tumor suppressor with respect to growth factor signaling (56), sulfatases’ ability to enhance Wnt signaling means that they have a more complicated relationship with tumor growth. Both SULF1 and SULF2 are upregulated in pancreatic adenocarcinomas, where they appear to be positive regulators of tumor growth (59, 60). Expressing inactive sulfatases or silencing SULF2 reduces Wnt signaling in pancreatic cancer cells and slows tumor growth. Sulfatases might also play a role in other WNT-dependent cancers. In fact, SULF1 is upregulated in most hepatocellular carcinomas (55), in breast cancer (61, 62), and in head and neck cancer (63). In these cases, inhibition or destruction of extracellular sulfatases could represent a therapeutic approach to treatment. As extracellular enzymes, SULF1 and SULF2 are attractive drug targets. But since each sulfatase can affect multiple signaling pathways, inhibitors of these enzymes could have significant off-target effects.
Less information is available about functional roles of SULF2. Analysis of Sulf1−/− Sulf2−/− cells suggested that SULF1 and SULF2 have partially redundant activities (45, 46), but conflicting data exist with respect to SULF2’s effects on FGF2 signaling. One set of data indicates that SULF2 reduces FGF2 signaling in tumor cells (58), while another report presented evidence in support of SULF2-promoted FGF2 signaling (64). In vitro experiments established that SULF2 is able to hydrolyze sulfates from heparin, thereby liberating a variety of heparin-associated growth factors and chemokines (65), but how this change could lead to increased FGF2 signaling remains unclear. One possibility is that growth factors are freed from the extracellular matrix and can diffuse to nearby cells where they induce FGF2 signaling.
The structure of extracellular HS can also be modified by the action of secreted heparanase (HPSE). HPSE hydrolytically cleaves HS into smaller fragments (10–20 sugar units) that display binding properties distinct from those of the intact polymer. Increased HPSE expression has been observed in human breast, colon, ovary, lung, prostate, and pancreas tumors (66) and appears to to play key roles in tumor metastasis and angiogenesis (67). For these reasons, HPSE is attracting attention as a potential target for cancer therapeutics. Because of space limitations, we are unable to provide a detailed discussion of HPSE’s effects on signaling, but point the reader to recent review articles on the topic (68–71).
Changes in sialidase expression levels are also associated with cancer, having been observed in primary samples and in cancer cell lines (reviewed in (72, 73). Different sialidases have been observed to promote or oppose malignant phenotypes. NEU3 is upregulated up to 100-fold in colon cancer (74) and also overexpressed in renal cell carcinomas and ovarian clear cell adenocarcinoma (75, 76). NEU3 expression causes cell-type specific effects on cell proliferation, apoptosis, and motility. These effects are probably due, at least in part, to NEU3’s ability to desialylate glycolipids: overexpression of NEU3 leads to decreased levels of certain sialylated glycolipids, such as GM3, and an increased level of the unsialylated glycolipid LacCer.
In the leukemic cell line K562, NEU3 silencing leads to slowed cell growth, increased susceptibility to apoptosis, and increased propensity to differentiate (77). The mechanisms by which NEU3 suppresses apoptosis are still being deciphered, but existing evidence suggests that this protein functions both by interacting directly with signaling molecules and by exerting its enzymatic activity on ganglioside substrates, which, in turn, interact with signaling molecules. One substrate of NEU3, GM3, interacts directly with the EGF receptor and reduces its ability to respond to EGF ligand, possibly by sequestering EGFR in specialized membrane microdomains (Figure 5, panel a) (78). In the presence of NEU3, GM3 is hydrolyzed to LacCer, relieving the inhibition of EGF signaling. In this way, NEU3 activity leads to increased EGF signaling and cell proliferation (79).
Figure 5. Sialidase regulation of signal transduction.
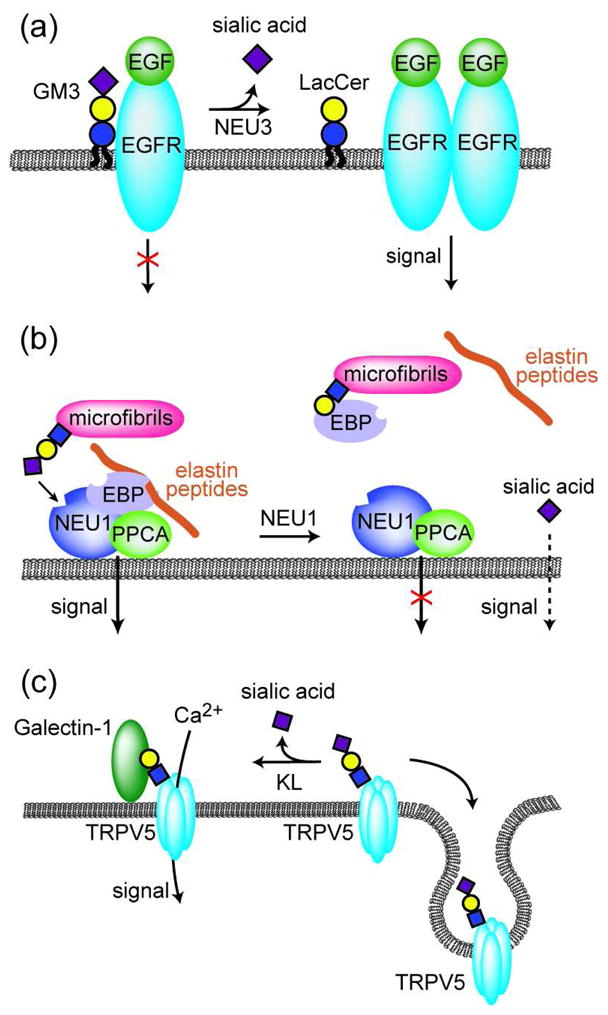
a) EGFR signaling is reduced in the presence of ganglioside GM3. NEU3-catalyzed conversion of GM3 to LacCer relieves this inhibition. b) NEU1 forms a cell surface receptor complex with PPCA and EBP. In the presence of elastin peptides, the receptor complex is activated leading to downstream ERK1/2 activation. In addition, the active complex enhances the catalytic activity of NEU1, leading to desialylation of microfibrillar glycoproteins and local release of elastin peptides. Liberated sialic acid may act as a second messenger that activates other signaling events. c) Sialylated TRPV5 is rapidly endocytosed from the cell membrane. Desialylation of TRPV5 by KL exposes underlying galactose residues that bind galectin-1, thereby retaining TRPV5 on the cell surface where functions in Ca2+ transport.
NEU3 also modulates integrin signaling pathways, leading to increased proliferation and motility (80). NEU3-mediated depletion of GM3 has been shown to block integrin-mediated adhesion to fibronectin, consistent with other work that showed functional and physical interactions between α5β1 integrin and GM3 (81, 82). NEU3 activity also promotes integrin-mediated adhesion to laminin (80). The net effect of these changes in adhesive properties is to stimulate cell proliferation. Additional work will be needed to clarify the molecular details of GM3’s and LacCer’s roles in these processes.
Sialidase activity in elastogenesis
NEU1 is often described as the lysosomal sialidase, because of its role in degradation of sialylated glycoproteins in the lysosome. Mutations to the NEU1 gene are a cause of human sialidosis, a rare inherited lysosomal storage disorder. Recent reports suggest that NEU1 may also function outside the cell, on the plasma membrane (83–85). One role of cell surface NEU1 is in the processes that lead to assembly of elastic fibers, important components of connective tissue. NEU1 associates with protective protein/cathepsin (PPCA) and elastin-binding protein (EBP), forming the cell-surface elastin receptor complex that mediates elastin fiber assembly (Figure 5, panel b) (83). NEU1 is proposed to remove terminal sialic acids from fibrillin glycoproteins, revealing galactose residues. The unmasked galactose residues can be recognized by EBP, which is a splice variant of galactosidase that has lost its catalytic activity but retains the ability to bind galactosylated glycoconjugates. Mice that lack the Neu1 gene display abnormal development of elastin fibers, similar to that seen in sialidosis patients (86). The elastin receptor complex is also integral to fibroblast’s ability to respond to elastin degradation: association of elastin peptides with elastin receptor complex activates an intracellular signal cascade that includes ERK1/2 activation and pro-MMP-1 production. Signal transduction depends on the catalytic activity of NEU1, but effects were also observed upon treatment of fibroblasts with sialic acid, suggesting that the sialic acid liberated by NEU1 could act as a second messenger (87). The putative receptor of free sialic acid remains to be identified.
Glycan remodeling in the immune system
Differentiation of myeloid precursor cells to mature granulocyte-series cells is associated with loss of the CD15s (sialyl Lewis X) epitope and increased expression of its unsialylated counterpart, CD15. Although they differ only by a sialic acid, the two glycans have very different functions. CD15s enables neutrophils to adhere to endothelial cells at sites of inflammation (88). Neutrophils then enter the subendothelial tissues through a process called extravasation. Once there, the neutrophil cell surface exchanges CD15s expression for CD15, and uses the unsialylated CD15 glycan to engage the dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) lectin presented on dendritic cells and thereby signal for dendritic cell maturation (89). Surprising new evidence indicates that the shift to CD15 expression results from cell surface desialylation of CD15s, rather than synthesis of new CD15 structures (84). The observed desialylation of CD15s occurs primarily on glycoproteins, rather than glycolipids, hinting that, in this case, NEU3 is unlikely to be the responsible sialidase. Support for this hypothesis comes from the observation that NEU1 mRNA levels increase during myeloid differentiation. Shifts from CD15s to CD15 expression also occur at other stages of hematopoiesis, but mechanisms behind the losses of sialylation have not yet been elucidated.
Cell surface remodeling of sialylated glycans is also observed in the repression of B cell receptor (BCR) signaling that occurs in mature B cells. This repression appears to be an essential aspect of immunological tolerance of self (90). In 1996, a gene encoding a sialic acid-specific 9-O-acetylesterase (SIAE) was shown to be expressed in mature, but not immature, B cells (91), but it was only this year that significance of Siae’s developmental regulation became clear. Cariappa et al. showed that SIAE regulates the acetylation state of α2,6-linked sialic acids attached to N-linked glycans and displayed on the surface of B cells (36). Such glycans are ligands for the cell surface receptor CD22, but 9-O-acetylation of the sialic acids interferes with binding. In mature B cells, the action of SIAE reveals the unacetylated, high-affinity CD22 ligand and activates inhibitory signaling through the Lyn-CD22-SHP-1 pathway (36, 90). In mice that are engineered to lack Siae, defects in inhibitory signaling and B cell development are observed (36). Strikingly, these animals produce elevated levels of autoantibodies, indicating that their ability to recognize and tolerate self has been compromised. Further work will be required to determine if inadequate SIAE activity also plays a role in human autoimmune diseases where self-tolerance is impaired (92). Since SIAE is an extracellular enzyme, a defect in its activity could conceivably be corrected by delivery of SIAE as a therapeutic protein.
A novel sialidase regulates calcium homeostasis
In 1997, the Kl gene was identified on the basis of effects on aging processes that it exerts in mice (93). Mice lacking Kl exhibit symptoms of premature aging and die early, while mice overexpressing Kl have an extended lifespan, living 20–30% longer than their normal counterparts (94). The KL protein localizes to the cell surface and includes a large extracellular domain that can be proteolytically released by ADAM10 and ADAM17, resulting in a soluble protein that is found in the blood, urine, and cerebrospinal fluid. The extracellular portion of KL contains two domains with weak similarity to each other and to β-glucosidases. Unexpectedly, both extracellular domains were found to have sialidase activity (32). KL regulates the activity of the Ca2+ channel known as transient receptor potential vanilloid 5 (TRPV5) through desialylation (Figure 5, panel c). Normally, sialylated TRPV5 is rapidly removed from the cell surface by endocytosis. Once desialylated by KL, TRPV5 displays exposed galactose residues that interact with galectin-1, facilitating the retention of TRPV5 at the cell surface, where it acts to increase calcium influx. This mechanism likely underlies KL’s observed ability to promote renal Ca2+ reabsorption and may protect the kidneys from damage (95). KL’s sialidase activity could play a role in its effects on other age-related physiological changes. For example, KL also regulates the cell surface retention and activity of Na+,K+-ATPase α1 subunit, although it is not yet known whether desialylation is the mechanism at work.
Sialidase activity stimulates axonal outgrowth
Sialylated molecules are abundant in the nervous system, suggesting that this might be a location where sialidases could play an important regulatory role. In fact, NEU3 activity in primary neurons leads to increased axonal growth (96). NEU3 induces TrkA-mediated signaling, leading to actin depolymerization and axonal growth. The mechanism by which NEU3 affects TrkA is likely through changes in cells’ ganglioside composition, such as desialylation of GD1a to produce increased levels of GM1. NEU3 has been shown to cause increases in ganglioside GM1, which is capable of enhancing TrkA dimerization and potentiating the effect of nerve growth factor (NGF) (97). NEU3 activity can also affect cellular proliferation and neurite outgrowth in neuroblastoma cells, but the mechanistic details of these changes remain unclear. Somewhat puzzlingly, both Neu3 silencing (98) and NEU3 overexpression (99) have been shown to stimulate neurite outgrowth. This apparent discrepancy may reflect the differences present in the cholinergic and adrenergic neuroblastoma cell lines in which these experiments were performed (100). NEU3 activity is enriched in membrane microdomains of neuroblastoma cells and co-segregates with GM1 and other lipid raft markers such as flotillin, src family kinases, and glycosylphosphatidylinositol (GPI)-anchored proteins (101). The localized distribution of NEU3 within a particular neurite specifies the site of axon generation (102). Intriguingly, a bacterial sialidase can have a similar effect in a living animals: delivery of Clostridium perfringens sialidase to a spinal cord injury site in a rat dramatically enhanced spinal axon outgrowth and might represent a therapy to improve recoveries from central nervous system injuries (103).
More glycan remodeling still undiscovered?
As examples of extracellular glycan remodeling become more and more common, we speculate that this phenomenon may emerge as a key mechanism for regulating signaling events, especially those that require rapid response times. Along with the cases described here, additional extracellular glycosidases have been reported. For example, Porwoll et al. showed that N-linked glycans attached to plasma membrane proteins can be demannosylated by an extracellular mannosidase, a change that would be expected to radically affect their ability to participate in signaling events (104, 105). Because fucose, like sialic acid and sulfate, is often a critical regulator of glycan binding events, it is not surprising that an extracellular human fucosidase has been discovered (106). This enzyme is found in seminal fluid (107), where it may play a role in fertilization and has also been observed on the plasma membrane in hepatocarcinoma (108). Recent work points to a role for fucosidase activity in leukocyte trafficking (109). In other cases, glycosidase activity has been observed but the responsible enzymes have not been identified. For example, a plasma membrane-associated β-hexosaminidase activity capable of removing N-acetylgalactosamine (GalNAc) from the GM2 ganglioside has been discovered in fibroblasts (110) and evidence for membrane-associated galactosidase and glucosidase activities has also been reported (22).
The ubiquity of extracellular glycosidase activity poses a challenge for production of protein-based pharmaceutics. Most therapeutic proteins and antibodies are glycosylated. Production standards require that the attached glycans be as homogenous as possible (111), but extracellular glycosidase activities are often an impediment to reaching this goal (112). For this reason, the biotechnology industry is likely to be a future source of information on the activities of extracellular glycosidases and strategies to interfere with their activity.
Finally, we hypothesize that roles for extracellular glycosidases may represent a mechanism by which bacteria induce signaling changes in host cells. Pathogenic bacteria commonly produce sialidases that allow them to scavenge host sialic acids (113). Intriguingly, commensal bacteria are also known to produce a variety of extracellular glycosidases that enable them to colonize the intestinal tract and to utilize complex oligosaccharides as energy sources (114). Understanding how glycosylation changes induced by bacteria affect host cell signaling is a relatively unexplored frontier.
Acknowledgments
We thank Seok-Ho Yu, Michelle Bond, and Chad Whitman for comments on the manuscript and gratefully acknowledge the support of the March of Dimes (5-FY06-913), the Welch Foundation (I-1686), and the National Institutes of Health (GM090271). This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. J. J. K is an Alfred P. Sloan Research Fellow.
References
- 1.Roseman S. Reflections on glycobiology. J Biol Chem. 2001;276:41527–41542. doi: 10.1074/jbc.R100053200. [DOI] [PubMed] [Google Scholar]
- 2.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 3.Lopez PH, Schnaar RL. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. 2009;19:549–557. doi: 10.1016/j.sbi.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haltiwanger RS. Regulation of signal transduction pathways in development by glycosylation. Curr Opin Struct Biol. 2002;12:593–598. doi: 10.1016/s0959-440x(02)00371-8. [DOI] [PubMed] [Google Scholar]
- 5.Stanley P. Regulation of Notch signaling by glycosylation. Curr Opin Struct Biol. 2007;17:530–535. doi: 10.1016/j.sbi.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehta NR, Lopez PHH, Vyas AA, Schnaar RL. Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grewal PK, Boton M, Ramirez K, Collins BE, Saito A, Green RS, Ohtsubo K, Chui D, Marth JD. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol Cell Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Dyken SJ, Green RS, Marth JD. Structural and mechanistic features of protein O-glycosylation linked to CD8+ T-cell apoptosis. Mol Cell Biol. 2007;27:1096–1111. doi: 10.1128/MCB.01750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujiki R, Chikanishi T, Hashiba W, Ito H, Takada I, Roeder RG, Kitagawa H, Kato S. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455–459. doi: 10.1038/nature07954. [DOI] [PubMed] [Google Scholar]
- 10.Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-Glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 11.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 12.Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. 2009;5:1087–1104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 13.Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 14.Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: An evolutionary perspective. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Chen X. Carbohydrate post-glycosylational modifications. Org Biomol Chem. 2007;5:865–872. doi: 10.1039/b700034k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottschalk A. Neuraminidase: the specific enzyme of influenza virus and Vibrio cholerae. Biochim Biophys Acta. 1957;23:645–646. doi: 10.1016/0006-3002(57)90389-x. [DOI] [PubMed] [Google Scholar]
- 17.Miyagi T, Konno K, Emori Y, Kawasaki H, Suzuki K, Yasui A, Tsuik S. Molecular cloning and expression of cDNA encoding rat skeletal muscle cytosolic sialidase. J Biol Chem. 1993;268:26435–26440. [PubMed] [Google Scholar]
- 18.Monti E, Preti A, Rossi E, Ballabio A, Borsani G. Cloning and characterization of NEU2, a human gene homologous to rodent soluble sialidases. Genomics. 1999;57:137–143. doi: 10.1006/geno.1999.5749. [DOI] [PubMed] [Google Scholar]
- 19.Bonten E, van der Spoel A, Fornerod M, Grosveld G, d’Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996;10:3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- 20.Miyagi T, Hata K, Hasegawa T, Aoyagi T. Differential effect of various inhibitors on four types of rat sialidase. Glycoconj J. 1993;10:45–49. doi: 10.1007/BF00731186. [DOI] [PubMed] [Google Scholar]
- 21.Liang F, Seyrantepe V, Landry K, Ahmad R, Ahmad A, Stamatos NM, Pshezhetsky AV. Monocyte differentiation up-regulates the expression of the lysosomal sialidase, Neu1, and triggers its targeting to the plasma membrane via major histocompatibility complex class II-positive compartments. J Biol Chem. 2006;281:27526–27538. doi: 10.1074/jbc.M605633200. [DOI] [PubMed] [Google Scholar]
- 22.Valaperta R, Chigorno V, Basso L, Prinetti A, Bresciani R, Preti A, Miyagi T, Sonnino S. Plasma membrane production of ceramide from ganglioside GM3 in human fibroblasts. FASEB J. 2006;20:1227–1229. doi: 10.1096/fj.05-5077fje. [DOI] [PubMed] [Google Scholar]
- 23.Tringali C, Papini N, Fusi P, Croci G, Borsani G, Preti A, Tortora P, Tettamanti G, Venerando B, Monti E. Properties of recombinant human cytosolic sialidase hsNEU2. J Biol Chem. 2004;279:3169–3179. doi: 10.1074/jbc.M308381200. [DOI] [PubMed] [Google Scholar]
- 24.Monti E, Bassi MT, Papini N, Riboni M, Manzoni M, Venerando B, Croci G, Preti A, Ballabio A, Tettamanti G, Borsani G. Identification and expression of NEU3, a novel human sialidase associated to the plasma membrane. Biochem J. 2000;349:343–351. doi: 10.1042/0264-6021:3490343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hata K, Wada T, Hasegawa A, Kiso M, Miyagi T. Purification and characterization of a membrane-associated ganglioside sialidase from bovine brain. J Biochem. 1998;123:899–905. doi: 10.1093/oxfordjournals.jbchem.a022022. [DOI] [PubMed] [Google Scholar]
- 26.Zanchetti G, Colombi P, Manzoni M, Anastasia L, Caimi L, Borsani G, Venerando B, Tettamanti G, Preti A, Monti E, Bresciani R. Sialidase NEU3 is a peripheral membrane protein localized on the cell surface and in endosomal structures. Biochem J. 2007;408:211–219. doi: 10.1042/BJ20070503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Yamaguchi K, Wada T, Hata K, Zhao XJ, Fujimoto T, Miyagi T. A close association of the ganglioside-specific sialidase Neu3 with caveolin in membrane microdomains. J Biol Chem. 2002;277:26252–26259. doi: 10.1074/jbc.M110515200. [DOI] [PubMed] [Google Scholar]
- 28.Comelli EM, Amado M, Lustig SR, Paulson JC. Identification and expression of Neu4, a novel murine sialidase. Gene. 2003;321:155–161. doi: 10.1016/j.gene.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Monti E, Bassi MT, Bresciani R, Civini S, Croci GL, Papini N, Riboni M, Zanchetti G, Ballabio A, Preti A, Tettamanti G, Venerando B, Borsani G. Molecular cloning and characterization of NEU4, the fourth member of the human sialidase gene family. Genomics. 2004;83:445–453. doi: 10.1016/j.ygeno.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi K, Hata K, Koseki K, Shiozaki K, Akita H, Wada T, Moriya S, Miyagi T. Evidence for mitochondrial localization of a novel human sialidase (NEU4) Biochem J. 2005;390:85–93. doi: 10.1042/BJ20050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seyrantepe V, Landry K, Trudel S, Hassan JA, Morales CR, Pshezhetsky AV. Neu4, a novel human lysosomal lumen sialidase, confers normal phenotype to sialidosis and galactosialidosis cells. J Biol Chem. 2004;279:37021–37029. doi: 10.1074/jbc.M404531200. [DOI] [PubMed] [Google Scholar]
- 32.Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci USA. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi WX, Chammas R, Varki A. Induction of sialic acid 9-O-acetylation by diverse gene products: implications for the expression cloning of sialic acid O- acetyltransferases. Glycobiology. 1998;8:199–205. doi: 10.1093/glycob/8.2.199. [DOI] [PubMed] [Google Scholar]
- 34.Chen HY, Challa AK, Varki A. 9-O-Acetylation of exogenously added ganglioside GD3. J Biol Chem. 2006;281:7825–7833. doi: 10.1074/jbc.M512379200. [DOI] [PubMed] [Google Scholar]
- 35.Takematsu H, Diaz S, Stoddart A, Zhang Y, Varki A. Lysosomal and cytosolic sialic acid 9-O-acetylesterase activities can be encoded by one gene via differential usage of a signal peptide-encoding exon at the N terminus. J Biol Chem. 1999;274:25623–25631. doi: 10.1074/jbc.274.36.25623. [DOI] [PubMed] [Google Scholar]
- 36.Cariappa A, Takematsu H, Liu H, Diaz S, Haider K, Boboila C, Kalloo G, Connole M, Shi HN, Varki N, Varki A, Pillai S. B cell antigen receptor signal strength and peripheral B cell development are regulated by a 9-O-acetyl sialic acid esterase. J Exp Med. 2009;206:125–138. doi: 10.1084/jem.20081399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 38.Gama CI, Hsieh-Wilson LC. Chemical approaches to deciphering the glycosaminoglycan code. Curr Opin Chem Biol. 2005;9:609–619. doi: 10.1016/j.cbpa.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ai X, Do AT, Kusche-Gullberg M, Lindahl U, Lu K, Emerson CP., Jr Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J Biol Chem. 2006;281:4969–4976. doi: 10.1074/jbc.M511902200. [DOI] [PubMed] [Google Scholar]
- 41.Papini N, Anastasia L, Tringali C, Croci G, Bresciani R, Yamaguchi K, Miyagi T, Preti A, Prinetti A, Prioni S, Sonnino S, Tettamanti G, Venerando B, Monti E. The plasma membrane-associated sialidase MmNEU3 modifies the ganglioside pattern of adjacent cells supporting its involvement in cell-to-cell interactions. J Biol Chem. 2004;279:16989–16995. doi: 10.1074/jbc.M400881200. [DOI] [PubMed] [Google Scholar]
- 42.Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP., Jr Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001;293:1663–1666. doi: 10.1126/science.293.5535.1663. [DOI] [PubMed] [Google Scholar]
- 43.Ai X, Do A-T, Lozynska O, Kusche-Gullberg M, Lindahl U, Emerson CP., Jr QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S, Ai X, Freeman SD, Pownall ME, Lu Q, Kessler DS, Emerson CP. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci USA. 2004;101:4833–4838. doi: 10.1073/pnas.0401028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holst CR, Bou-Reslan H, Gore BB, Wong K, Grant D, Chalasani S, Carano RA, Frantz GD, Tessier-Lavigne M, Bolon B, French DM, Ashkenazi A. Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. PLoS One. 2007;2:e575. doi: 10.1371/journal.pone.0000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ratzka A, Kalus I, Moser M, Dierks T, Mundlos S, Vortkamp A. Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Dev Dyn. 2008;237:339–353. doi: 10.1002/dvdy.21423. [DOI] [PubMed] [Google Scholar]
- 47.Lamanna WC, Baldwin RJ, Padva M, Kalus I, Dam GT, Kuppevelt THV, Gallagher JT, Figura KV, Dierks T, Merry CLR. Heparan sulfate 6-O-endosulfatases: discrete in vivo activities and functional co-operativity. Biochem J. 2006;400:63–73. doi: 10.1042/BJ20060848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao G-Q. Consequences of knocking out BMP signaling in the mouse. Genesis. 2003;35:43–56. doi: 10.1002/gene.10167. [DOI] [PubMed] [Google Scholar]
- 49.Paine-Saunders S, Viviano BL, Economides AN, Saunders S. Heparan sulfate proteoglycans retain Noggin at the cell surface. J Biol Chem. 2002;277:2089–2096. doi: 10.1074/jbc.M109151200. [DOI] [PubMed] [Google Scholar]
- 50.Viviano BL, Paine-Saunders S, Gasiunas N, Gallagher J, Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J Biol Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- 51.Hu Z, Wang C, Xiao Y, Sheng N, Chen Y, Xu Y, Zhang L, Mo W, Jing N, Hu G. NDST1-dependent heparan sulfate regulates BMP signaling and internalization in lung development. J Cell Sci. 2009;122:1145–1154. doi: 10.1242/jcs.034736. [DOI] [PubMed] [Google Scholar]
- 52.Freeman SD, Moore WM, Guiral EC, Holme AD, Turnbull JE, Pownall ME. Extracellular regulation of developmental cell signaling by XtSulf1. Dev Biol. 2008;320:436–445. doi: 10.1016/j.ydbio.2008.05.554. [DOI] [PubMed] [Google Scholar]
- 53.Lamanna WC, Frese MA, Balleininger M, Dierks T. Sulf loss influences N-, 2-O-, and 6-O-Sulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J Biol Chem. 2008;283:27724–27735. doi: 10.1074/jbc.M802130200. [DOI] [PubMed] [Google Scholar]
- 54.Lai J, Chien J, Staub J, Avula R, Greene EL, Matthews TA, Smith DI, Kaufmann SH, Roberts LR, Shridhar V. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem. 2003;278:23107–23117. doi: 10.1074/jbc.M302203200. [DOI] [PubMed] [Google Scholar]
- 55.Lai JP, Chien JR, Moser DR, Staub JK, Aderca I, Montoya DP, Matthews TA, Nagorney DM, Cunningham JM, Smith DI, Greene EL, Shridhar V, Roberts LR. hSulf1 sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology. 2004;126:231–248. doi: 10.1053/j.gastro.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 56.Lai J, Sandhu DS, Shire AM, Roberts LR. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer. 2008;39:149–158. doi: 10.1007/s12029-009-9058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Narita K, Staub J, Chien J, Meyer K, Bauer M, Friedl A, Ramakrishnan S, Shridhar V. HSulf-1 inhibits angiogenesis and tumorigenesis in vivo. Cancer Res. 2006;66:6025–6032. doi: 10.1158/0008-5472.CAN-05-3582. [DOI] [PubMed] [Google Scholar]
- 58.Dai Y, Yang Y, MacLeod V, Yue X, Rapraeger AC, Shriver Z, Venkataraman G, Sasisekharan R, Sanderson RD. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem. 2005;280:40066–40073. doi: 10.1074/jbc.M508136200. [DOI] [PubMed] [Google Scholar]
- 59.Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE. 2007;2:e392. doi: 10.1371/journal.pone.0000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abiatari I, Kleeff J, Li J, Felix K, Buchler MW, Friess H. Hsulf-1 regulates growth and invasion of pancreatic cancer cells. J Clin Pathol. 2006;59:1052–1058. doi: 10.1136/jcp.2005.031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grigoriadis A, Mackay A, Reis-Filho J, Steele D, Iseli C, Stevenson B, Jongeneel CV, Valgeirsson H, Fenwick K, Iravani M, Leao M, Simpson A, Strausberg R, Jat P, Ashworth A, Neville AM, O’Hare M. Establishment of the epithelial-specific transcriptome of normal and malignant human breast cells based on MPSS and array expression data. Breast Cancer Res. 2006;8:R56. doi: 10.1186/bcr1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castro N, Osorio C, Torres C, Bastos E, Mourao-Neto M, Soares F, Brentani H, Carraro D. Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma. Breast Cancer Res. 2008;10:R87. doi: 10.1186/bcr2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kudo Y, Ogawa I, Kitajima S, Kitagawa M, Kawai H, Gaffney PM, Miyauchi M, Takata T. Periostin promotes invasion and anchorage-independent growth in the metastatic process of head and neck cancer. Cancer Res. 2006;66:6928–6935. doi: 10.1158/0008-5472.CAN-05-4540. [DOI] [PubMed] [Google Scholar]
- 64.Lai JP, Sandhu DS, Yu C, Han T, Moser CD, Jackson KK, Guerrero RB, Aderca I, Isomoto H, Garrity-Park MM, Zou H, Shire AM, Nagorney DM, Sanderson SO, Adjei AA, Lee JS, Thorgeirsson SS, Roberts LR. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology. 2008;47:1211–1222. doi: 10.1002/hep.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uchimura K, Morimoto-Tomita M, Bistrup A, Li J, Lyon M, Gallagher J, Werb Z, Rosen SD. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006;7:2. doi: 10.1186/1471-2091-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McKenzie E, Tyson K, Stamps A, Smith P, Turner P, Barry R, Hircock M, Patel S, Barry E, Stubberfield C, Terrett J, Page M. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem Biophys Res Commun. 2000;276:1170–1177. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 67.Goldshmidt O, Zcharia E, Abramovitch R, Metzger S, Aingorn H, Friedmann Y, Schirrmacher V, Mitrani E, Vlodavsky I. Cell surface expression and secretion of heparanase markedly promote tumor angiogenesis and metastasis. Proc Nat Acad Sci USA. 2002;99:10031–10036. doi: 10.1073/pnas.152070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fux L, Ilan N, Sanderson RD, Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34:511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vlodavsky I, Goldshmidt O, Zcharia E, Atzmon R, Rangini-Guatta Z, Elkin M, Peretz T, Friedmann Y. Mammalian heparanase: involvement in cancer metastasis, angiogenesis and normal development. Semin Cancer Biol. 2002;12:121–129. doi: 10.1006/scbi.2001.0420. [DOI] [PubMed] [Google Scholar]
- 71.Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11:427–452. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miyagi T, Wada T, Yamaguchi K, Hata K. Sialidase and malignancy: A minireview. Glycoconj J. 2004;20:189–198. doi: 10.1023/B:GLYC.0000024250.48506.bf. [DOI] [PubMed] [Google Scholar]
- 73.Miyagi T, Wada T, Yamaguchi K, Shiozaki K, Sato I, Kakugawa Y, Yamanami H, Fujiya T. Human sialidase as a cancer marker. Proteomics. 2008;8:3303–3311. doi: 10.1002/pmic.200800248. [DOI] [PubMed] [Google Scholar]
- 74.Kakugawa Y, Wada T, Yamaguchi K, Yamanami H, Ouchi K, Sato I, Miyagi T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc Nat Acad Sci USA. 2002;99:10718–10723. doi: 10.1073/pnas.152597199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ueno S, Saito S, Wada T, Yamaguchi K, Satoh M, Arai Y, Miyagi T. Plasma membrane-associated sialidase is up-regulated in renal cell carcinoma and promotes interleukin-6-induced apoptosis suppression and cell motility. J Biol Chem. 2006;281:7756–7764. doi: 10.1074/jbc.M509668200. [DOI] [PubMed] [Google Scholar]
- 76.Nomura H, Tamada Y, Miyagi T, Suzuki A, Taira M, Suzuki N, Susumu N, Irimura T, Aoki D. Expression of NEU3 (plasma membrane-associated sialidase) in clear cell adenocarcinoma of the ovary: Its relationship with T factor of pTNM classification. Oncol Res. 2006;16:289–297. doi: 10.3727/000000006783981035. [DOI] [PubMed] [Google Scholar]
- 77.Tringali C, Lupo B, Cirillo F, Papini N, Anastasia L, Lamorte G, Colombi P, Bresciani R, Monti E, Tettamanti G, Venerando B. Silencing of membrane-associated sialidase Neu3 diminishes apoptosis resistance and triggers megakaryocytic differentiation of chronic myeloid leukemic cells K562 through the increase of ganglioside GM3. Cell Death Differ. 2009;16:164–174. doi: 10.1038/cdd.2008.141. [DOI] [PubMed] [Google Scholar]
- 78.Yoon SJ, Nakayama K, Hikita T, Handa K, Hakomori SI. Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc Natl Acad Sci U S A. 2006;103:18987–18991. doi: 10.1073/pnas.0609281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anastasia L, Papini N, Colazzo F, Palazzolo G, Tringali C, Dileo L, Piccoli M, Conforti E, Sitzia C, Monti E, Sampaolesi M, Tettamanti G, Venerando B. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J Biol Chem. 2008;283:36265–36271. doi: 10.1074/jbc.M805755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kato K, Shiga K, Yamaguchi K, Hata K, Kobayashi T, Miyazaki K, Saijo S, Miyagi T. Plasma-membrane-associated sialidase (NEU3) differentially regulates integrin-mediated cell proliferation through laminin- and fibronectin-derived signalling. Biochem J. 2006;394:647–656. doi: 10.1042/BJ20050737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Toledo MS, Suzuki E, Handa K, Hakomori S. Effect of ganglioside and tetraspanins in microdomains on interaction of integrins with fibroblast growth factor receptor. J Biol Chem. 2005;280:16227–16234. doi: 10.1074/jbc.M413713200. [DOI] [PubMed] [Google Scholar]
- 82.Gopalakrishna P, Rangaraj N, Pande G. Cholesterol alters the interaction of glycosphingolipid GM3 with alpha5beta1 integrin and increases integrin-mediated cell adhesion to fibronectin. Exp Cell Res. 2004;300:43–53. doi: 10.1016/j.yexcr.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 83.Hinek A, Pshezhetsky AV, von Itzstein M, Starcher B. Lysosomal sialidase (neuraminidase-1) is targeted to the cell surface in a multiprotein complex that facilitates elastic fiber assembly. J Biol Chem. 2006;281:3698–3710. doi: 10.1074/jbc.M508736200. [DOI] [PubMed] [Google Scholar]
- 84.Gadhoum SZ, Sackstein R. CD15 expression in human myeloid cell differentiation is regulated by sialidase activity. Nat Chem Biol. 2008;4:751–757. doi: 10.1038/nchembio.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uemura T, Shiozaki K, Yamaguchi K, Miyazaki S, Satomi S, Kato K, Sakuraba H, Miyagi T. Contribution of sialidase NEU1 to suppression of metastasis of human colon cancer cells through desialylation of integrin beta 4. Oncogene. 2009;28:1218–1229. doi: 10.1038/onc.2008.471. [DOI] [PubMed] [Google Scholar]
- 86.Starcher B, d’Azzo A, Keller PW, Rao GK, Nadarajah D, Hinek A. Neuraminidase-1 is required for the normal assembly of elastic fibers. Am J Physiol Lung Cell Mol Physiol. 2008;295:L637–L647. doi: 10.1152/ajplung.90346.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duca L, Blanchevoye C, Cantarelli B, Ghoneim C, Dedieu S, Delacoux F, Hornebeck W, Hinek A, Martiny L, Debelle L. The elastin receptor complex transduces signals through the catalytic activity of its Neu-1 subunit. J Biol Chem. 2007;282:12484–12491. doi: 10.1074/jbc.M609505200. [DOI] [PubMed] [Google Scholar]
- 88.Phillips ML, Nudelman E, Gaeta FC, Perez M, Singhal AK, Hakomori S, Paulson JC. ELAM-1 mediates cell adhesion by recognition of a carbohydrate ligand, sialyl-LeX. Science. 1990;250:1130–1132. doi: 10.1126/science.1701274. [DOI] [PubMed] [Google Scholar]
- 89.van Gisbergen KPJM, Sanchez-Hernandez M, Geijtenbeek TBH, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med. 2005;201:1281–1292. doi: 10.1084/jem.20041276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gross AJ, Lyandres JR, Panigrahi AK, Prak ETL, DeFranco AL. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol. 2009;182:5382–5392. doi: 10.4049/jimmunol.0803941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guimaraes MJ, Bazan JF, Castagnola J, Diaz S, Copeland NG, Gilbert DJ, Jenkins NA, Varki A, Zlotnik A. Molecular cloning and characterization of lysosomal sialic acid O-acetylesterase. J Biol Chem. 1996;271:13697–13705. doi: 10.1074/jbc.271.23.13697. [DOI] [PubMed] [Google Scholar]
- 92.Pillai S, Cariappa A, Pirnie SP. Esterases and autoimmunity: the sialic acid acetylesterase pathway and the regulation of peripheral B cell tolerance. Trends Immunol. 2009;30:488–493. doi: 10.1016/j.it.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y-I. Mutation of the mouse Klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 94.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54:810–817. doi: 10.1161/HYPERTENSIONAHA.109.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rodriguez JA, Piddini E, Hasegawa T, Miyagi T, Dotti CG. Plasma membrane ganglioside sialidase regulates axonal growth and regeneration in hippocampal neurons in culture. J Neurosci. 2001;21:8387–8395. doi: 10.1523/JNEUROSCI.21-21-08387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Farooqui T, Franklin T, Pearl DK, Yates A. Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J Neurochem. 1997;68:2348–2355. doi: 10.1046/j.1471-4159.1997.68062348.x. [DOI] [PubMed] [Google Scholar]
- 98.Valaperta R, Valsecchi M, Rocchetta F, Aureli M, Prioni S, Prinetti A, Chigorno V, Sonnino S. Induction of axonal differentiation by silencing plasma membrane-associated sialidase Neu3 in neuroblastoma cells. J Neurochem. 2007;100:708–719. doi: 10.1111/j.1471-4159.2006.04279.x. [DOI] [PubMed] [Google Scholar]
- 99.Proshin S, Yamaguchi K, Wada T, Miyagi T. Modulation of neuritogenesis by ganglioside-specific sialidase (Neu 3) in human neuroblastoma NB-1 cells. Neurochem Res. 2002;27:841–846. doi: 10.1023/a:1020269326825. [DOI] [PubMed] [Google Scholar]
- 100.von Reitzenstein C, Kopitz J, Schuhmann V, Cantz M. Differential functional relevance of a plasma membrane ganglioside sialidase in cholinergic and adrenergic neuroblastoma cell lines. Eur J Biochem. 2001;268:326–333. doi: 10.1046/j.1432-1033.2001.01883.x. [DOI] [PubMed] [Google Scholar]
- 101.Kalka D, von Reitzenstein C, Kopitz J, Cantz M. The plasma membrane ganglioside sialidase cofractionates with markers of lipid rafts. Biochem Biophys Res Commun. 2001;283:989–993. doi: 10.1006/bbrc.2001.4864. [DOI] [PubMed] [Google Scholar]
- 102.Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, Abad-Rodriguez J. Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat Neurosci. 2005;8:606–615. doi: 10.1038/nn1442. [DOI] [PubMed] [Google Scholar]
- 103.Yang LJS, Lorenzini I, Vajn K, Mountney A, Schramm LP, Schnaar RL. Sialidase enhances spinal axon outgrowth in vivo. Proc Nat Acad Sci USA. 2006;103:11057–11062. doi: 10.1073/pnas.0604613103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porwoll S, Loch N, Kannicht C, Nuck R, Grunow D, Reutter W, Tauber R. Cell surface glycoproteins undergo postbiosynthetic modification of their N-glycans by stepwise demannosylation. J Biol Chem. 1998;273:1075–1085. doi: 10.1074/jbc.273.2.1075. [DOI] [PubMed] [Google Scholar]
- 105.Porwoll S, Fuchs H, Tauber R. Characterization of a soluble class I alpha-mannosidase in human serum. FEBS Lett. 1999;449:175–178. doi: 10.1016/s0014-5793(99)00422-6. [DOI] [PubMed] [Google Scholar]
- 106.Khunsook S, Bean BS, McGowan SR, Alhadeff JA. Purification and characterization of plasma membrane-associated human sperm alpha-L-fucosidase. Biol Reprod. 2003;68:709–716. doi: 10.1095/biolreprod.102.004465. [DOI] [PubMed] [Google Scholar]
- 107.Khunsook S, Alhadeff JA, Bean BS. Purification and characterization of human seminal plasma α-L-fucosidase. Mol Hum Reprod. 2002;8:221–227. doi: 10.1093/molehr/8.3.221. [DOI] [PubMed] [Google Scholar]
- 108.Li C, Qian J, Lin JS. Purification and characterization of alpha-L-fucosidase from human primary hepatocarcinoma tissue. World J Gastroenterol. 2006;12:3770–3775. doi: 10.3748/wjg.v12.i23.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ali S, Jenkins Y, Kirkley M, Dagkalis A, Manivannan A, Crane IJ, Kirby JA. Leukocyte extravasation: An immunoregulatory role for α-L-fucosidase? J Immunol. 2008;181:2407–2413. doi: 10.4049/jimmunol.181.4.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mencarelli S, Cavalieri C, Magini A, Tancini B, Basso L, Lemansky P, Hasilik A, Li YT, Chigorno V, Orlacchio A, Emiliani C, Sonnino S. Identification of plasma membrane associated mature beta-hexosaminidase A, active towards GM2 ganglioside, in human fibroblasts. FEBS Lett. 2005;579:5501–5506. doi: 10.1016/j.febslet.2005.08.081. [DOI] [PubMed] [Google Scholar]
- 111.Jenkins N, Parekh RB, James DC. Getting the glycosylation right: Implications for the biotechnology industry. Nat Biotechnol. 1996;14:975–981. doi: 10.1038/nbt0896-975. [DOI] [PubMed] [Google Scholar]
- 112.Gramer MJ. Detecting and minimizing glycosidase activities that can hydrolyze sugars from cell culture-produced glycoproteins. Mol Biotechnol. 2000;15:69–75. doi: 10.1385/MB:15:1:69. [DOI] [PubMed] [Google Scholar]
- 113.Severi E, Hood DW, Thomas GH. Sialic acid utilization by bacterial pathogens. Microbiology. 2007;153:2817–2822. doi: 10.1099/mic.0.2007/009480-0. [DOI] [PubMed] [Google Scholar]
- 114.Katayama T, Fujita K, Yamamoto K. Novel bifidobacterial glycosidases acting on sugar chains of mucin glycoproteins. J Biosci Bioeng. 2005;99:457–465. doi: 10.1263/jbb.99.457. [DOI] [PubMed] [Google Scholar]