

Abstract
Background
Aberrant endoplasmic reticulum (ER) stress is associated with several cardiovascular diseases including atherosclerosis. The mechanism by which aberrant ER stress develops is poorly understood. This study investigated whether dysfunction of AMP-activated protein kinase (AMPK) causes aberrant ER stress and atherosclerosis in vivo.
Methods and Results
Human umbilical vein endothelial cells (HUVEC) and mouse aortic endothelial cells (MAEC) from AMPK-deficient mice were used to assess the level of ER stress using western blotting. Reduction of AMPKα2 expression significantly increased the level of ER stress in HUVEC. In addition, MAEC from AMPKα2 knockout mice (AMPKα2−/−) had higher expression of markers of ER stress and increased levels of intracellular Ca2+. These phenotypes were abolished by adenovirally overexpressing constitutively active AMPK mutants (Ad-AMPK-CA) or by transfecting sarco-endoplasmic reticulum calcium ATPase (SERCA). Inhibition of SERCA induced ER stress in endothelial cells. Furthermore, reduction of AMPKα expression suppressed SERCA activity. In addition, SERCA activity was significantly reduced concomitantly with increased oxidation of SERCA in MAEC from AMPKα2−/− mice. Both of these phenotypes were abolished by adenovirally overexpressing Ad-AMPK-CA. Furthermore, tempol, which restored SERCA activity and decreased oxidized SERCA levels, markedly reduced the level of ER stress in MAEC from AMPKα2−/− mice. Finally, oral administration of tauroursodeoxycholic acid (TUDCA), a chemical chaperone that inhibits ER stress, significantly reduced both ER stress and aortic lesion development in LDL receptor and AMPKα2 deficient mice.
Conclusions
These results suggest that AMPK functions as a physiological suppressor of ER stress by maintaining SERCA activity and intracellular Ca2+ homeostasis.
Keywords: AMPK, SERCA, oxidation, ER stress, atherosclerosis
Introduction
The endoplasmic reticulum (ER), which serves as a center for lipid synthesis, protein folding, and maturation in eukaryotic cells, is a major signal transducing organelle that senses and responds to changes in homeostasis.1 The accumulation of unfolded protein aggregates leads to the activation of transmembrane sensors/transducers, including inositol-requiring enzyme (Ire)1α, PERK (RNA-dependent protein kinase–like endoplasmic reticulum kinase), and activating transcription factor (ATF) 6. These sensors regulate several signaling pathways, which results in changes in gene expression and protein synthesis. Altered protein folding and ER stress occur during pathological conditions, such as ischemia, hypoxia, heat shock, proteasome inhibition, glycosylation inhibition, oxidative stress, andCa2+ depletion of ER stores, which are collectively referred to as ER stress.2, 3 Increasing evidence suggests that aberrant ER stress plays important roles in the etiology of numerous diseases, such as diabetes mellitus, obesity, atherosclerosis, cancer, and neurodegenerative disorders.3–6
Disruption of the ER stress response and/or the activation of the unfolded protein response (UPR) can play a significant role in the developmentand progression of atherosclerotic lesions. 7, 8 ER stress-inducing agents canpromote cellular responses in vascular cells that mimic the hallmark featuresof atherosclerosis, including apoptosis, cholesterol accumulation, and activation of inflammatory pathways.8–11 Furthermore, ER stress is markedly increased in endothelial cells subjected to atherosclerosis-prone shear stress.12 Increased endothelial expression of GRP78 was also observed in atheroprone versus atheroprotective regions in C57BL6 mice.12 ER stress occurs in the atherosclerotic lesions of hyperglycemic apolipoprotein (Apo) E knockout mice and is reported to be associated with acute coronary syndrome.13 ER stress has emerged as a new adaptive system that determines the survival fate of cells; therefore, it may contribute tothe erosion or rupture of atherosclerotic plaques.14 However, the mechanism by which ER stress is regulated under physiological conditions is unknown and a casual role for ER stress in the development of atherosclerosis remains to be established.
The AMP-activated protein kinase (AMPK), an evolutionarily conserved energy sensor, has been shown to play a critical role in controlling systemic energy balance and metabolism.15, 16 AMPK is activated in response to stresses including hypoxia,17 oxidants (peroxynitrite and hydrogen peroxide),18, 19 hyperosmolarity, exercise (in muscle), adipokines (adiponectin and leptin),20 and drugs (metformin and thiazolidinediones).21 Activation of AMPK leads to the phosphorylation of a number of target molecules, and AMPK has been reported to exert multiple protective effects in vascular cells by inhibiting inflammation, oxidant production, vascular smooth muscle cell proliferation, insulin resistance,22 and reduces the risk for developing obesity and type 2 diabetes.15, 23, 24 Interestingly, several recent studies 17, 25–27 have shown that AMPK activation protects against hypoxic injury to cardiac tissue by suppressing ER stress. However, the role of AMPK in ER stress and whether this affects atherosclerosis remains unknown. The present study was aimed to establish whether AMPK functions as a physiological suppressor of aberrant ER stress and whether AMPK inhibition contributed to aberrant ER stress and atherosclerosis in vivo. Our data suggest that AMPKα2 deletion increases SERCA oxidation, intracellular Ca2+ accumulation, and consequent ER stress and atherosclerosis.
Material and Methods
A detailed description of the Materials and Methods including isolation and culture of mouse aortic endothelial cells +, intracellular Ca2+ levels, AMPK activity and SERCA activity, radiometric Ca2+ and dynamic SERCA function assays, biotinlated-iodocetamide (b-IAM) labeling of SERCA cysteine 674, and assays for aortic lesions is provided in the on-line supplemental materials section.
Methods
Animals
Male AMPKα1 knockout (AMPKα1−/−), male AMPKα2 knockout (AMPKα2−/−), and their genetic controls (C57BL6 WT mice) were breed in the University of Oklahoma Health Science Center. Mice were housed in temperature-controlled cages under a 12-h light-dark cycle and given free access to water and normal chow. The mice were euthanized with inhaled isoflurane. Aortas were then removed and immediately frozen in liquid nitrogen or incubated with different agents. AMPKα2−/− mice that had been backcrossed onto a C57BL/6 background were crossed with LDLr−/− mice (The Jackson Laboratory), also on the C57BL/6 background, to generate LDLr−/−/AMPKα2−/− mice. LDLr−/− littermates or C57BL/6 WT mice were served as controls. Atherosclerosis was accelerated by feeding mice atherogenic rodent diet containing 1.3% cholesterol and 0.5% cholic acid (Harlan Teklad, TD. 02028). This diet was administered beginning at 8–12 weeks of age in LDLr−/− or LDLr−/−/AMPKα2−/− mice with or without TUDCA (0.5 g/kg/day in drinking water) for 10 consecutive weeks. The animal protocol was reviewed and approved by the University of Oklahoma Institute Animal Care and Use Committee.
Cell culture
Human umbilical vein endothelial cells (HUVEC) were grown in Eagle’s Basal Medium (EBM) (Clonetics Inc. Walkersville, MD) supplemented with 2% fetal bovine serum, penicillin (100 u/ml), and streptomycin (100μg/ml). In all experiments, cells between passages 3 and 8 were used. All cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were grown until they reached 70 to 80% confluence before being treated with the indicated agents.
For isolation of mouse aortic endothelial cells (MAECs), aortas were collected from mice and washed twice with PBS at 4°C. Aortas were then carefully stripped of fat and connective tissue, cut into 3 mm long sections, and soaked in a 0.2% collagenase solution at 37°C with shaking to detach the endothelial cells. The solution containing the MAECs was centrifuged at 1000 rpm for 15 min at 4°C, and the resulting pellet was washed with PBS. The cells were then seeded onto culture plates and grown in EBM. Endothelial cells were identified based on their positive expression for eNOS, ICAM-1, and VCAM-1. MAECs between passages 3 and 5 were used for siRNA transfection.
siRNA transfection in endothelial cells
Transient transfection of siRNA was carried out according to Santa Cruz’s protocol 28. Briefly, the siRNAs were dissolved in ddH2O to prepare a 10 μM stock solution. HUVEC and MAEC grown in six-well plates were transfected with siRNA in transfection medium (Gibcol) containing RNAiMax (Invitrogen). For each transfection, 100 μl of transfection medium containing 4 μl of siRNA was gently mixed with 100 μl of transfection medium containing 4 μl of transfection reagent. After 30-min incubation at room temperature, the siRNA-lipid complexes were added to the cells in 1 ml of transfection medium. The cells were incubated with this mixture for 6 h at 37°C. After incubation, the transfection medium was replaced with normal medium, and cells were further cultured for 48 h.
Adenoviral infection
Ad-GFP, a replication-defective adenoviral vector expressing GFP, served as a control for all adenoviral experiments. The AMPK-DN adenoviral vector expressed a mutated form of AMPK in which Lysine 45 was substituted with arginine (K45R), as previously described.29 To generate the AMPK-CA adenoviral vector, we subcloned a rat cDNA encoding residues 1 – 312 of AMPK, which contained an aspartic acid residue substituted for Threonine 172 (T172D), into a shuttle vector (pShuttle CMV [cytomegalovirus]) (Stratagene, CA).
HUVEC were infected with Ad-GFP, Ad-AMPK-DN, or Ad-AMPK-CA overnight in medium supplemented with 2% fetal calf serum. The cells were then washed and incubated in fresh endothelium growth medium without fetal calf serum for an additional 12 h prior to experimentation. These conditions typically produced an infection efficiency of at least 80%, as determined by GFP expression.
Detection of superoxide anions by dihydroethidine (DHE) staining of isolated aortas
Aortas were harvested, washed in cold PBS, embedded in tissue freezing medium (Polysciences, Inc.), and cryosectioned into 8-μm thick sections. Frozen sections were mounted onto Superfrost Plus slides (Fisher Scientific) and stained with DHE (2 μM). Fluorescence of 2-hydroxyethidium, the sole product of the reaction between O2.− and DHE, was imaged using an Olympus fluorescence microscope.
Immunohistochemistry
The aortic arch was dissected, fixed in 4% paraformaldehyde for 16 h, and embedded in paraffin. Four-micrometer thick sections were deparaffinized, rehydrated, and microwaved in citrate buffer for antigen retrieval. Sections were successively incubated in endogenous peroxidase and alkaline phosphatase block buffer (DAKO), protein block buffer, and primary antibodies, which were incubated with sections overnight at 4°C. After rinsing in wash buffer, sections were incubated with labeled polymer-horseradish peroxidase anti-mouse or anti-rabbit antibodies and DAB chromogen. Alternatively, they were incubated with polymer-alkaline phosphatase anti-mouse or anti-rabbit antibody and Permanent Red chromogen (EnVision™ G|2 Doublestain System, DAKO). After the final wash, the sections were counterstained with hematoxylin.
Western blot analysis
Cell lysates and tissue homogenates were subjected to western blotting analysis, as previously described19.
Detection of ROS
Tissue and cell O2− levels were assessed using the DHE fluorescence/HPLC assay with minor modifications 30. Briefly, tissue and cells were incubated with DHE (10 μM) for 30 min, homogenized, and subjected to methanol extraction. HPLC was performed using a C-18 column (mobile phase: gradient of acetonitrile and 0.1% trifluoroacetic acid) to separate and quantify oxyethidium (product of DHE and O2−) and ethidium (product of DHE auto-oxidation). O2− production was determined by assessing the conversion of DHE into oxyethidine.
Statistical analysis
Data represent mean ± standard error of the mean (SEM). Statistical analysis was performed using Student t test (2 groups) or one-way ANOVA with Bonferroni’s procedure for multiple comparison tests (≥ 3 groups) using GraphPad Prism 4 (GraphPad Software, Inc). Values of p<0.05 were considered significant.
The authors had full access to and take full responsibility for the integrity of the data presented in this manuscript. All authors have read and agreed to the data presented in this manuscript. There are no conflicts of interest to disclose.
Results
Detection of AMPKα2 expression in endothelial cells
We have previously31 reported that AMPKα1βγ1 is a predominant AMPK isoform in cultured endothelial cells. The relative contribution of AMPKα2 to total AMPK activity in endothelial cells is unknown. HUVEC stained positively with isoform-specific antibodies for AMPKα1 and AMPKα2 (Figure 1A). To confirm the specificity of these antibodies, HUVEC were transfected with control siRNA or siRNA specific to AMPKα1, AMPKα2, or non-selective AMPKα. Transfection of non-selective AMPKα siRNA reduced the levels of AMPKα protein by 88.7±4.5%, as compared to the control siRNA (Figure 1B). Transfection of AMPKα1-specific siRNA suppressed AMPKα levels by 77.8%, whereas AMPKα2 siRNA reduced AMPKα levels by 22.2% (Figure 1B). These results implied that AMPKα2 is a minor AMPKα isoform expressed in HUVEC, as it only accounted for approximately 22% of the total AMPKα.
Figure 1. Reduction of AMPKα2 expression induces ER stress in endothelial cells.

A&B. Western blot analysis of AMPKα2 in HUVEC. The blot is representative of at least three blots from three independent experiments; ♣p<0.05, AMPKα1 or α2 or AMPKα siRNA vs. control siRNA, respectively; #p<0.05, AMPKα2 siRNA vs. AMPKα1 siRNA; †p<0.05, AMPKα siRNA vs. AMPKα1 or α2 siRNA. n=4. C&D. Genetic inhibition of AMPKα2 induces ER stress in HUVEC. ♣p<0.05 vs. control siRNA; #p<0.05 vs. AMPKα1 siRNA, †p<0.05 vs. AMPKα1 or AMPK α2 siRNA; n=4.
Reduction of AMPKα expression in endothelial cells increases ER stress
Several recent studies 17, 25–27 have shown that AMPK activation protects against hypoxic injury in cardiac tissue by suppressing ER stress, so we determined whether reduction of AMPKα1 or AMPKα2 expression induced ER stress in HUVEC. Confluent HUVEC were transfected with either non-selective AMPKα siRNA, AMPKα1-specific siRNA, or AMPK α2-specific siRNA. Transfection of AMPKα1 siRNA caused a modest but significant increase (p<0.05) in ER stress, as evidenced by increased expression of p-PERK, p-elf2α, p-JNK, and XBP-1, as compared to HUVEC transfected with control siRNA (Figure 1C and 1D). Transfection of AMPKα2 siRNA or non-selective AMPKα siRNA induced greater ER stress response (increased p-PERK, p-elf2α, p-JNK, and XBP-1) than that observed in HUVEC transfected with control siRNA or AMPKα1-specific siRNA (Figure 1D). Thus, these results suggest that AMPKα2 is the major isoform that regulates ER stress in HUVEC.
Assessment of AMPKα2 protein and activity in isolated mouse aortic endothelial cells
To further examine the potential contribution of both AMPKα1 and AMPKα2 activity to ER stress in endothelial cells, mouse aortic endothelial cells (MAEC) were isolated from AMPKα1−/− and AMPKα2−/− mice. These isolated primary endothelial cells were first confirmed by using specific antibodies against AMPKα1 or AMPKα2 or AMPKα (pan-) (Figure 2A). We assessed the relative distribution of AMPKα1 and AMPKα2 in MAEC. As compared to MAEC from wild type (WT) mice, the levels of AMPKα in AMPKα1−/− mice (i.e. AMPKα2 protein) were reduced to 21.7% (Figure 2A-B). In contrast, the AMPKα protein expression in AMPKα2−/− mice (i.e., AMPKα1 protein) was equal to 78.3% of that observed in WT mice (Figure 2A–B).
Figure 2. AMPKα2-dependent ER stress in isolated mouse aortic endothelial cells.


A. Characterization of MAEC from WT, AMPKα1 KO, and AMPKα2 KO. AMPKα, AMPKα1, and AMPKα2 were detected by using the specific antibodies in western blots. The blot is a representative of 6 blots from 6 individual experiments. B. Effects of AMPKα1 deletion and AMPKα2 deletion on the levels of phospho-ACC at Ser79 in MAEC. The blot is a representative of three blots obtained from three individual experiments. C. Distribution of AMPKα1 and AMPKα2 in isolated of MAEC. AMPKα was first immunoprecipitated from MAEC of WT, AMPKα1, and AMPKα2 followed by western blot analysis of AMPK. The blot is a representative of four blots from four independent experiments. ♣p<0.05 vs. WT; #p<0.05 vs. AMPKα1−/−; D. Assays of AMPK activity in MAEC from WT, AMPKα1−/− or AMPKα2−/− primary cultured endothelial cells. ♣P<0.05 vs. WT; #p<0.05 vs. AMPKα1−/−; n=4. E&F. Increased levels of ER stress in MAEC from AMPKα1−/− or AMPKα2−/− mice. ♣P<0.05 vs. WT; #p<0.05 vs. AMPKα1−/−; n=8. G&H. Adenoviral overexpression of Ad-AMPK-CA alleviates ER stress in MAEC from AMPKα2−/− mice. ♣p<0.05 vs. WT; #P<0.05 vs. AMPKα2−/− or AMPKα2−/− infected with Ad-GFP; n=5.
We next determined the AMPK activity in the MAEC isolated from AMPKα1−/− and AMPKα2−/− mice. AMPK is known to phosphorylate ACC at Ser79 (p-ACC) and the levels of p-ACC can be used as an index for AMPK activity in endothelial cells. As shown in Figure 2B, p-ACC was markedly reduced in MAEC isolated from AMPKα1 KO than those in WT, confirming that AMPKα1 is a predominant isform of AMPKα in endothelial cells. In contrast, a modest reduction of p-ACC was also found seen in those from AMPKα2 KO (Figure 2B), implying that AMPKα2 also contributed to maintaining total AMPK activity in endothelial cells.
To further confirm the relative contributions of AMPKα1 and AMPKα2 to total AMPK, AMPKα was immunoprecipitated from MAEC of AMPKα1−/− and AMPKα2−/− mice by using a non-selective antibody against AMPKα. As shown in Figure 2C, AMPKα1 appeared to be a major isoform whereas AMPKα2 accounted for ~20% of total AMPKα in MAEC from WT. AMPK activity was assayed by measuring 32P-ATP incorporation into the SAMS peptide. The AMPK activity in MAEC from AMPKα1−/− mice, which represented AMPKα2 activity, was equal to 22.5% of the total AMPK activity observed in MAEC from WT mice (Figure 2D). In contrast, the AMPK activity measured in MAEC from AMPKα2−/− mice, which represented AMPKα1 activity, was approximately 77.5% of that observed in MAEC from WT mice (Figure 2D). Thus, these results indicate that AMPKα1 accounts for 78% of the total AMPK in MAEC, whereas AMPKα2 accounts for 22%.
Increased ER stress in AMPKα2−/− mouse aortic endothelial cells
We determined the levels of ER stress markers in cultured MAEC isolated from AMPKα1−/− and AMPKα2−/− mice. As depicted in Figures 2E and 2F, the levels of ER stress markers (p-PERK, GRP78, XBP-1, and ATF6) were markedly elevated in the MAEC isolated from both the AMPKα1−/− and AMPKα2−/− mice, as compared to MAEC obtained from WT mice (Figure 2F). Notably, the increased expression of ER stress markers was significantly greater (p<0.05) in MAEC from AMPKα2−/− mice, as compared to those isolated from AMPKα1−/− mice, which is consistent with the previous data (Figures 1C and 1D). Taken together, our results suggest that AMPKα2 might play a more important role than AMPKα1 in regulating ER stress in endothelial cells.
Adenoviral overexpression of a constitutively active form of AMPK reduces ER stress in mouse aortic endothelial cells isolated from AMPKα2−/− mice
To establish a causal relationship between loss of AMPKα2 expression and an aberrant ER stress response in endothelial cells, we investigated whether reconstituting AMPK expression reduced the ER stress response in the MAEC derived from AMPKα2−/− mice. MAEC from AMPKα2−/− and WT mice were infected with adenoviruses encoding GFP (Ad-GFP) or constitutively active AMPK mutants (Ad-AMPK-CA). Overexpression of Ad-AMPK-CA, but not Ad-GFP, markedly reduced the ER stress response (p-PERK, GRP78, XBP-1, and ATF6) in MAEC from AMPKα2−/− mice (Figure 2G and 2H), suggesting that activation of AMPK can effectively suppress the ER stress response in AMPKα2−/− mice.
Deletion of AMPKα2 increases the intracellular Ca2+ levels and calmodulin kinase II activity
Previous studies 32, 33 have demonstrated that elevation of intracellular Ca2+ is a common mechanism for aberrant ER stress and UPR activation, therefore we assessed whether inhibition of AMPK increased ER stress by altering intracellular Ca2+ levels. AMPKα1 siRNA or AMPKα2 siRNA or non-specific AMPKα siRNA markedly increased the intracellular Ca2+ levels in HUVEC (Figure 3A). Furthermore, intracellular Ca2+ levels were also elevated in AMPKα2−/−endothelial cells (p<0.05), as compared to MAEC isolated from WT mice (Figure 3B). Consistent with this result, the activity of calmodulin kinase II, a Ca2+-dependent kinase, was significantly greater in MAEC from AMPKα2−/− mice, as compared to those obtained from WT mice (p<0.05) (Figure 3C). Taken together, these results suggest that reduction of AMPKα2 activity increased the intracellular Ca2+ levels.
Figure 3. AMPKα deletion causes a calcium-dependent ER stress response in endothelial cells.

A. Genetic inhibition of AMPK increases the intracellular Ca2+ levels in HUVEC. ♣p<0.05 vs. control siRNA; n=6. B. Elevation of intracellular Ca2+ in cultured MAEC from AMPKα2−/− mice. ♣p<0.05 vs. WT; n=8. C. Increased activity of Ca2+–dependent CAMKII in MAEC from AMPKα2−/− mice. ♣p<0.05 vs. WT; n=4. D. Reduction of ER stress by BAPTA in MAEC isolated from AMPKα2−/− mice. The blot is a representative of four blots from four independent experiments.
Chelation of intracellular Ca2+ suppresses the ER stress response in MAEC from AMPKα2−/− mice
We determined whether the increased intracellular Ca2+ levels contributed to an aberrant ER stress response in MAEC from AMPKα2−/− mice. We assessed whether BAPTA, an intracellular Ca2+ chelator, suppressed the ER stress response in MAEC. The addition of BAPTA attenuated the ER stress response in MAEC from AMPKα2−/− mice (Figure 3D). Taken together, these data support that the AMPK inhibition-induced ER stress response was mediated by an increase in intracellular Ca2+ levels.
AMPK inhibition reduces Ca2+ clearance and intracellular Ca2+ storage in endothelial cells
Ionomycin causes a rapid release of Ca2+ from the ER. Under normal conditions, Ca2+ is subsequently reabsorbed back into the ER primarily through the function of the sarco-endoplasmic reticulum calcium ATPase (SERCA). Therefore, the function of SERCA can be assayed by measuring ionomycin-induced Ca2+ clearance and storage. Since inhibition of AMPKα2 causes an elevation in intracellular Ca2+ levels, we reasoned that inhibition of AMPKα2 might suppress SERCA activity. To assess the role of SERCA, Indo-1/AM loaded endothelial cells were suspended in a nominally Ca2+-free solution to eliminate Ca2+ influx and the intracellular Ca2+ stores were depleted by adding ionomycin (10 μM). Ionomycin caused a rapid rise in intracellular Ca2+ levels, which subsequently returned to basal levels within 100 seconds (Figure 4A). The AMPK inhibitor Compound C markedly attenuated the amount of Ca2+ released in response to ionomycin and prolonged the time required for intracellular Ca2+ normalization (Figure 4A). MAEC from AMPKα2−/− mice exhibited impaired Ca2+ clearance and storage, as compared to MAEC from WT mice (Figures 4B). Similarly, gene silencing of both AMPKα isoforms in HUVEC caused similar effects on Ca2+ dynamics (Figure 4C). Taken together, these results suggest that inhibition of AMPK or reduction in AMPKα2 expression inhibits SERCA-dependent Ca2+ clearance and storage in endothelial cells.
Figure 4. AMPK inhibition impairs ionomycin-induced Ca2+ release and return transients.
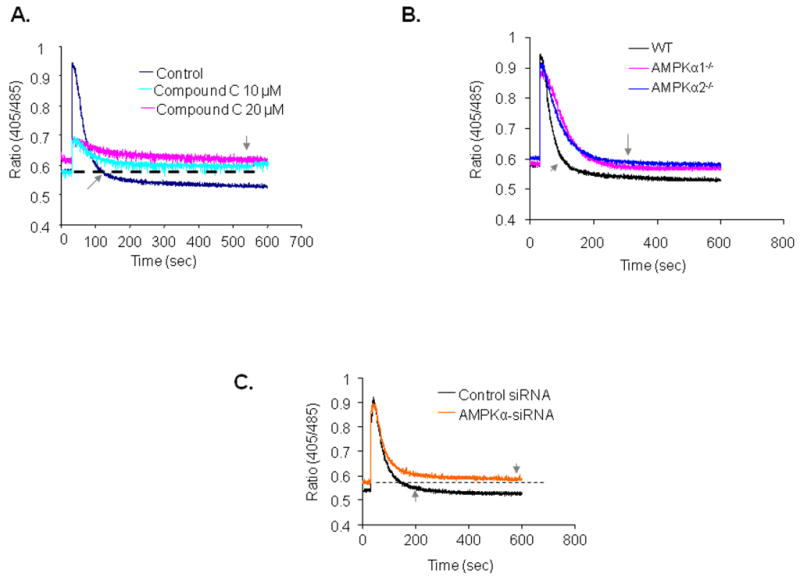
After being loaded with Indo-1/AM dye, the cells were stimulated with ionomycin (10 μM) which induces Ca2+ release from the ER. The small arrow indicates the timing to return to basal Ca2+ levels. A. Effects of Compound C on ionomycin-induced Ca2+ release and intracellular Ca2+ stores. B. Defective Ca2+ release and stores in MAEC derived from AMPKα1−/− mice (red), AMPKα2−/− mice (blue) and WT mice (black), n=3. C. Genetic inhibition of AMPK impairs ionomycin-induced Ca2+ release and intracellular Ca2+ stores. HUVEC were transfected with control siRNA or AMPK-specific siRNA. The ratiometric Ca2+ dynamics were monitored 36 h after the siRNA transfections with AMPK-siRNA (orange) or control siRNA (black), n=3.
SERCA inhibition increases intracellular Ca2+ levels and the ER stress response in endothelial cells
There is evidence that inhibition of SERCA causes ER stress.34 Since AMPK inhibition reduced SERCA activity, we determined whether overexpression of SERCA reduced the ER stress response caused by loss of AMPKα2 expression. The human SERCA2b gene (c-terminally myc-tagged), which functionally compensates for SERCA3 loss in the endothelium,35 was transfected into MAEC by electroporation. Overexpression of SERCA2b was confirmed using western blotting with an anti-myc antibody (Figure 5A). As expected, transfection of SERCA2b reduced the levels of intracellular Ca2+ in MAEC from AMPKα2−/− mice, whereas it had no effect in MAEC from WT mice (Figure 5B). In addition, SERCA2b transfection significantly attenuated the ER stress response in MAEC from AMPKα2−/− mice (Figure 5C and 5D).
Figure 5. SERCA-mediated ER stress in MAEC from AMPKα2−/− mice.

A. Transfection of SERCA2b into MAEC from WT and AMPKα2−/− mice. The blot is representative of four blots obtained from four independent experiments. B. Transfection of SERCA2b into MAEC from AMPKα2−/− mice normalizes intracellular Ca2+ levels. ♣p<0.05 vs. WT; #p<0.05 vs. AMPKα2−/− alone. C&D. Overexpression of SERCA2 suppresses ER stress in MAEC from AMPKα2−/− mice. The blot is representative of three blots obtained from three independent experiments. ♣p<0.05 vs. WT + Ad-GFP; #p<0.05 vs. AMPKα2−/− + Ad-GFP. E. Reduction of SERCA2 expression by siRNA induces ER stress in HUVEC. After the siRNA transfection, the cells were exposed to calcimycin (5 μM) for 1 h. ER stress markers were monitored using western blotting. The blot is a representative of three blots obtained from three independent experiments.
We next determined whether reduction of SERCA2 expression altered the levels of intracellular Ca2+ and the ER stress response in HUVEC. As shown in Figure 5E, transfection of SERCA2-specific siRNA, but not control siRNA, significantly reduced SERCA2 expression in HUVEC. This increased the ER stress response under basal conditions (Figure 5E). The levels of ER stress in response to calcimycin were greater in HUVEC transfected with SERCA-specific siRNA than those transfected with control siRNA (Figure 5E). Taken together, these results suggest that pharmacological or genetic inhibition of SERCA induced ER stress in endothelial cells.
Deletion of AMPKα2 suppresses SERCA activity but increases its oxidation
We further examined the mechanism by which AMPK regulated SERCA activity. Reduction of AMPK expression using AMPKα-specific siRNA markedly reduced SERCA activity in MAEC, as compared to those transfected with control siRNA (Figure 6A). SERCA activity was also markedly reduced in MAEC from AMPKα2−/− mice, relative to WT mice (p<0.05) (Figure 6B).
Figure 6. AMPKα2 deletion inhibits SERCA activity but increases its oxidation status in endothelial cells.


A. Reduction of AMPK expression with AMPKα-specific siRNA decreases SERCA activity; ♣p<0.05 vs. control siRNA; n=4. B. Adenoviral overexpression of AMPK-CA normalizes SERCA activity in MAEC from AMPKα2−/− mice. ♣p<0.05 vs. WT, n=4; #p<0.05 vs. AMPKα2−/− +Ad-GFP; n=4; C. Adenoviral overexpression of AMPK-CA prevents the oxidation of SERCA in MAEC from AMPKα2−/− mice. ♣p<0.05 vs. WT; # p<0.05 vs. Ad-GFP; n=4. D. Increased ER stress in response to calcimycin in MAEC from AMPKα2−/− mice. MAEC from WT and AMPKα2−/− mice were exposed to calcimycin at the indicated concentrations for 2 h. Both ER stress markers and SERCA2 expression were monitored using western blotting, as described in the Materials and Methods. The blot is representative of three blots from three independent experiments. E. Anti-oxidant Tempol prevents SERCA from oxidation in MAEC from AMPKα2−/− mice. ♣p<0.05 vs. WT; #p<0.05 vs. AMPKα2−/−; n=4. F. The antioxidant Tempol normalizes SERCA activity in MAEC from AMPKα2−/− mice. MAEC from WT and AMPKα2−/−mice were treated with Tempol (10 μM) for 16 h. SERCA activity was monitored as described in the Materials and Methods. ♣p<0.05 vs. WT; #p<0.05 vs. AMPKα2−/−; n=4. G. Tempol supplementation reduces ER stress in MAEC from AMPKα2−/− mice. ER stress markers were monitored in MAEC from WT or AMPKα2−/− treated with Tempol (10 μM) for 16 h. The blot is representative of four blots obtained from four independent experiments;
Oxidation of Cys674 in SERCA is reported to inhibit SERCA activity,36 so we determined whether inhibition of AMPK increases the oxidation of SERCA at Cys674 using the biotinylated–iodoacetamide (b-IAM) labeling technique.37 MAEC from AMPKα2−/− mice exhibited decreased levels of b-IAM-SERCA and no change in SERCA expression, as compared to MAEC from WT mice. These data suggested that there was increased oxidation of SERCA in MAEC from AMPKα2−/− mice (Figure 6C).
We tested whether this reduction of SERCA activity in AMPKα2−/− mice was due to a reduction in SERCA2 and SERCA3 expression. The levels of SERCA2 and SERCA3 expression were similar in the MAEC isolated from both AMPKα2−/− and WT mice (data not shown). In addition, AMPK inhibition either pharmacologically (Compound C) or genetically (AMPK siRNA or AMPK depletion) did not affect the expression of SERCA2/SERCA3 in endothelial cells (data not shown).
Adenoviral overexpression of a constitutively active AMPK reduces ER stress in mouse aortic endothelial cells isolated from AMPKα2−/− mice
To establish a causative role for AMPK in SERCA inhibition, it was important to demonstrate whether overexpression of Ad-AMPK-CA could restore SERCA activity. Overexpression of Ad-AMPK-CA, but not Ad-GFP, significantly increased SERCA activity in MAEC from AMPKα2−/− mice (Figure 6B). MAEC from AMPKα2−/− mice, but not MAEC from WT mice, exhibited hypersensitivity to calcimycin (Figure 6D), suggesting that the Ca2+ pump was abnormal in the MAEC from AMPKα2−/− mice.
AMPK inhibition increases the oxidation of SERCA2 in endothelial cells
Since AMPKα2 deletion suppresses SERCA activity without altering SERCA protein expression, we determined whether AMPKα2 deletion suppressed SERCA activity by increasing its oxidation status. The number of reactive thiols (b-IAM) in SERCA2 was significantly reduced (~47%) in MAEC from AMPKα2−/− mice, as compared to MAEC from WT mice (Figure 6E). Furthermore, tempol, a potent anti-oxidant, significantly increased the amount of b-IAM-SERCA and SERCA activity in MAEC from AMPKα2−/− mice (Figure 6E–F).
Anti-oxidants normalize SERCA activity and ER stress in endothelial cells
We assayed the effects of anti-oxidant on SERCA oxidation and ER stress in MAEC from AMPKα2−/− mice. MAEC from WT and AMPKα2−/− mice were exposed to tempol (10 μM) for 24 h. Tempol significantly increased SERCA activity (Figure 6E) but reduced the expression of ER stress markers (p-PERK, p-elf2α, and ATF6) (Figure 6G).
Deletion of AMPKα2 promotes the development of aortic lesions in LDLr−/− mice
To investigate the contribution of AMPKα2 to atherogenesis, we compared the aortic lesion size in double knockout LDLr−/−/AMPKα2−/− mice receiving an 8-week atherogenic diet with age- and gender-matched LDLr−/− and C57BL6 WT mice receiving the same diet. Atherosclerotic plaques in the aortic root were visualized using Oil Red O staining. The area of aortic plaques in the aortic root was significantly greater in the LDLr−/−/AMPKα2−/− mice, as compared to the LDLr−/− mice (Figure 7A).
Figure 7. Increased atherosclerosis in the aortic roots of LDLr−/−/AMPKα2−/− mice.


A. Atherosclerotic lesion size in the aortic root in LDLr−/− and LDLr−/−/AMPKα2−/− mice, as determined by Oil Red O staining. The lesion area is expressed as a percentage of the total analyzed area of the aortic root. ♣P<0.05 vs. LDLr−/−, n=5 or 6 for each group. B. Histological features of an atherosclerotic lesion in the aortic arch of male LDLr−/− and LDL−/−/AMPKα2−/− mice. Cross sections of the aortic arch from LDLr−/− and LDLr−/−/AMPKα2−/− mice were stained with hematoxylin and eosin (upper panel), or with an antibody against α-smooth muscle actin (bottom). C. Immunohistochemical staining for CD68 (upper) and F4/80 (low) in the aortic roots. D. Immunohistochemical staining with an antibody against 3-NT in the aortic roots. ♣P<0.05 vs. LDLr−/−, n=5 or 6 for each group. E. Immunohistochemical staining with an antibody against 3-NT in the aortic arches. F. Immunohistochemical staining for malondialdehyde (MDA) and HNE. G. Immunohistochemical staining for ER stress markers (ATF6, KDEL and XBP-1) in the aortic roots of LDLr−/− and LDLr−/−/AMPKα2−/− mice. H. Immunohistochemical staining of ER stress markers (ATF6, KDEL and XBP-1) in the non-atherosclerotic aortic arches of LDLr−/− and LDLr−/−/AMPKα2−/− mice.
Histological characterization of aortic lesions in LDLr−/− and LDL−/−/AMPKα2−/− mice
To further characterize the histological features of aortic lesions, we performed hematoxylin and eosin (H&E) and immunohistochemical staining using antibodies against macrophages (CD68 and F4/80) and vascular smooth muscle cells (α-actin) in the aortic roots and arches. In aortic roots, the advanced lesions were greater in LDLR−/−/AMPKα2−/− mice than those in LDLR−/− mice (Figure 7B, upper panel). However, the α-smooth muscle actin staining in the aortic roots was similar in the LDLr−/−/AMPKα2−/− and LDLr−/− mice (Figure 7B, low panel). CD68 and F4/80 positive staining area in the aortic roots was more extensive in LDLr−/−/AMPKα2−/− mice (Figure 7C), likely due to the larger lesion areas seen in those mice. The collagen content (stained with Masson’s trichrome) did not differ between the mice (data not shown).
Deletion of AMPKα2 increases aortic inflammatory responses and oxidative stress
We investigated whether the enhancement of aortic lesions induced by AMPKα2 deletion was associated with an increase in inflammatory responses and/or the production of ROS, such as superoxide anions (O2. −) or peroxynitrite (ONOO−). The aortic O2. − levels, as measured by dihydroethidium (DHE) fluorescence, were also significantly higher in LDLr−/−/AMPKα2−/− mice, as compared to LDLr−/− mice (data not shown). Furthermore, the aortic levels of 3-nitrotyrosine (3-NT), a stable marker of reactive nitrogen species, were greater in the aortic roots of LDLr−/−/AMPKα2−/− mice, as compared to their LDLr−/− counterparts (Figure 7D). The levels of 3-NT-positive proteins were also markedly greater in non-atherosclerotic aortic arches of LDLr−/−/AMPKα2−/− mice, as compared to those in the LDLr−/− mice (Figure 7E). Immunostaining for malondialdehyde and 4-hydroxy-nonenal (HNE), which are two products of lipid peroxidation, was markedly increased in LDLr−/−/AMPKα2−/− mice, as compared to LDLr−/− mice (Figure 7F). Taken together, these results suggest that deletion of AMPKα2 increases oxidative stress.
Deletion of AMPKα2 increases ER stress
As the markers of ER stress were found to be elevated in human atherosclerotic plaques and AMPKα2 deletion increased the number of aortic lesions, we assessed the levels of markers of ER stress [ATF6, KDEL (GRP78/94), and XBP-1] in aortas from both LDLr−/− and LDLr−/−/AMPKα2−/− mice. The levels of ATF6, KDEL (GRP78/94), and XBP-1, which are well characterized ER stress markers, were markedly elevated in the aortic roots of LDLr−/−/AMPKα2−/− mice, as compared to those in LDLr−/− mice (Figure 7G). Notably, there was intense staining mainly observed in the endothelial cells of the aortic arches from both LDLr−/−/AMPKα2−/− and LDLr−/− mice (Figure 7H). Importantly, the level of staining for ATF6, KDEL (GRP78/94), and XBP-1 in the non-atherosclerotic aortic arches of LDLr−/−/AMPKα2−/− mice was markedly greater than that observed in their LDLr−/− counterparts (Figure 7H). These data suggest that deletion of AMPKα2 promotes ER stress in endothelial cells in vivo.
Chronic administration of TUDCA inhibits ER stress in vivo
Tauroursodeoxycholic acid (TUDCA) is a potent chemical chaperone that inhibits ER stress38, therefore we determined whether chronic administration of TUDCA suppressed ER stress in vivo. ER stress markers were monitored using western blotting. The ER stress response was significantly higher in LDLr−/−/AMPKα2−/− mice, as compared to LDLr−/− mice (Figure 8A and 8B). TUDCA markedly attenuated the ER stress response in both groups of mice (Figure 8A and 8B), indicating that TUDCA could effectively suppress ER stress in vivo.
Figure 8. Chronic administration of TUDCA reduces ER stress and aortic lesions in mice deficient for both LDLr−/− and AMPKα2.

LDLr−/− and LDLr−/−/AMPKα2−/− mice were fed a high fat diet for 10 weeks, with or without TUDCA treatment. Each group contained six to eight mice. A&B. Chronic administration of TUDCA attenuates ER stress in vivo. ♣p<0.05 vs. LDLr−/−; #p<0.05 vs. LDLr−/−/AMPKα2−/−; C&D. Chronic administration of TUDCA suppresses aortic lesions in LDLr−/−and LDLr/AMPKα2−/− mice. ♣p<0.05 LDLr−/−/AMPKα2−/− vs. LDLr−/−; #p<0.05 LDLr−/− vs. LDLr−/−+TUD; LDLr−/−/AMPKα2−/− vs LDLr−/−/AMPKα2−/−+TUD, respectively; n=6–8. E. Comparison of the effects of TUDCA in LDLr−/− and LDLr−/−/AMPKα2−/− mice, ♣p<0.05 vs. LDLr−/−, n=6–8.
TUDCA suppresses high fat diet-enhanced atherosclerosis in vivo
An earlier study 39 showed an increased ER stress response in human atherosclerotic lesions, although this study did not establish a causal role for ER stress in the development of atherosclerosis. We determined whether chemical inhibition of ER stress using TUDCA altered the development of aortic lesions in LDLr−/−/AMPKα2−/− and LDLr−/− mice. TUDCA markedly reduced the development of aortic lesions in LDLr−/− and LDLr−/−/AMPKα2−/− mice (Figure 8C and 8D). The TUDCA-induced reduction in the area of aortic lesions in LDLr−/−/AMPKα2−/− mice was significantly greater than that observed in LDLr−/− mice (25% vs. 38%, n=6–8, p=0.003, Figure 8E).
The effects of TUDCA are independent of serum lipid or blood glucose levels
We compared the metabolic parameters between WT, LDLr−/−, and LDLr−/−/AMPKα2−/−mice. There were no differences observed between LDLr−/−/AMPKα2−/− and LDLr−/− mice (Table 1). In addition, the body weights of LDLr−/−/AMPKα2−/− and LDLr−/− mice were similar at the completion of the 8-week atherogenic diet, with neither group exhibiting an increase in their body weight (data not shown). Furthermore, TUDCA had no effect on cholesterol, triglyceride, and glucose serum levels in either LDLr−/− or LDLr−/−/AMPKα2−/− mice (Table 1). Taken together, these results suggest that loss of AMPKα2 expression promoted ER stress and its associated atherosclerosis, and the protective effects of TUDCA against aortic atherosclerosis are most likely due to its suppressive effects on ER stress in vivo.
Table 1.
Effects of tauroursodeoxycholic acid on blood glucose and lipid profiles in LDLr−/− and LDLr−/−/AMPKα2−/− mice
| LDLr−/− | LDLr−/−+TUD | LDLr−/−/AMPKα2−/− | LDLr−/−/AMPKα2−/−+TUD | |
|---|---|---|---|---|
| Cholesterol (mmol/L) | 43.1±0.8 | 42.6±1.4 | 41.3±3.9 | 41.3±1.3 |
| Triglyceride (mmol/L) | 3.6±0.1 | 3.8±0.1 | 3.7±0.5 | 3.9±0.3 |
| Glucose (mg/dL) | 170±16 | 172±11 | 172±9 | 171±4 |
Non-fasting; n=6–8, no statistic difference among groups.
Discussion
This study provides the first evidence for AMPK as an important regulator of intracellular Ca2+ levels and ER homeostasis in endothelial cells, and these functions are mediated through its suppression of SERCA oxidation. Pharmacological or genetic inhibition of AMPK in cultured endothelial cells increased the levels of ER stress and intracellular Ca2+, and concomitantly reduced SERCA activity. Overexpression of SERCA2b attenuated the ER stress caused by AMPKα2 inhibition. Aortas isolated from mice deficient for both LDLr and AMPKα2 exhibited increased levels of ER stress markers and aortic lesion development, as compared to LDLr−/−mice. Importantly, the chronic administration of TUDCA significantly reduced the expression of ER stress markers and attenuated the development of aortic lesions in LDLr−/−/AMPKα2−/− mice. These results imply that AMPK might confer its protective effects against atherosclerosis by inhibiting the ER stress response in endothelial cells.
The most important finding is that loss of AMPKα2 expression increases ER stress, which accelerates atherosclerosis in vivo. Aortas from LDLr−/−/AMPKα2−/− mice exhibited greater aortic lesion development and ER stress, as compared to LDLr−/− mice. Administration of TUDCA, a chemical chaperone with clinical potential,38 significantly suppressed both the development of aortic lesions and ER stress in LDLr−/−/AMPKα2−/− mice. Thus, these results indicate that AMPK might be important for maintaining ER homeostasis and that aberrant ER stress might contribute to the initiation and progression of atherosclerosis. This finding is consistent with a previous study that reported on the benefits of using chemical chaperones to treat obesity-induced type 2 diabetes.38
An additional important finding is that SERCA oxidation mediates an increase in ER stress when AMPK is inhibited. We determined that the activity of SERCA is reduced and its function is compromised in AMPKα2 deficient endothelial cells. In addition, inhibition of AMPK suppressed SERCA activity and impaired its function. Importantly, adenoviral overexpression of AMPK-CA restored SERCA activity without altering the expression of SERCA2/SERCA3. Furthermore, we found that SERCA is oxidized and inhibited when AMPK activity is reduced. In MAEC isolated from AMPKα2 KO mice, ~ 50% of total SERCA2 is oxidized. Similarly, similar degree of SERCA inhibition by SERCA-specific siRNA effectively increased the detection of ER stress markers in MAEC, suggesting that a reduction of SERCA in AMPKα2 KO mice can lead to an imbalance in intracellular Ca2+ levels and induced ER stress. Finally, overexpression of SERCA2b in AMPKα2−/− endothelial cells resulted in the maintenance of intracellular Ca2+ homeostasis with a concomitant reduction in ER stress. Based on these data, we conclude that aberrant ER stress in AMPKα2−/− mice might be related to an increase in SERCA oxidation. Indeed, there is evidence that SERCA is modified by several oxidants, including NO, H2O2, and ONOO−, under certain physiological and pathological conditions, including diabetes and aging.40–43 Several thiol residues in SERCA are potential targets for oxidation.36, 42, 44, 45 In AMPKα−/−endothelial cells, SERCA activity was significantly reduced. It has been previously reported 36 that nitric oxide (NO) physiologically stimulates SERCA through S-glutathiolation to decrease intracellular Ca2+ levels and relax cardiac, skeletal, and vascular smooth muscle. This modification of SERCA is blocked by irreversible oxidation of the relevant cysteine thiols during atherosclerosis. Oxidation at Cys674 is the most important thiol residue required for SERCA activity.36 We have recently reported that AMPK regulates endogenous NO levels and found that NO production in AMPKα2−/− endothelial cells is reduced by 44.5% relative to WT cells (p<0.05), and A23187-stimulated NO production is reduced by more than two-fold.46 Since treatment with an anti-oxidant, Tempol, protected SERCA activity without affecting its expression, the reduction of SERCA activity is most likely due to oxidative modification. Taken together, we conclude that AMPK-mediated suppression of ER stress is likely to occur via its inhibition of oxidants-mediated SERCA oxidation.
How AMPK suppresses SERCA oxidation and oxidative stress warrants further investigation. There is overwhelming evidence suggesting that AMPK might suppress oxidative stress. For example, a study from Ido et al. 47 showed that incubation with the AMPK activator, AICAR, completely prevents hyperglycemia-driven oxidative stress and apoptosis, suggesting that AMPK plays an important role in protecting endothelial cells against the adverse effects of sustained hyperglycemia. Ouslimani et al. 48 reported that metformin, another AMPK activator, decreases ROS production in aortic endothelial cells, which is partially due to decreased ROS derived from the mitochondrial respiratory chain. Kukidome et al. provided further direct evidence that activation of AMPK reduces hyperglycemia-induced mitochondrial ROS production;49 they showed that induction of manganese superoxide dismutase (MnSOD) and promotion of mitochondrial biogenesis occurs through the activation of the AMPK-PGC1α pathway in HUVEC. In a previous paper 50, we showed that AMPK activation prevents diabetes-induced PGIS nitration. We also determined that AMPK activation suppresses the expression of the NAD(P)H oxidase subunits (p47phox, p67phox, p91phox and NOX-4) (Wang et al., unpublished data). Thus, AMPK activation appears to not only increase the expression of antioxidant enzymes such as MnSOD and uncoupling proteins but also inhibits the expression of ROS-producing enzymes, such as NA(D)PH oxidase.
In summary, the data in this study has established a causal link between ER stress and atherosclerosis. In addition, our data tentatively suggest that AMPKα2 might play a more important role than AMPKα1, which is a predominant isoform in endothelial cells. However, this dominant role of AMPKα2 may be limited to cultured endothelial cells, as we only used this model system for these studies. Whether or not AMPKα1 deletion increases ER stress and atherosclerosis remains to be established because the knockout of AMPKα1 causes splenomegaly and severe anemia in AMPKα1−/− and LDLr−/−/AMPKα1−/− mice (Zhang, et al., unpublished data), which may itself increase the ER stress response and associated atherosclerosis by inducing anemia-associated inflammation and low oxygen tension.
Clinical Perspective.
The endoplasmic reticulum (ER) is an organelle that has an essential role in multiple cellular processes, such as the folding of secretory and membrane proteins, calcium homeostasis, and lipid biosynthesis. A variety of insults can interfere with ER function, leading to the accumulation of unfolded and misfolded proteins in the ER. When ER transmembrane sensors detect the accumulation of unfolded proteins, the unfolded protein response (UPR) is initiated to cope with the resulting ER stress. If ER stress is prolonged or overwhelming, however, it can induce cell death. Recent studies have suggested that the UPR and ER-initiated apoptosis play a crucial role in both atherosclerosis and plaque rupture. Aberrant ER stress in atherosclerotic plaques could lead to the death of macrophages and smooth muscle cells, which would contribute to plaque instability. In the present study, we report that chronic inhibition of ER stress suppressed aortic lesions in mice deficient of low density lipoprotein receptor (LDLr−/−). Further, we have for the first time established a causal link between aberrant ER stress and AMPK dysfunction in vivo. Overall, our results indicate that aberrant ER stress and dysfunctional AMPK might contribute to the initiation and progression of atherosclerosis. The study of AMPK, ER stress, and atherosclerosis takes on added importance since metformin was recently shown to exert its therapeutic effect in diabetes by activating AMPK and most importantly, metformin has been shown to improve vascular function and to dramatically reduce cardiovascular endpoints and mortality in type II diabetic patients in large scale clinical trials.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported by funding from the following agencies: NIH RO1 (HL074399, HL079584, HL080499, HL08920, and HL096032), the Juvenile Diabetes Research Foundation (JDRF), Oklahoma Center for the Advancement of Science and Technology, the American Diabetes Association, and the Travis Endowed Chair of the University of Oklahoma Health Science Center (all to M.H. Zou). Dr. M.H. Zou is a recipient of the National Established Investigator Award of the American Heart Association.
- AICAR
aminoimidazole carboxamide ribonucleotide
- AMPK
5′-AMP activated-kinase
- Ad-AMPK-CA
adenovirus encoding constitutively active AMPK
- Ad-AMPK-DN
adenovirus encoding dominant negative AMPK
- DHE
dihydroethidine
- Ad-GFP
adenovirus encoding green fluorescent protein
- HUVEC
human umbilical vein endothelial cells
- ONOO−
peroxynitrite
- ER
endoplasmic reticulum
- HNE
4-hydroxy-nonenal
- SERCA
Sarco/(endo)plasmic reticulum Ca2+ ATPase
- LDLr
low density lipoprotein receptor
- ROS
reactive oxygen species
- TUDCA
tauroursodeoxycholic acid
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
Footnotes
Conflict of Interest Disclosures
None
References
- 1.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 4.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 5.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 7.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2008 doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowes AJ, Khan MI, Shi Y, Robertson L, Werstuck GH. Valproate attenuates accelerated atherosclerosis in hyperglycemic apoE-deficient mice: evidence in support of a role for endoplasmic reticulum stress and glycogen synthase kinase-3 in lesion development and hepatic steatosis. Am J Pathol. 2009;174:330–342. doi: 10.2353/ajpath.2009.080385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werstuck GH, Khan MI, Femia G, Kim AJ, Tedesco V, Trigatti B, Shi Y. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes. 2006;55:93–101. [PubMed] [Google Scholar]
- 10.Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, Austin RC. Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: implications in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:2623–2629. doi: 10.1161/01.ATV.0000189159.96900.d9. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Werstuck GH, Lhotak S, de Koning AB, Sood SK, Hossain GS, Moller J, Ritskes-Hoitinga M, Falk E, Dayal S, Lentz SR, Austin RC. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–213. doi: 10.1161/01.CIR.0000134487.51510.97. [DOI] [PubMed] [Google Scholar]
- 12.Feaver RE, Hastings NE, Pryor A, Blackman BR. GRP78 upregulation by atheroprone shear stress via p38-, alpha2beta1-dependent mechanism in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1534–1541. doi: 10.1161/ATVBAHA.108.167999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009 doi: 10.1172/JCI37262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanson M, Auge N, Vindis C, Muller C, Bando Y, Thiers JC, Marachet MA, Zarkovic K, Sawa Y, Salvayre R, Negre-Salvayre A. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ Res. 2009;104:328–336. doi: 10.1161/CIRCRESAHA.108.183749. [DOI] [PubMed] [Google Scholar]
- 15.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 17.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou MH, Hou XY, Shi CM, Nagata D, Walsh K, Cohen RA. Modulation by peroxynitrite of Akt-and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem. 2002;277:32552–32557. doi: 10.1074/jbc.M204512200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song P, Guzman M, Wu J, Zou MH. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102:328–337. doi: 10.1161/CIRCRESAHA.107.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 21.Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WGt, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 22.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 23.Zou MH, Wu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin Exp Pharmacol Physiol. 2008;35:535–545. doi: 10.1111/j.1440-1681.2007.04851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frederich M, Zhang L, Balschi JA. Hypoxia and AMP independently regulate AMP-activated protein kinase activity in heart. Am J Physiol Heart Circ Physiol. 2005;288:H2412–2421. doi: 10.1152/ajpheart.00558.2004. [DOI] [PubMed] [Google Scholar]
- 27.Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation. 2008;117:832–840. doi: 10.1161/CIRCULATIONAHA.107.713115. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 29.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, Neumann D, Schlattner U, Zou MH. Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Xie Z, Reece R, Pimental D, Zou MH. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol. 2006;26:2688–2695. doi: 10.1161/01.ATV.0000249394.94588.82. [DOI] [PubMed] [Google Scholar]
- 31.Schulz E, Anter E, Zou MH, Keaney JF., Jr Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation. 2005;111:3473–3480. doi: 10.1161/CIRCULATIONAHA.105.546812. [DOI] [PubMed] [Google Scholar]
- 32.Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 33.Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, Pinton P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium. 2008;43:184–195. doi: 10.1016/j.ceca.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 35.Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 36.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 37.Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008;44:361–369. doi: 10.1016/j.yjmcc.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 40.Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, Lazar H, Menzoian J, Knyushko TV, Bigelow D, Schoneich C, Cohen RA. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol. 2006;290:H2220–2227. doi: 10.1152/ajpheart.01293.2005. [DOI] [PubMed] [Google Scholar]
- 41.Schertzer JD, Plant DR, Ryall JG, Beitzel F, Stupka N, Lynch GS. Beta2-agonist administration increases sarcoplasmic reticulum Ca2+-ATPase activity in aged rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E526–533. doi: 10.1152/ajpendo.00399.2004. [DOI] [PubMed] [Google Scholar]
- 42.Gutierrez-Martin Y, Martin-Romero FJ, Inesta-Vaquera FA, Gutierrez-Merino C, Henao F. Modulation of sarcoplasmic reticulum Ca(2+)-ATPase by chronic and acute exposure to peroxynitrite. Eur J Biochem. 2004;271:2647–2657. doi: 10.1111/j.1432-1033.2004.04193.x. [DOI] [PubMed] [Google Scholar]
- 43.Adachi T, Matsui R, Xu S, Kirber M, Lazar HL, Sharov VS, Schoneich C, Cohen RA. Antioxidant improves smooth muscle sarco/endoplasmic reticulum Ca(2+)-ATPase function and lowers tyrosine nitration in hypercholesterolemia and improves nitric oxide-induced relaxation. Circ Res. 2002;90:1114–1121. doi: 10.1161/01.res.0000019757.57344.d5. [DOI] [PubMed] [Google Scholar]
- 44.Cohen RA, Adachi T. Nitric-oxide-induced vasodilatation: regulation by physiologic s-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc Med. 2006;16:109–114. doi: 10.1016/j.tcm.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Dremina ES, Sharov VS, Davies MJ, Schoneich C. Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem Res Toxicol. 2007;20:1462–1469. doi: 10.1021/tx700108w. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem. 2008;283:27452–27461. doi: 10.1074/jbc.M802578200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–167. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]
- 48.Ouslimani N, Peynet J, Bonnefont-Rousselot D, Therond P, Legrand A, Beaudeux JL. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism. 2005;54:829–834. doi: 10.1016/j.metabol.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 49.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–127. [PubMed] [Google Scholar]
- 50.Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. 2008;57:3222–3230. doi: 10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.