

Abstract
Macrophages have a central role in the pathogenesis of cryptococcosis since they are an important line of defense, serve as a site for fungal replication, and also can contribute to tissue damage. The objective of this study was to investigate the interaction of macrophages with cells from smooth-colony variants (SM) and mucoid-colony variants (MC) arising from phenotypic switching of Cryptococcus neoformans. Alveolar macrophages (AMs) isolated from SM- and MC-infected mice exhibited differences in gene and surface expression of PD-L1, PD-L2, and major histocompatibility class II (MHC-II). PD-L1 and PD-L2 are the ligands for PD1 and are differentially regulated in Th1- and Th2-type cells. In addition, macrophage activation in SM- and MC-infected mice was characterized as alternatively activated. Flow cytometric and cytokine analysis demonstrated that MC infection was associated with the emergence of Th17 cells and higher levels of interleukin-17 (IL-17) in lung tissue, which were reduced by AM depletion. In conclusion, our results indicate that macrophages play a significant role in maintaining damage-promoting inflammation in the lung during MC infection, which ultimately results in death.
Cryptococcosis is an infection that occurs commonly in AIDS patients, as well as other immunocompromised patients, and is caused by Cryptococcus neoformans. Most affected patients present with subacute or chronic meningoencephalitis. C. neoformans is an encapsulated yeast that can undergo phenotypic switching in vivo, which affects the outcome of chronic cryptococcosis (15, 17). The model C. neoformans strain RC-2 switches from a smooth-parental-colony (SM) morphology to a hypervirulent mucoid-colony (MC) morphology. Phenotypic switching involves the emergence of morphological colony variants whose cells differ in the cell wall and polysaccharide capsule, including the biochemical composition of the major capsular polysaccharide, glucuronoxylomannan (GXM) (14, 15). Recent work in our laboratory demonstrated that the murine immune responses to infection with MC and SM cells are qualitatively different. Enhanced recruitment of CD8 cells, NK cells, and macrophages and an early polarized Th1-type cell response were observed in the lungs of MC-infected mice and were associated with altered cytokine production. The finding that interleukin-10 (IL-10) affected the survival of SM-infected mice but not MC-infected mice (20) further supported the conclusion that phenotypic switching alters cryptococcal virulence by changing the host-pathogen interaction in a way that is manifested through different immune responses. Histological analysis of MC-infected lungs demonstrated that there was enhanced macrophage recruitment and suggested that this recruitment resulted in damage to the alveolar tissue and decreased survival.
The majority of individuals with chronic cryptococcosis have defects in cellular immunity (9, 11, 35, 46). The high incidence of cryptococcosis only in HIV-infected patients with low CD4 counts underscores the importance of T-cell-based defenses. Furthermore, the crucial role of T cells in host defense has been supported by findings with animal models (22, 32). Alveolar macrophages (AMs) also are important effector cells against C. neoformans (45). They are the primary phagocytic cells, and together with dendritic cells they facilitate antigen presentation (31, 39, 47, 50). In addition, C. neoformans is a facultative intracellular pathogen that can reside in a macrophage (12); hence, this type of cell is also a niche to which the pathogen adapts.
The interaction of macrophages with a pathogen such as C. neoformans leads to activation, which can be classified as either classical or alternative activation (16, 18). Macrophages infected with C. neoformans are alternatively activated, but the role of macrophage activation during infection is unknown (3, 4, 37, 38). In African trypanosomiasis macrophages are activated classically early and alternatively late in infection, which leads to progression of the disease (4, 38). Given that there is a marked difference in virulence between the SM and MC variants and that persistent MC infection is associated with enhanced macrophage recruitment, the objective of this study was to further explore macrophage activation and function by examining infection with phenotypic switch variants.
MATERIALS AND METHODS
C. neoformans strain.
RC-2 is a variant of serotype D strain 24067, which was originally obtained from the American Tissue Type Collection (Rockville, MD). The RC-2 strain was streaked to obtain single colonies and maintained on Sabouraud dextrose agar (SDA) plates. The RC-2 strain can produce two types of colonies on agar, smooth (SM) and mucoid (MC), both of which are characteristic of C. neoformans (13, 15).
Animal studies.
BALB/c and BALB/c/SCID mice that were 6 to 12 weeks old were obtained from the National Cancer Institute (Bethesda, MD), and C57BL/6J mice that were 6 to 8 weeks old were obtained from Jackson Laboratory (Bar Harbor, ME). TgE26 breeder mice were a generous gift from C. Terhorst (Harvard Institutes of Medicine). The two switch variants were streaked onto SDA plates, and single colonies were selected and grown in broth overnight, diluted 1:50, and grown again overnight. Dilutions of each infecting suspension were plated onto SDA plates to ensure that comparable numbers of yeast cells were injected. Anesthetized mice were infected by intratracheal (i.t.) inoculation of 106 (high dose) or 1 × 104 to 5 × 104 (low dose) C. neoformans cells in 50 μl sterile nonpyrogenic phosphate-buffered saline (PBS) using a 26-gauge needle as described previously (20). Mice were observed daily for signs of disease. Mice that were moribund and unable to reach water were killed in accordance with institutional regulations. Mice were killed, and the organ fungal burden was determined by homogenizing lung tissue in 10 ml PBS and plating 100-μl portions of different dilutions of the homogenate on SDA (Difco Laboratories, Detroit, MI). Colonies were counted after 72 to 96 h (one colony was defined as 1 CFU). Experiments were done with 5 to 10 mice per group and repeated at least once.
AM depletion.
AMs were depleted using dichloromethylene diphosphonate (Cl2MDP) encapsulated in liposomes (Cl2MDP-liposomes) as described previously (45). Cl2MDP-liposomes and PBS-liposomes were prepared as described previously (49) and were a gift from Roche Diagnostics GmbH, Mannheim, Germany. At time zero, mice were given 60 μl of Cl2MDP-liposomes or PBS-liposomes intranasally. Two days later mice were infected i.t. with SM and MC cells as described above. To ensure ongoing depletion of AMs, Cl2MDP-liposomes or PBS-liposomes were injected intranasally weekly until the time of death. AM depletion was confirmed by flow cytometric analysis of isolated lung leukocytes with CD11b (clone M1/70; eBioscience, San Diego CA), as well as by immunohistochemical analysis of lung tissue with macrophage-specific antibody (Ab) Mac-3 (Pharmingen).
Lung leukocyte isolation and flow cytometric analysis of leukocyte subsets.
Single-cell suspensions from infected lungs were obtained by mincing excised lungs of individual C57BL/6J mice, as previously described (21). Then the lung tissues were enzymatically digested for 30 min in 15 ml of digestion buffer (RPMI with 5% fetal calf serum [FCS], antibiotics, collagenase, and DNase). After a series of washes and further dispersion of undigested lung components, erythrocytes were lysed with red blood cell lysing buffer (Sigma-Aldrich Company). The total cell suspension was collected by centrifugation, and the cells were counted with a hemocytometer. Antibodies against PD-L1 (clone MIH5), PD-L2 (clone TY25), major histocompatibility class II (MHC-II) (clone M5/114.15.2), F4/80 (clone BM8), CD4 (clone GK1.5), and interleukin-17A (IL-17A) (clone eBio17B7) were all purchased from eBioscience (San Diego, CA). The percentage of PD-L1-, PD-L2-, and MHC-II-positive (F4/80-positive) macrophages was determined by flow cytometry. F4/80-positive macrophages were first gated equally between the SM and MC isolated leukocyte suspensions. Cells were blocked with Fc blocker for 5 min, and then they were stained with a specific Ab (1 μg/106 cells) for 45 min at 4°C. To determine the percentages of IL-17-producing CD4+ T cells, an intracellular staining protocol using CD4 and IL-17A antibodies was followed as recommended by the manufacturer. Flow cytometric analysis results were analyzed using Flojow software, version 8.5.
Gene expression of macrophage RNA of SM- and MC-infected mice.
Macrophages were isolated from suspensions of leukocytes from infected mouse lungs (right lungs) by using adherence on cell culture plates. Leukocytes were pelleted and resuspended in RPMI with 10% fetal calf serum (FCS) and then incubated in a CO2 incubator for 2 h. The purity of cell layers was verified by staining them with Giemsa stain and with a fluorescein isothiocyanate (FITC)-conjugated MAC3 monoclonal antibody (MAb) and 4′,6′-diamidino-2-phenylindole (DAPI). As shown in Fig. S1 in the supplemental material, the majority of cells were macrophages. Each plate was washed, and macrophages were scrapped off with ice-cold PBS. Cells were mechanically disrupted in RNeasy lysis buffer (RLT buffer; Qiagen) with 0.5-mm zirconia-silica beads (1:1, vol/vol; BioSpec Products, Inc.) in a bead mill homogenizer. RNA was isolated using an RNeasy mini kit (Qiagen) according to the manufacturer's instructions. For real-time PCR (RT-PCR) analysis macrophage RNA was prepared by using an RNeasy miniprep kit (Qiagen). Prior to cDNA synthesis with the SuperScript II enzyme system (Invitrogen), contaminating genomic DNA was removed from RNA by using a message clean DNase kit (Genehunter). Gene expression compared to actin gene expression was calculated using ΔCt, as follows: 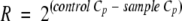 and then
and then 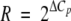 . Both actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as controls. The primers used in these experiments are listed in Table 1.
. Both actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as controls. The primers used in these experiments are listed in Table 1.
TABLE 1.
Primers used for RT-PCR
| Primer | Sequence |
|---|---|
| B7-H1 (PD-L1) Forward | CGTGAGTGGGAAGAGAAGTG |
| B7-H1 (PD-L1) Reverse | ACATCATTCGCTGTGGCGTT |
| B7-DC (PD-L2) Forward | GCTTTATTCACCGTGACAGC |
| B7-DC (PD-L2) Reverse | TTCAGTGCATTCTCTGCGGTCA |
| YM1 Forward | GCCCTCCTAAGGACAAACAT |
| YM1 Reverse | CCTTGGAATGTCTTTCTCCA |
| MHC II H2-Ab Forward | GTTCATGGGCGAGTGCTAC |
| MHC II H2-Ab Reverse | ACTCCTCCCGGTTGTAGATG |
| iNOS Forward | GAAACGCTTCACTTCCAATG |
| iNOS Reverse | GCTCTGTTGAGGTCTAAAGGTCCG |
| Actin Forward | TGGAATCCTGTGGCATCCATGAAAC |
| Actin Reverse | CACTGTGTTGGCATAGAGGTC |
| GAPDH Forward | AGATTGTTGCCATCAACGAC |
| GAPDH Reverse | TTGACTGTGCCGTTGAATTT |
| Arginase Forward | CCTGTGTCCTTTCTCCTGAA |
| Arginase Reverse | CAGATATGCAGGGAGTCACC |
Measurement of cytokine levels in tissue and arginase activity in AMs.
Five mice were killed on days 7 and 14, and each left lung was homogenized in 2 ml of PBS in the presence of protease inhibitors (Complete Mini; Boehringer Mannheim, Indianapolis, IN). The homogenate was centrifuged at 6,000 × g for 10 min to remove cell debris, and the supernatant was frozen at −80°C until it was tested. The supernatants of the lung homogenates were assayed to determine IL-17 IL-6, IL-10, IL-12, IL-2, IL-4, tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein (MCP), and MIP-1α concentrations using enzyme-linked immunosorbent assay (ELISA) kits (R & D Systems Inc., Minneapolis, MN, or BD Biosciences). AMs were isolated from SM- and-MC infected mice by bronchoalveolar lavage (BAL) as described previously (15). AMs were then plated in 96-well plates and incubated overnight, and each supernatant was collected for quantification of cytokines and arginase activity. Arginase activity was determined as described previously (10, 38).
Lymphocyte T-cell proliferation assay.
AMs were isolated from SM- and MC-infected C57BL/6J mice by BAL with Hanks' balanced salt solution (Mediatech, Inc.). AMs (3 × 104 cells) were adhered to 96-well round-bottom plates with or without 3 × 105 heat-inactivated SM and MC cells for 2 h at 37°C in the presence of 5% CO2 in RPMI with 10% FCS. The layer of cells was washed to remove nonadherent fungal cells. Approximately 2 × 105 T cells (isolated from noninfected mice) in RMPI with 10% FCS were added to the mixture of AMs and cells. The cells were coincubated for 48 h, and T-cell proliferation was measured using a ViaLight high-sensitivity cell proliferation/cytotoxicity kit according to the manufacturer's instructions (Lonza Rockland, Inc.). T cells were isolated from the spleens of uninfected wild-type C57BL/6J mice using an EasySep mouse T-cell enrichment kit (Stemcell Technologies). Each sample was examined in quadruplicate, and as a positive control AMs were stimulated with concanavalin A (ConA). In some experiments antigens (Ags) were switched, and AMs isolated from SM-infected mice were coincubated with heat-inactivated MC cells and visa versa.
Immunohistochemistry.
Formalin-fixed, paraffin-embedded lung sections were used for histological analysis. These sections were stained with standard hematoxylin and eosin (H&E). Macrophages were identified by staining them with a macrophage-specific Mac-3 (PharMingen) MAb as previously described (15). Purified mouse Ab against arginase was purchased from BD Bioscience. Immunostaining of arginase and Mac-3 was performed with an M.O.M immunodetection kit (Vector Laboratories). Negative control experiments were performed without the primary Ab. Immunostaining was developed with a DAB substrate kit (Vector Laboratories). Staining was visualized at a magnification ×100 using a Zeiss microscope linked to a digital camera.
Statistics.
Standard statistical analyses, including a t test, a Mann-Whitney test, a Fisher exact test, a Kaplan-Meyer test, and a log rank regression analysis, were performed using SPSS version 11 for Mac and EXCEL of Microsoft Office for Mac 2004.
RESULTS
Virulence of SM and MC in immunocompromised mice.
To investigate the contribution of macrophages to the differences in the immunological responses to SM and MC, we first examined the virulence of SM and MC in a pulmonary infection model using T-cell-deficient SCID and TgE26 mice. The latter mice lack both NK and T cells and therefore exhibit altered macrophage activation. These experiments demonstrated that the virulence of MC was greater than the virulence of SM in both mouse strains (SCID and TgE26) (Fig. 1). SCID mice infected with SM cells died 32 days earlier than control BALB/c mice, whereas the survival data for SCID and control mice infected with MC cells were comparable (median length of survival, 33 days) (Fig. 1A). Similar differences in survival were observed for TgE26 mice. SM-infected TgE26 mice also died 17 days earlier than control C57/BL mice (median lengths of survival, 34 and 51 days; P = 0.02), whereas the difference in survival was not as great for MC-infected TgE26 mice and C57/BL control mice; TgE26 mice died 3 days earlier than control C57/BL mice (median lengths of survival, 22 and 19 days; P = 0.003) (Fig. 1B). Immunohistological analysis demonstrated that there was abundant recruitment of macrophages into the lung tissue of both MC-infected mouse strains that was associated with enhanced inflammation (Fig. 1C).
FIG. 1.

(A and B) Differences in survival of SM- and MC-infected SCID (A) and TgE26 (B) mice. Mice (5 to 10 mice per group) were infected i.t. as described in Materials and Methods Levels of survival were compared by using a log rank test. (C) Macrophage-specific staining with MAC3 (Mac) of SM- and MC-infected SCID mice (day14; magnification, ×20) and TgE26 mice (day 21; magnification, ×10) demonstrated that there was enhanced recruitment of macrophages in MC-infected SCID and TgE26 lung tissue compared to SM-infected lung tissue. HE, hematoxylin and eosin stain.
Cytokine levels in lung homogenates from SM-, MC-, and sham-infected SCID mice were determined by ELISA at day 14. Significant differences in protein levels between SM- and MC-infected SCID mice were detected only for IL-12, IL-10, MCP, and MIP-1α on day 14 after infection, whereas comparable tissue levels were observed for IL-6, IL-2, IL-4, and TNF-α (Table 2). The MIP-1α and MCP levels were about 3-fold higher in MC-infected mice than in SM-infected mice, whereas for IL-10 and IL-12 the levels of expression were found to be significantly higher in SM-infected mice than in MC-infected SCID mice.
TABLE 2.
Expression of cytokines and chemokines in SM- and MC-infected lungs of SCID mice on day 14
| Concn (pg/ml) ina: |
||||
|---|---|---|---|---|
| Cytokine or chemokine | Sham-inoculated mice | SM-infected SCID mice | MC-infected SCID mice | P valueb |
| IL-6 | ND | 350 ± 75 | 434 ± 108 | 0.02 |
| IL-10 | 88 ± 65 | 522 ± 218 | 278 ± 79 | <0.001 |
| IL-12 | 578 ± 187 | 979 ± 529 | 432 ± 396 | 0.005 |
| IL-2 | 60 ± 10 | ND | ND | NS |
| IL-4 | 46 ± 29 | 95 ± 44 | 81 ± 35 | NS |
| TNF-α | 1,037 ± 37 | 2,672 ± 1,501 | 2,772 ± 1,652 | NS |
| MCP | ND | 41 ± 28 | 110 ± 108 | 0.03 |
| MIP-1α | 29 ± 13 | 40 ± 31 | 150 ± 116 | 0.003 |
The data are medians ± standard deviations for the results of three experiments with five mice in each experiment. ND, not detected.
The P value was determined by comparing the protein expression in SM-infected SCID mice and the protein expression in MC-infected SCID mice using a t test. NS, not significant.
Taken together, these data suggested that activation and recruitment of macrophages are different in SM- and MC-infected mice that lack T cells and that macrophages themselves may contribute to a harmful immune response.
Macrophage activation in SM- and MC-infected mice.
Next, we examined if macrophage activation differed in SM- and MC-infected C57/BL6 mice. In both types of infection macrophage activation was achieved via an alternative pathway, as expected, but differences in the level and quality were noted. At 7 days postinfection, the arginase mRNA levels determined by RT-PCR were significantly higher (P = 0.01) in AMs isolated from MC-infected mice than in AMs isolated from SM-infected mice (Fig. 2A). Immunohistochemical analysis of lung tissue from SM- and MC-infected mice with an arginase-specific Ab confirmed that there were differences in arginase production and alternative macrophage activation (Fig. 2B). In addition, data for in vitro cytokine production by AMs derived from SM- and MC-infected lung tissue at days 7 and 14 demonstrated that there were qualitative differences in SM- and MC-infected macrophages. A trend toward increased levels of IL-6 and MCP was found in AMs from MC-infected mice (Table 3). A significantly higher level of arginase was detected in the supernatant of AMs derived from MC-infected macrophages at day 14 (for MC-infected macrophages, 6.25 ± 1.89 μg/ml; for SM-infected macrophages, 3.64 ± 0.49 μg/ml) (Fig. 2C), whereas the inducible nitric oxide synthase (iNOS) levels were very low. The higher levels of IL-6 and MCP produced by MC-infected macrophages were consistent with previously reported elevated levels of these cytokines in lung tissue (20) and also with the results for SCID mice, as shown in Table 2. In summary, these findings indicate that activation of SM- and MC-infected AMs, although achieved via the alternative pathway, results in qualitative differences in arginase and cytokine production.
FIG. 2.

Activation of AMs isolated from SM- and MC-infected C57BL/6J mice (five mice per group) confirmed that there was alternative activation. (A) RT-PCR demonstrated that at day 7 (d-7) there were significantly higher levels of arginase (ARG) mRNA (A) in AMs derived from MC-infected mice than in AMs isolated from SM-infected mice. The data were obtained by comparison with the abundance of actin. (B and C) Immunohistochemical analysis with arginase-specific Abs of lungs (day 14) (B) and in vitro production of arginase from the supernatant of isolated AMs from SM- and MC-infected mice (C) both confirmed that there was increased arginase production in MC-infected AMs. There was a significant difference between samples obtained from SM-infected mice and samples obtained from MC-infected mice (P < 0.05) as determined by a t test. The error bars indicate standard deviations.
TABLE 3.
Cytokine analysis of SM- and MC-infected AM supernatantsa
| Concn (pg/ml) inb: |
|||
|---|---|---|---|
| No. of days after i.t. infection | Cytokine | SM-infected AM supernatant | MC-infected AM supernatant |
| 7 | IL-6 | 1,677 ± 1,086 | 5,722 ± 4,273 |
| MCP-1 | 1,979 ± 1,411 | 3,284 ± 3,591 | |
| 14 | IL-6 | 1,864 ± 601 | 2,632 ± 2,965 |
| MCP-1 | 1,780 ± 1,219 | 2,498 ± 4,007 | |
In vivo-infected AMs were recovered by BAL.
The data are medians ± standard deviations.
Surface expression and gene expression of PD-L1, PD-L2, and MHC-II markers in SM- and MC-infected AMs.
Next, expression of the macrophage activation-associated surface markers PD-L1, PD-L2, and MHC-II was measured by RT-PCR and flow cytometry using AMs isolated from SM- and MC-infected mice (Fig. 3). At both 7 and 14 days postinfection PD-L2 gene expression and MHC-II gene expression were significantly higher (P < 0.03) in AMs isolated from MC-infected mice than in AMs isolated from SM-infected mice (Fig. 3B and C). At 14 days postinfection PD-L1 gene expression was downregulated in MC and thus significantly (P = 0.01) higher in AMs from SM-infected mice (Fig. 3A). Flow cytometry confirmed some of these differences, as a higher level of PD-L2 surface expression was observed for AMs from MC-infected mice than for AMs from SM-infected mice at 7 days postinfection. MHC-II surface expression exhibited the same trend (Fig. 3D).
FIG. 3.

(A to C) mRNA levels in AMs isolated from SM- and MC-infected C57BL/6J mice (five mice per group) as measured by RT-PCR showed that there were significant differences in PD-L1 (A), PD-L2 (B), and MHC-II (C) levels at 7 days (d-7) and 14 days postinfection. The data were obtained by comparison with the abundance of actin. (D) Flow cytometric analysis of AMs isolated from sham-, SM-, and MC-infected mice. The preparations were stained with PD-L1, PD-L2, and MHC-II Abs as described in Material and Methods. SM7 and MC7, SM- and MC-infected mice at 7 days postinfection, respectively; SM14 and MC14, SM- and MC-infected mice at 14 days postinfection, respectively. *, significant difference between SM and MC samples (P < 0.05) as determined by a t test (n = 5). The error bars indicate standard deviations.
Characterization of macrophage function in the setting of SM and MC infection.
We then investigated parameters associated with macrophage function, specifically Ag presentation in SM- and MC-infected macrophages. The ATP levels of AMs isolated at 7 and 14 days postinfection revealed that both macrophage populations were metabolically active and alive (Fig. 4A). ConA-stimulated uninfected donor T cells proliferated significantly better (P < 0.01) in the presence of AMs isolated from SM-infected lungs than in the presence of AMs isolated from MC-infected lungs (Fig. 4B). A difference was not observed if proliferation was induced by a specific Ag, namely heat-inactivated SM and MC cells. In this case the T-cell proliferation induced by SM cells in the presence of AMs derived from SM-infected mice at both times was significantly less than the MC-induced T-cell proliferation with the corresponding AMs (Fig. 4C). The difference was eliminated when the SM and MC Ags were switched (Fig. 4D). We concluded from these data that AMs from both SM- and MC-infected lung tissue can support T-cell proliferation. The increased T-cell proliferation induced by MC-infected AMs in the presence of MC Ag is not a result of differences in macrophage activation but likely is a result of differences in epitope distribution between the SM and MC switch variants.
FIG. 4.

T-cell proliferation in the presence of AMs derived from SM- and MC-infected C57Bl/6J mice at 7 days (d-7) and 14 days postinfection. Proliferation was measured in quadruplicate by using a ViaLight high-sensitivity cell proliferation kit as described in Materials and Methods. The following preparations were used in this experiment: negative controls consisting of macrophages and T cells (isolated from noninfected mice) alone (donor T-cells alone and AM alone); ConA-stimulated T-cells in the presence of SM and MC macrophages (AM + ConA stimulated T-cells); cultures with Ag-specific T-cell proliferation induced by heat-inactivated SM and MC cells in the presence of the corresponding AMs from SM- and MC-infected mice (AM + Ag stimulated T-cells); and cultures with T-cell proliferation with the Ags (heat-inactivated SM and MC cells) reversed (AM + Reverse Ag stimulated T-cells). *, significant difference between SM and MC samples (P < 0.05) as determined by a t test. The error bars indicate standard deviations. AM, alveolar macrophage.
Characterization of Th17 cells in SM- and MC-infected mice.
Previous studies have demonstrated that persistence of an Aspergillus or Candida infection is associated with the emergence of IL-17-producing Th17 cells (44, 51). Our previous studies demonstrated that the levels of IL-21, transforming growth factor β (TGF-β), and IL-6 in lung tissue are higher in MC-infected mice (20). These cytokines are critical for the emergence of Th17 cells. Further characterization of lung-associated lymphocytes by flow cytometry, as well as measurement of cytokine levels by ELISA, demonstrated that MC-infected lung tissue contained significantly higher percentages of Th17 cells after 7 and 14 days (Fig. 5A). Consistent with this difference in T-cell recruitment, quantification of IL-17 in lung tissue by ELISA revealed higher IL-17 levels in MC-infected lungs (Fig. 5B). In summary, these data demonstrate that the persistence of MC infection in mice was associated with the presence of Th17 cells.
FIG. 5.

Differences in CD4+ and CD4+ IL-17A+ T-cell recruitment between SM- and MC-infected C57BL/6J mice. (A) Flow cytometric analysis of lung leukocytes with Abs specific for CD4+ cells and intracellular staining for CD4+ IL-17A+ cells (five mice per group) at 7 days (d-7) and 14 days postinfection. (B) Cytokine IL-17 production as measured by ELISA was higher in MC-infected lungs (five mice per group). *, significant difference between SM and MC samples (P < 0.05) as determined by a t test. The error bars indicate standard deviations.
Effect of macrophage depletion on the outcome for SM- and MC-infected mice.
To examine the hypothesis that macrophage activation was linked to greater damage as a result of an overstimulated immune response, we depleted AMs in SM- and MC-infected mice using liposomal clodronate. Depletion of AMs was performed once a week throughout the course of infection. Histological analysis of mouse lung tissue at the time of death demonstrated that there was granuloma formation in the lung tissue of SM-infected control and AM-depleted mice. It was noteworthy that granuloma formation was more pronounced in the sham-treated mice. In MC-infected mice, however, there was less disruption of alveolar microarchitecture in the lungs of AM-depleted mice than in the lungs of control infected mice (Fig. 6B) despite the presence of abundant yeast cells.
FIG. 6.

(A) H&E staining of lung tissue from mice at the time of death, showing differences in the inflammatory response between SM- and MC-infected C57BL/6J mice. There was less inflammatory response in AM-depleted MC-infected mice than in MC-infected wild-type mice. In contrast to the findings for SM-infected mice, there was no difference in the inflammatory response between AM-depleted mice and wild-type control mice. (B) Cytokine IL-17 production as measured by ELISA was lower in lungs of MC-infected AM-depleted mice at 14 days postinfection (d-14) (five mice per group). (C) Flow cytometric analysis of lung leukocytes showing lower levels of CD4+ IL-17A+ cells in AM-depleted MC-infected mice than in MC-infected control mice. *, significant difference between SM and MC samples (P < 0.05) as determined by a t test. The error bars indicate standard deviations.
Next we measured the production of the IL-17 cytokine in the lung tissue of SM- and MC-infected control and AM-depleted mice. There was a low level of IL-17 production and no difference in the lungs of SM-infected control mice and AM-depleted mice at 14 days postinfection. In contrast, at day 14 postinfection the level of IL-17 production was significantly lower in MC-infected AM-depleted mice than in control MC-infected mice (P = 0.01) (Fig. 6A). Also, the emergence of Th17-type cells was less pronounced in the lungs of AM-depleted MC-infected mice than in the lungs of MC-infected control mice (Fig. 6C). In summary, these results also support our conclusion that MC-infected macrophages contribute to the persistence of the inflammatory response by signaling or maintaining Th17 cells and/or by producing IL-17 cytokine themselves.
DISCUSSION
This study demonstrated the effect of phenotypic switching on the macrophage-pathogen interaction and its contribution to the damage-promoting host response that is elicited by the MC switch variant. The role of macrophages in chronic cryptococcosis is complex because these cells are involved in many different aspects of the immune response. As primary effector cells of the innate immune response they kill C. neoformans, and as Ag-presenting cells they directly contribute to the cell-mediated immune response (31). However, macrophages can become a safe harbor for intracellular C. neoformans and as such can promote evasion of the immune response and intracellular dissemination (1, 8, 12, 48). Macrophages are a potential niche where C. neoformans can multiply undisturbed and then either lyse or exit from the host macrophages (2). Because of these opposing functions in host defense and pathogenesis, it is evident that the balance of these diverse functions greatly affects the outcome of infection. Our data show that phenotypic switching disturbs this balance, which in turn affects the outcome and virulence. When the MC switch variant emerges during chronic infection, the macrophage-pathogen interaction is altered and macrophages in pulmonary infiltrates contribute to damage and pathogenesis, particularly if large numbers are recruited and they are overstimulated. Even though MC-infected macrophages continue to promote T-cell proliferation, additional cytokines, like IL-6, are produced by overactivated macrophages, which contributes to shifting the CD4+ effector T-cell population to an IL-17-producing Th17-cell response that promotes persistence of a fungal infection rather than clearing the infection.
Our data also support the concept of the “damage response” framework of microbial pathogenesis (6, 7). They highlight the close link between the innate immune response and the cell-mediated immune response and demonstrate how phenotypic switching can alter pathogenesis and the outcome of a disease by affecting the innate immune response.
Both the fact that the MC variant exhibited enhanced virulence in the absence of T cells and even NK cells and the fact that decreased survival was associated with increased recruitment of macrophages to lung tissue and damage to the alveolar microarchitecture indicated that these cells are differentially stimulated by SM and MC infection. Although the recommended C57/BL6 strain is not an ideal control strain for TgE26 mice, the survival data clearly indicate that the survival of SM-infected TgE26 mice and the survival of control mice differ, whereas the survival times for MC-infected mice are more comparable. In addition, the cytokine profiles for lung tissue of MC-infected SCID mice clearly differ from the cytokine profiles for lung tissue of SM-infected SCID mice, which have higher IL-12 and IL-10 levels but lower IL-6 and MCP levels.
Taken together, these findings further support the notion that MC-infected AMs and SM-infected AMs differ in their activation levels. The level of arginase expression, a hallmark of alternative activation, was higher in MC-infected AMs. Also, the level of expression of PD-L2 (B7-DC), another marker for alternative activated macrophages (33) and a ligand for PD-1 that inhibits T-cell activation (30), was also higher for MC-infected AMs. As expected, the PD-L1 RNA levels were higher than the PD-L2 RNA levels (33). Furthermore, the level of MHC-II expression, which is a nonspecific marker of macrophage activation in C. neoformans (26), was higher in MC-infected AMs as determined by both flow cytometry and gene expression analysis.
Alternative activation of macrophages in protozoan infections has been well studied and leads to persistence of parasitic infection. The studies demonstrated that l-ornithine, the product of the arginase pathway, allows generation of polyamines, which are needed for parasite replication (4, 5, 23, 24, 28, 29, 41). This could not be demonstrated with C. neoformans (data not shown).
AMs derived from both SM- and MC-infected lungs presented an Ag appropriately and induced T-cell proliferation. Swapping SM Ag for MC Ag alleviated the observed differences in T-cell proliferation. However, further characterization of T-cell populations in infected lungs by flow cytometry demonstrated that higher numbers of Th17 cells emerged in MC-infected lungs, whereas much lower numbers of these cells were present in SM-infected lungs. This finding was supported by the higher levels of IL-17 in MC-infected lung tissue than in SM-infected lung tissue.
The emergence of Th17 cells has not been directly associated with persistent cryptococcosis. However, studies performed with IL-23p19−/− mice indicated that the production of the IL-17 cytokine was associated with the production of IL-23 during C. neoformans infection (27). In other fungi, such as Candida and Aspergillus, a chronic inflammatory response has been linked to activation of the Th17 pathway (44, 51). Interestingly, in a cryptococcal infection with H99 the development of a robust Th1-Th17 response was shown to result in containment of H99 in the lungs of IL-4/13−/− mice, compared to the unopposed growth of H99 in the lungs of wild-type mice (52). This effect, however, was observed only in IL-4/13−/− mice, which also exhibited classical activation of macrophages. In our infection model the macrophages are alternatively activated with some evidence, but not exclusive evidence, of Th1 polarization (20).
Accordingly, previous studies that investigated the cell-mediated immune response in SM- and MC-infected C57BL/6J and IL-10−/− mice (20) determined the levels of both proinflammatory cytokines and anti-inflammatory cytokines (namely, IL-6, TGF-β, and IL-21) in lung tissue of MC-infected mice. This is important because IL-6, IL-21, and TGF-β in combination prime and sustain Th17 cells, whereas IL-10, which was consistently downregulated in MC-infected mice, negatively regulates the expression of Th17 cytokines (19). In this study, enhanced production of IL-6 was observed in macrophages derived from MC-infected mice, which supports the notion that differential macrophage activation in SM and MC infections is linked to the emergence of Th17 cells and the outcome.
To further explore the differences in macrophage activation, we depleted AMs in SM- and MC-infected mice using Cl2MDP-liposomes. Lung tissue of AM-depleted MC-infected mice exhibited less damage (loss of alveolar microarchitecture) than lung tissue of control mice. In addition, macrophage depletion also resulted in lower levels of IL-17 in tissue and Th17 cells, which is consistent with our interpretation that overactivation is linked to IL-17.
Which specific phenotypic change in the MC variant alters macrophage activation remains unclear. Several studies have demonstrated that the biophysical and biochemical properties of the SM and MC polysaccharide capsules differ (34), as do their abilities to suppress T-cell proliferation (40). Other workers demonstrated that internalization of GXM by monocyte-derived macrophages was associated with modulation of the expression of MHC-II, induction of IL-10, increased expression of CD40 and CD86 by blocking of Toll-like receptor 4, and upregulation of the Fas ligand on macrophages (36, 42, 43). Given that recent data obtained in our laboratory identified genes that are differentially regulated in the SM and MC switch variants, the goal of future studies is to genetically modify the parent SM strain and investigate the contribution of individual gene functions to macrophage activation (25).
We concluded from our data that phenotypic switching results in enhanced virulence because the MC switch variant alters macrophage activation, which shifts the T-cell response to a Th17-type T-cell response, which ultimately contributes to persistence and death. As this shift appears to be a relatively common theme in chronic mycosis, efforts to develop immune modulatory therapies that neutralize this damage-promoting aspect of the host inflammatory response should be supported.
Supplementary Material
Acknowledgments
We thank C. Terhorst (Harvard Institutes of Medicine) for providing the TgE26 mice.
This work was funded by grant RO1 AI059681-05.
Editor: G. S. Deepe, Jr.
Footnotes
Published ahead of print on 4 January 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Alvarez, M., and A. Casadevall. 2007. Cell-to-cell spread and massive vacuole formation after Cryptococcus neoformans infection of murine macrophages. BMC Immunol. 8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez, M., and A. Casadevall. 2006. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr. Biol. 16:2161-2165. [DOI] [PubMed] [Google Scholar]
- 3.Arora, S., Y. Hernandez, J. R. Erb-Downward, R. A. McDonald, G. B. Toews, and G. B. Huffnagle. 2005. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. J. Immunol. 174:6346-6356. [DOI] [PubMed] [Google Scholar]
- 4.Baetselier, P. D., B. Namangala, W. Noel, L. Brys, E. Pays, and A. Beschin. 2001. Alternative versus classical macrophage activation during experimental African trypanosomosis. Int. J. Parasitol. 31:575-587. [DOI] [PubMed] [Google Scholar]
- 5.Bronte, V., and P. Zanovello. 2005. Regulation of immune responses by l-arginine metabolism. Nat. Rev. Immunol. 5:641-654. [DOI] [PubMed] [Google Scholar]
- 6.Casadevall, A., and L. Pirofski. 2001. Host-pathogen interactions: the attributes of virulence. J. Infect. Dis. 184:337-344. [DOI] [PubMed] [Google Scholar]
- 7.Casadevall, A., and L. A. Pirofski. 2003. The damage-response framework of microbial pathogenesis. Nat. Rev. Microbiol. 1:17-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charlier, C., F. Dromer, C. Leveque, L. Chartier, Y. S. Cordoliani, A. Fontanet, O. Launay, and O. Lortholary. 2008. Cryptococcal neuroradiological lesions correlate with severity during cryptococcal meningoencephalitis in HIV-positive patients in the HAART era. PLoS One 3:e1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chayakulkeeree, M., T. H. Rude, D. L. Toffaletti, and J. R. Perfect. 2007. Fatty acid synthesis is essential for survival of Cryptococcus neoformans and a potential fungicidal target. Antimicrob. Agents Chemother. 51:3537-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corraliza, I. M., M. L. Campo, G. Soler, and M. Modolell. 1994. Determination of arginase activity in macrophages: a micromethod. J. Immunol. Methods 174:231-235. [DOI] [PubMed] [Google Scholar]
- 11.Dromer, F., S. Mathoulin, B. Dupont, and A. Laporte. 1996. Epidemiology of cryptococcosis in France: a 9-year survey (1985-1993). French Cryptococcosis Study Group. Clin. Infect. Dis. 23:82-90. [DOI] [PubMed] [Google Scholar]
- 12.Feldmesser, M., Y. Kress, P. Novikoff, and A. Casadevall. 2000. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect. Immun. 68:4225-4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franzot, S. P., J. Mukherjee, R. Cherniak, L. C. Chen, J. S. Hamdan, and A. Casadevall. 1998. Microevolution of a standard strain of Cryptococcus neoformans resulting in differences in virulence and other phenotypes. Infect. Immun. 66:89-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fries, B. C., D. L. Goldman, R. Cherniak, R. Ju, and A. Casadevall. 1999. Phenotypic switching in Cryptococcus neoformans results in changes in cellular morphology and glucuronoxylomannan structure. Infect. Immun. 67:6076-6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fries, B. C., C. P. Taborda, E. Serfass, and A. Casadevall. 2001. Phenotypic switching of Cryptococcus neoformans occurs in vivo and influences the outcome of infection. J. Clin. Invest. 108:1639-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goerdt, S., O. Politz, K. Schledzewski, R. Birk, A. Gratchev, P. Guillot, N. Hakiy, C. D. Klemke, E. Dippel, V. Kodelja, and C. E. Orfanos. 1999. Alternative versus classical activation of macrophages. Pathobiology 67:222-226. [DOI] [PubMed] [Google Scholar]
- 17.Goldman, D. L., B. C. Fries, S. P. Franzot, L. Montella, and A. Casadevall. 1998. Phenotypic switching in the human pathogenic fungus Cryptococcus neoformans is associated with changes in virulence and pulmonary inflammatory response in rodents. Proc. Natl. Acad. Sci. U. S. A. 95:14967-14972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon, S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3:23-35. [DOI] [PubMed] [Google Scholar]
- 19.Gu, Y., J. Yang, X. Ouyang, W. Liu, H. Li, J. Yang, J. Bromberg, S. H. Chen, L. Mayer, J. C. Unkeless, and H. Xiong. 2008. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur. J. Immunol. 38:1807-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerrero, A., and B. C. Fries. 2008. Phenotypic switching in Cryptococcus neoformans contributes to virulence by changing the immunological host response. Infect. Immun. 76:4322-4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez, Y., S. Arora, J. R. Erb-Downward, R. A. McDonald, G. B. Toews, and G. B. Huffnagle. 2005. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J. Immunol. 174:1027-1036. [DOI] [PubMed] [Google Scholar]
- 22.Huffnagle, G. B., M. F. Lipscomb, J. A. Lovchik, K. A. Hoag, and N. E. Street. 1994. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J. Leukoc. Biol. 55:35-42. [DOI] [PubMed] [Google Scholar]
- 23.Iniesta, V., J. Carcelen, I. Molano, P. M. Peixoto, E. Redondo, P. Parra, M. Mangas, I. Monroy, M. L. Campo, C. G. Nieto, and I. Corraliza. 2005. Arginase I induction during Leishmania major infection mediates the development of disease. Infect. Immun. 73:6085-6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iniesta, V., L. C. Gomez-Nieto, and I. Corraliza. 2001. The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J. Exp. Med. 193:777-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jain, N., L. Li, Y. P. Hsueh, A. Guerrero, J. Heitman, D. L. Goldman, and B. C. Fries. 2009. Loss of allergen 1 confers a hypervirulent phenotype that resembles mucoid switch variants of Cryptococcus neoformans. Infect. Immun. 77:128-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawakami, K., S. Kohno, N. Morikawa, J. Kadota, A. Saito, and K. Hara. 1994. Activation of macrophages and expansion of specific T lymphocytes in the lungs of mice intratracheally inoculated with Cryptococcus neoformans. Clin. Exp. Immunol. 96:230-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleinschek, M. A., U. Muller, S. J. Brodie, W. Stenzel, G. Kohler, W. M. Blumenschein, R. K. Straubinger, T. McClanahan, R. A. Kastelein, and G. Alber. 2006. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J. Immunol. 176:1098-1106. [DOI] [PubMed] [Google Scholar]
- 28.Kreider, T., R. M. Anthony, J. F. Urban, Jr., and W. C. Gause. 2007. Alternatively activated macrophages in helminth infections. Curr. Opin. Immunol. 19:448-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kropf, P., J. M. Fuentes, E. Fahnrich, L. Arpa, S. Herath, V. Weber, G. Soler, A. Celada, M. Modolell, and I. Muller. 2005. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. 19:1000-1002. [DOI] [PubMed] [Google Scholar]
- 30.Latchman, Y., C. R. Wood, T. Chernova, D. Chaudhary, M. Borde, I. Chernova, Y. Iwai, A. J. Long, J. A. Brown, R. Nunes, E. A. Greenfield, K. Bourque, V. A. Boussiotis, L. L. Carter, B. M. Carreno, N. Malenkovich, H. Nishimura, T. Okazaki, T. Honjo, A. H. Sharpe, and G. J. Freeman. 2001. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2:261-268. [DOI] [PubMed] [Google Scholar]
- 31.Levitz, S. M. 1994. Macrophage-Cryptococcus interactions. Immunol. Ser. 60:533-543. [PubMed] [Google Scholar]
- 32.Lincoln, M. R., A. Montpetit, M. Z. Cader, J. Saarela, D. A. Dyment, M. Tiislar, V. Ferretti, P. J. Tienari, A. D. Sadovnick, L. Peltonen, G. C. Ebers, and T. J. Hudson. 2005. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat. Genet 37:1108-1112. [DOI] [PubMed] [Google Scholar]
- 33.Loke, P., and J. P. Allison. 2003. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. U. S. A. 100:5336-5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McFadden, D. C., B. C. Fries, F. Wang, and A. Casadevall. 2007. Capsule structural heterogeneity and antigenic variation in Cryptococcus neoformans. Eukaryot. Cell 6:1464-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell, T., and J. Perfect. 1995. Cryptococcosis in the era of AIDS—100 years after the discovery of Cryptococcus neoformans. Clin. Microbiol. Rev. 8:515-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monari, C., E. Pericolini, G. Bistoni, A. Casadevall, T. R. Kozel, and A. Vecchiarelli. 2005. Cryptococcus neoformans capsular glucuronoxylomannan induces expression of fas ligand in macrophages. J. Immunol. 174:3461-3468. [DOI] [PubMed] [Google Scholar]
- 37.Muller, U., W. Stenzel, G. Kohler, C. Werner, T. Polte, G. Hansen, N. Schutze, R. K. Straubinger, M. Blessing, A. N. McKenzie, F. Brombacher, and G. Alber. 2007. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J. Immunol. 179:5367-5377. [DOI] [PubMed] [Google Scholar]
- 38.Namangala, B., P. De Baetselier, W. Noel, L. Brys, and A. Beschin. 2001. Alternative versus classical macrophage activation during experimental African trypanosomosis. J. Leukoc. Biol. 69:387-396. [PubMed] [Google Scholar]
- 39.Pietrella, D., C. Corbucci, S. Perito, G. Bistoni, and A. Vecchiarelli. 2005. Mannoproteins from Cryptococcus neoformans promote dendritic cell maturation and activation. Infect. Immun. 73:820-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pietrella, D., B. Fries, P. Lupo, F. Bistoni, A. Casadevall, and A. Vecchiarelli. 2003. Phenotypic switching of Cryptococcus neoformans can influence the outcome of the human immune response. Cell. Microbiol. 5:513-522. [DOI] [PubMed] [Google Scholar]
- 41.Raes, G., A. Beschin, G. H. Ghassabeh, and P. De Baetselier. 2007. Alternatively activated macrophages in protozoan infections. Curr. Opin. Immunol. 19:454-459. [DOI] [PubMed] [Google Scholar]
- 42.Retini, C., T. R. Kozel, D. Pietrella, C. Monari, F. Bistoni, and A. Vecchiarelli. 2001. Interdependency of interleukin-10 and interleukin-12 in regulation of T-cell differentiation and effector function of monocytes in response to stimulation with Cryptococcus neoformans. Infect. Immun. 69:6064-6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Retini, C., A. Vecchiarelli, C. Monari, F. Bistoni, and T. R. Kozel. 1998. Encapsulation of Cryptococcus neoformans with glucuronoxylomannan inhibits the antigen-presenting capacity of monocytes. Infect. Immun. 66:664-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romani, L., T. Zelante, A. De Luca, F. Fallarino, and P. Puccetti. 2008. IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J. Immunol. 180:5157-5162. [DOI] [PubMed] [Google Scholar]
- 45.Shao, X., A. Mednick, M. Alvarez, N. van Rooijen, A. Casadevall, and D. L. Goldman. 2005. An innate immune system cell is a major determinant of species-related susceptibility differences to fungal pneumonia. J. Immunol. 175:3244-3251. [DOI] [PubMed] [Google Scholar]
- 46.Singh, N., B. D. Alexander, O. Lortholary, F. Dromer, K. L. Gupta, G. T. John, R. del Busto, G. B. Klintmalm, J. Somani, G. M. Lyon, K. Pursell, V. Stosor, P. Munoz, A. P. Limaye, A. C. Kalil, T. L. Pruett, J. Garcia-Diaz, A. Humar, S. Houston, A. A. House, D. Wray, S. Orloff, L. A. Dowdy, R. A. Fisher, J. Heitman, M. M. Wagener, and S. Husain. 2007. Cryptococcus neoformans in organ transplant recipients: impact of calcineurin-inhibitor agents on mortality. J. Infect. Dis. 195:756-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Syme, R. M., J. C. Spurrell, E. K. Amankwah, F. H. Green, and C. H. Mody. 2002. Primary dendritic cells phagocytose Cryptococcus neoformans via mannose receptors and Fcgamma receptor II for presentation to T lymphocytes. Infect. Immun. 70:5972-5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tucker, S. C., and A. Casadevall. 2002. Replication of Cryptococcus neoformans in macrophages is accompanied by phagosomal permeabilization and accumulation of vesicles containing polysaccharide in the cytoplasm. Proc. Natl. Acad. Sci. U. S. A. 99:3165-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Rooijen, N., and A. Sanders. 1994. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 174:83-93. [DOI] [PubMed] [Google Scholar]
- 50.Wozniak, K. L., J. M. Vyas, and S. M. Levitz. 2006. In vivo role of dendritic cells in a murine model of pulmonary cryptococcosis. Infect. Immun. 74:3817-3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zelante, T., A. De Luca, P. Bonifazi, C. Montagnoli, S. Bozza, S. Moretti, M. L. Belladonna, C. Vacca, C. Conte, P. Mosci, F. Bistoni, P. Puccetti, R. A. Kastelein, M. Kopf, and L. Romani. 2007. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur. J. Immunol. 37:2695-2706. [DOI] [PubMed] [Google Scholar]
- 52.Zhang, Y., F. Wang, K. C. Tompkins, A. McNamara, A. V. Jain, B. B. Moore, G. B. Toews, G. B. Huffnagle, and M. A. Olszewski. 2009. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent Cryptococcus neoformans H99. Am. J. Pathol. 175:2489-2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.