

Abstract
Uropathogenic Escherichia coli (UPEC) is the leading cause of urinary tract infections (UTIs). A murine UTI model has revealed an infection cascade whereby UPEC undergoes cycles of invasion of the bladder epithelium, intracellular proliferation in polysaccharide-containing biofilm-like masses called intracellular bacterial communities (IBC), and then dispersal into the bladder lumen to initiate further rounds of epithelial colonization and invasion. We predicted that the UPEC K1 polysaccharide capsule is a key constituent of the IBC matrix. Compared to prototypic E. coli K1 strain UTI89, a capsule assembly mutant had a fitness defect in functionally TLR4+ and TLR4− mice, suggesting a protective role of capsule in inflamed and noninflamed hosts. K1 capsule assembly and synthesis mutants had dramatically reduced IBC formation, demonstrating the common requirement for K1 polysaccharide in IBC development. The capsule assembly mutant appeared dispersed in the cytoplasm of the bladder epithelial cells and failed to undergo high-density intracellular replication during later stages of infection, when the wild-type strain continued to form serial generations of IBC. Deletion of the sialic acid regulator gene nanR partially restored IBC formation in the capsule assembly mutant. These data suggest that capsule is necessary for efficient IBC formation and that aberrant sialic acid accumulation, resulting from disruption of K1 capsule assembly, produces a NanR-mediated defect in intracellular proliferation and IBC development. Together, these data demonstrate the complex but important roles of UPEC polysaccharide encapsulation and sialic acid signaling in multiple stages of UTI pathogenesis.
Uropathogenic Escherichia coli (UPEC) is the leading cause of urinary tract infection (UTI), and health care costs for UTI exceed $1.5 billion per year in the United States alone (22). Most UTIs occur in the bladder (cystitis), but more-invasive infections lead to infection of the kidneys (pyelonephritis) and dissemination to the bloodstream (urosepsis) and central nervous system (meningitis). UPEC that expresses K1 capsule, a linear α2-8-linked sialic acid homopolymer, is commonly associated with each of these infections (29, 36). Cystitis is a common syndrome for the treatment of which antibiotics are widely administered. However, recurrent UTI often occurs despite appropriate antibiotic therapy. In addition, a disturbing trend toward increasingly antibiotic-resistant UPEC has been occurring over the past decade (11, 30). Thus, defining new targets for therapy through elucidation of the molecular basis for cystitis, as for other aspects of UTIs, has become increasingly important.
UPEC produces cystitis through complex interactions with the host. In mouse models, UPEC adheres to the bladder epithelium by type 1 pili. UPEC invades the epithelium through caveolae/lipid raft-mediated and clathrin-mediated endocytic pathways (20, 21, 40). In addition, UPEC has been shown to enter the bladder epithelium by cyclic AMP (cAMP)-responsive fusiform vesicles where elevations in intraepithelial cAMP can result in exocytosis of the vesicles and expulsion of the bacteria into the bladder lumen (7). To subvert the host response, however, UPEC has been shown in mouse models to escape into the cytoplasm of the infected cell and replicate in biomasses called intracellular bacterial communities (IBC) (2, 40, 43). The presence of IBC has been shown in numerous different mouse strains, as well as in urine sediment from humans experiencing acute and recurring UTIs (23, 53). IBC expand to fill the infected host epithelial cell. The IBC is transient in nature, and soon after its maturation, the bacterial community disperses, exiting the infected cell to invade naïve epithelium. Prior work has shown that the cycle of colonization, invasion, IBC formation, and dispersal is cyclical, producing multiple generations of IBC (31). Failure to initiate or complete IBC formation has been shown to attenuate cystitis (32, 33, 72).
The formation of IBC has similarities to that of biofilm communities previously modeled on abiotic surfaces. Bacteria embedded in the IBC have cell-associated appendages such as type 1 pili and antigen 43 (3). The matrix also stains by periodic acid Schiff, suggesting the presence of polysaccharides (3). However, the specific type of polysaccharide present in the IBC matrix is not known. The majority of UPEC isolates produce group 2 capsules, such as the prototype K1 (29, 52). In addition, the UPEC genome encodes colanic acid, β-1,6-N-acetyl-d-glucosamine, and cellulose, all of which have known roles in biofilm formation in vitro (16, 17, 69). We sought to determine if the common K1 polysaccharide contributes to IBC formation.
Like other bacterial polysaccharide capsules, K1 capsule has two classical roles in defense against host innate immunity: (i) inhibition of phagocytosis by granulocytes/monocytes (70) and (ii) serum resistance (47). More-recent studies have demonstrated that K capsules may be involved in nonspecific adherence and also in biofilm formation on artificial surfaces (48, 67). Like that of other group 2 capsules, the biogenesis of the K1 capsule requires three genetic regions located at a shared locus for their synthesis, assembly, and exportation (71). Region II gene products (neu) direct the synthesis and modification of the primary monosaccharides used in capsule assembly, thus determining the actual capsule antigen (K) type. Region I (kps) and III (kpsMT) gene products are involved in capsule assembly and export (9, 58, 71).
Although epidemiological studies suggest that UPEC is widely encapsulated, with K1 being a leading capsular subtype, few details are known about the complete molecular roles of encapsulation during the pathogenesis of E. coli UTI. Several prior studies have revealed that strains with capsule mutations have reduced fitness, but the specific molecular defects in pathogenesis were not demonstrated (6, 46). Similarly, a recent study found that mutation of K2 capsule synthesis in UPEC strain CFT073, a urosepsis blood isolate, resulted in decreased survival in the mouse urinary tract (10). The capsule mutant strain was most attenuated in the kidneys and urine; however, complementation of the capsule synthesis mutant resulted in a significant increase in the number of bacteria recovered from the bladder, suggesting a role for the K2 capsule as a virulence factor in upper and lower UTIs. These studies argue that polysaccharide capsules are important virulence factors during UTI. However, the precise function of capsule in the pathogenic cascade has not been previously studied. Furthermore, previous studies used non-K1 pyelonephritis and sepsis isolates as prototypic strains where specific evaluation of the molecular pathogenesis of cystitis may be better performed using clinical cystitis isolates, particularly carrying the prevalent K1 capsule type.
In our present study, we sought to elucidate the role of the K1 capsule of prototypic cystitis isolate UTI89 (12, 44) during cystitis. We hypothesized that the polysialic acid K1 capsule may not only protect UPEC from innate immunity but also form an IBC matrix component, facilitating aggregation of the bacterial communities, which in turn preclude infiltration of host inflammatory mediators and environmental stressors. We discovered that K1 capsule is an important virulence determinant in functionally TLR4+ and TLR4− mice and that the K1 capsule is significant in the maintenance of IBC morphology. We further demonstrated that IBC formation was partially restored in a capsule assembly mutant by abrogation of NanR-sialic acid regulation, suggesting a unique role for sialic acid signaling during intracellular UPEC growth. Together, these data suggest that both K1 capsule and sialic acid-dependent regulation have novel roles in the intracellular pathogenesis of cystitis caused by UPEC K1.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. In this study, a spontaneous streptomycin-resistant variant of cystitis strain UTI89 was used as the parent strain for construction of UTI89 kpsF::EP185. Bacteria were routinely grown in Luria-Bertani (LB) medium, except as otherwise indicated. Inocula for animal experiments were grown in LB medium at 37°C for 18 h under static conditions.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or feature(s)a | Reference or source |
|---|---|---|
| Strains | ||
| UTI89 | Clinical cystitis isolate | 44 |
| UTI89 attλ::PSSH10-1 | Promoterless gfp; spectinomycin resistance cassette at lambda integration site | 73 |
| MG1655 | K-12 reference strain | 8 |
| MG1655 attλ::PSSH10-1 | Promoterless gfp; spectinomycin resistance cassette at lambda integration site | 73 |
| UTI89 kpsF:: EP185 | Insertion of pEP185.2 in kpsF; polar inactivation of region I capsule genes | This study |
| UTI89 Δkps | ΔkpsFEDUCS::FRTΨ | This study |
| MG1655 attHK022::COM-GFP | tac-gfp in single copy at the HK022 phage integration site, Kanr | 73 |
| UTI89 Δkps attHK022::COM-GFP | ΔkpsFEDUCS::FRTΨtac-gfp; kanamycin resistance cassette | This study |
| MG1655 attλ::kps-gfp | PRegI::gfp at lambda integration site; Specr | This study |
| UTI89 attλ::kps-gfp | PRegI::gfp at lambda integration site; Specr | This study |
| UTI89 Δneu | ΔneuDBACES::FRTΨ | This study |
| UTI89 ΔcdiA | ΔcdiA::kan | This study |
| UTI89 Δkps ΔcdiA | ΔkpsFEDUCS::FRTΨ ΔcdiA::kan | This study |
| JW3195.1 | ΔnanR::kan | 5 |
| UTI89 Δkps ΔnanR | ΔkpsFEDUCS::FRTΨ ΔnanR::kan | This study |
| Plasmids | ||
| pPSSH10 | Promoterless gfp, Specr | 73 |
| pINT | Lambda integrase, Ampr | 49 |
| pCOM-GFP | tac-gfp, Ampr, Kanr | 66 |
| pKD4 | FRT-Kan-FRT, Ampr | 18 |
| pCP20 | FLP, Ampr | 13 |
| pEP185.2 | SacB, Cmr | 19 |
| pBR329 | Ampr, Cmr, Tetr | 15 |
| pSX50 | kpsFEDUCS, Ampr, Cmr | 61 |
| pGA122 | kps-gfp in pPSSH10-1 | This study |
| pBAC-LacZ | Mini F′ single-copy plasmid, Cmr | Addgene plasmid 13422 |
| pBAC-LacZ Reg I kps | EcoRI fragment of pSX50 containing kpsFEDUCS, Cmr | This study |
| pCR-nanR | nanR from UTI89 in pCR-BLUNT II, Kanr | This study |
| pBAC-LacZ nanR | nanR from pCR-nanR in pBAC-LAC, Cmr | This study |
Superscript Ψ Indicates residual interrupting scar after removal of antibiotic cassette by FLP recombinase.
Construction of a kps-gfp reporter strain.
A K1 capsule region I promoter transcriptional fusion to gfp was constructed as follows. The capsule region I promoter region (−722 to +37, relative to the kpsF GTG start codon) was amplified by PCR using primers PReg1#1 and PReg1#2, as indicated in Table 2. The product of this reaction (∼760 bp) was digested with ClaI and BamHI and ligated into the corresponding sites in pPSSH10-1 (73). The ligation reaction product was transformed into E. coli DH5α λ pir, and the resultant plasmid was named pGA122. E. coli MG1655 pINT-TS was transformed with pGA122 by electroporation according to standard methods. After outgrowth at 37°C, the culture was incubated at 42°C for 30 min and then plated on LB medium plates supplemented with 37.5 μg/ml spectinomycin. This resulted in the integration of pGA122 in single copy into the att λ integration site of MG1655, creating strain MG1655 attλ::kps-gfp. PCR was used to verify the location and copy number of the insert. This construct was P1 transduced into UTI89 as described above. The resultant strain, UTI89 attλ::kps-gfp, contained a kps-gfp expression construct integrated into the att site in UTI89.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′) | Reference |
|---|---|---|
| kpsF::EP185 #1 | GACCAGAGCTCTACTATCGA | This study |
| kpsF::EP185 #2 | GCAGGTACCATTCGCCATAT | This study |
| kps KO #1 | GTTACAACCCATTGATTTAGCATAAATAAATTATAGTGGGTTCGGGTTTGTTGTGTAGGCTGGAGCTGCTTC | This study |
| kps KO #2 | TGGAAATGATTTTTTGGCTACTTAAAATTCAAAAGATATTGACTTGAAATATGGGAATTAGCCATGGTCC | This study |
| kps flank #1 | ATGTTCCCGGTGGTCAACATGCTTCCAGCACTCCTT | This study |
| kps flank #2 | CCTCTTTGCACGATAAAAGGATTTTCTTG | This study |
| neu KO #1 | CCTATAGTGGTTACATTCCAATATTATGCCTTGGAAATATTTAACTGAGACATATCGTGTAGGCTGGAGCTGCTTC | This study |
| neu KO #2 | CTACTTTAAGATTTAATTTTACGACTGGTACTGTAATAGAATATAAAATGTAGCTTTTAATGGGAATTAGCCATGGTCC | This study |
| neu flank #1 | GTGCGGAACTGTCAAACCCGCTCAC | This study |
| neu flank #2 | AGACGCAGGTTAATTGGGTTTATTATGGG | This study |
| PRegI #1 | GACAATCGATTATCGGCGCA | This study |
| PRegI #2 | TTGGATCCTATCAGCTCCTT | This study |
| cdiA KO #1 | GCTCTTTTGTCATATTGATGTTTCCAATCAATTTACGTAAGGTAAGTGTAGGCTGGAGCTGCTTC | This study |
| cdiA KO #2 | ACGGTTTACTGGCGCGTTGCCGTCGCGTTTTAAGGGATTATTACCATGGGAATTAGCCATGGTCC | This study |
| cdiA flank #1 | GGTTGACCGGCCAGTCTGCTATGA | This study |
| cdiA flank #2 | CCAGAAGAACGGCTGCGTATTGC | This study |
| nanR flank #1 | CTTGCTTCTTTAACTGAAGCTGAACGTGAA | This study |
| nanR flank #2 | TCTGCTTCACTAAAGTGGCATTATTTCT | This study |
Insertional interruption of K1 capsule region I genes.
A polar disruption of the K1 genes for assembly (region I) was made by creating a large insertion in the first gene of the region, kpsF, in cystitis isolate UTI89. An approximately 450-bp fragment from the 5′ end of kpsF was amplified by PCR using primers kpsF::EP185 #1 and kpsF::EP185 #2 (Table 2). This fragment was digested with restriction enzymes SacI and KpnI and ligated into compatibility-cut suicide vector pEP185.2 (37). The ligation reaction product was transformed into E. coli strain SM10 λ pir (41) using standard transformation protocols. The resultant plasmid, named pGA108, was transferred to UTI89 by conjugation. Briefly, 500-μl volumes of both UTI89 (streptomycin resistant) and SM10 λ pir/pGA108 (kanamycin and chloramphenicol resistant) were mixed and incubated on an LB medium plate at 37°C. After an overnight incubation, the plate was rinsed with 3 ml of LB broth and serial dilutions were plated on LB medium plates supplemented with streptomycin and chloramphenicol. The resultant kpsF insertional mutant is referred to as UTI89 kpsF::EP185.
Construction of K1 capsule region I and region II deletion strains.
To construct defined isogenic strains of UTI89 with complete deletions of the K1 capsule region I or II coding sequences (kpsFEDUCS and neuDBACES), a modification of the method of Murphy and Campellone was used (45). A linear knockout fragment was constructed by PCR using pKD4 (18) as a template with primers kps KO #1 and #2 for the region I deletion and neu KO #1 and #2 for the region II deletion. The amplicons were electroporated into UTI89/pKM208 expressing the red recombinase machinery, and the deletion strains were selected on LB medium/kanamycin (50 μg/ml) plates. The mutants were confirmed using kps flank #1 and #2 or neu flank #1 and #2. The antibiotic resistance cassettes, where necessary, were excised by FLP recombination (18). The confirmed strains were designated UTI89 Δkps and UTI89 Δneu, respectively. For competition experiments, kanamycin resistance and constitutive gfp expression cassettes were introduced into UTI89 Δkps by P1 transduction of the locus from MG1655 attHK022::COM-GFP, resulting in strain UTI89 Δkps attHK022::COM-GFP.
P1 transduction of nanR::kan into UTI89.
To construct UTI89 Δkps ΔnanR, generalized P1 transduction was performed using standard techniques with phage stock derived from JW3195.1 (5). JW3195.1 has a complete replacement of nanR with a kanamycin resistance cassette. The phage lysate was incubated with UTI89 Δkps prepared in MC buffer and recovered on L agar/kanamycin. PCR analysis confirmed the presence of nanR::kan in the transductants.
Construction of single-copy complementation plasmids.
Control experiments demonstrated poor retention of several base plasmids in vitro during shaken and static growth in the absence of antibiotic pressure, including pBR329 and pACYC184. Thus, these plasmid systems were avoided as vectors intended to complement mutations in vivo for >6 h during murine UTI modeling. Complementation of the kps region I deletion and nanR was performed by cloning the respective genes into pBAC-LacZ (Table 1) as follows. The K1 region I genes were excised from pSX50 by EcoRI restriction digestion, the ends were filled with Klenow (exo−) plus nucleotides, and the genes were ligated into pBAC-LacZ digested with SnaBI, yielding the vector pBAC-LacZ RegI kps. The nanR gene from UTI89 genomic DNA was amplified by PCR with Primestar polymerase (Takara) using primers nanR flank #1 and #2 (5′-CTTGCTTCTTTAA CTGAAGCTGAACGTGAA-3′ and 5′-TCTGCTTCACTAAAGTGGCATTATTTCT-3′), with 1 cycle of 95°C for 1 min, followed by 25 cycles of 95°C for 15 s, 57°C for 5 s, and 72°C for 2.5 min, and ending with 72°C for 10 min. The1.5-kb amplicon was cloned into pCR-BLUNT II (Invitrogen) to obtain pCR-nanR. The fully sequenced insert was subcloned as a Klenow-treated EcoRI digestion product into NotI-digested, Klenow-filled pBAC-LacZ to produce pBAC-LacZ nanR.
Murine UTI model.
Mouse infections were carried out by intraurethral inoculation as previously described (26). The inoculum, unless stated otherwise, was ∼2 × 107 in 50 μl of sterile phosphate-buffered saline (PBS). For bacterial titers, bladders were harvested and homogenized in 1 ml of 0.02% Triton X-100/PBS using an electric handheld tissue homogenizer. Serial dilutions were plated on appropriate medium. For mounting of whole bladder tissues, mice were infected with either UTI89 or isogenic mutants containing pCOM-GFP, which constitutively expresses green fluorescent protein (GFP). Bladders were extracted, splayed open on silica resin-coated dishes using dissecting pins, stained with wheat germ agglutinin-Alexa Fluor 633 (Molecular Probes) for 15 min, washed three times with PBS, and fixed with 3% paraformaldehyde. Fixed samples were mounted and visualized on a Zeiss LSM510 laser scanning confocal microscope. For the coinfection and kps-gfp expression studies, splayed bladders were fixed with 3% paraformaldehyde, washed three times with PBS, and stained with the nuclear stain TO-PRO-3 iodide (Molecular Probes) in 0.1% Triton X-100 for 15 min.
Coinfections were carried out as described above, except that the competitive strains were mixed from the prepared suspensions (∼5 × 108 CFU/ml) at a 1:1 ratio. Fifty microliters of the mixture was used per transurethral infection.
All animal studies were conducted with approval from and in accordance with the Committee for Animal Studies at Washington University School of Medicine and the Duke University Institutional Animal Care and Use Committee.
IBC enumeration.
Infected bladder tissue was resected, splayed, and fixed for 1 h at room temperature (RT) in 3% paraformaldehyde. For confocal microscopy, the fixed tissues were stained for 10 min at RT with a 1:1,000 dilution of TO-PRO-3 iodide (Molecular Probes) in PBS and directly evaluated in whole mount. For Lac staining, the tissue was subsequently washed gently two times with PBS, resuspended in Lac wash buffer, and stained as previously described (26, 33).
Analysis of IBC morphology.
Determination of the number of bacteria in IBC was carried out using the Volocity software (Improvision), in which the volume of GFP emission was measured across IBC Z-section series captured by a Zeiss LSM510 laser scanning confocal microscope. The Volocity software calculates the GFP volume by counting the green pixels within a given area of the region defined as an IBC within the image. Using LSM 5 Image Browser (Zeiss), we measured the area occupied by the IBC on each Z slice, and we divided this area by the slice width to determine the volume of the IBC on that slice. The total IBC volume was determined by adding the volumes of the individual slices. IBC density was calculated by dividing the GFP volume by the total IBC volume (μm3GFP/μm3TOTAL).
K antigen radial immunodiffusion and K1 phage sensitivity.
For radial immunodiffusion to detect K1 polysaccharide, bacteria were grown for 6 h with shaking at 37°C in LB medium and pelleted at 6,000 × g for 5 min at RT. The cells were adjusted to an equal optical density at 600 nm in sterile PBS and sonicated for complete disruption. The whole-cell lysates were distributed into wells cut in 1% agarose containing 5% horse H46 anti-meningococcus type B antiserum and 0.01% sodium azide. The meningococcus type B capsule is antigenically identical to the E. coli K1 capsule. The plates were incubated for ≥24 h at 30°C until a well-delineated precipitin ring was present for the positive control (wild-type [WT] strain).
K1 phage sensitivity was determined by growing the test strains overnight at 37°C with shaking and streaking lawns onto L agar. In the center of the fresh lawns, 10 μl of a fresh K1F phage lysate was placed. The plates were incubated upright at 37°C for ∼6 h. Strains with zones of clearing at the site of the K1F phage were determined to be sensitive.
Statistical analysis.
Statistical analyses using the nonparametric Mann-Whitney U test, Fisher's exact test, or Student's t test were performed using GraphPad Prism (GraphPad Software) where indicated in Results. Statistical significance was defined by attaining P values of ≤0.05. Errors are expressed as the standard deviation of the mean unless indicated otherwise.
RESULTS
Loss of capsule results in reduced fitness in functionally TLR4+ and TLR4− hosts during cystitis.
Although a classical role of polysaccharide capsules is protection against neutrophil phagocytosis and killing, the specific role of the K1 capsule in UTI is not well established. The conserved features of group II capsule assembly and export make it an attractive target for future chemotherapeutics, and therefore we sought to determine if interruption of capsule biogenesis through interruption of assembly would attenuate UPEC in vivo. We generated a polar interruption of the region 1 gene locus at kpsF in prototypic cystitis strain UTI89 (44) through an insertional mutation (Fig. 1A). The mutant (UTI89 kpsF::EP185) had reduced K1 antigen latex agglutination and was resistant to K1-specific bacteriophage lysis (data not shown), demonstrating the expected loss of cell surface-associated capsule. The resulting acapsular and parental WT strains were used to infect female C3H/HeN mice (TLR4+) via transurethral catheterization to initiate cystitis. The total bacterial burden in the bladder was evaluated at multiple times postinoculation (p.i.). UTI89 kpsF::EP185 had a significant loss of fitness by 6 h p.i. and 24 h p.i., with the trend continuing through to 2 weeks p.i. (Fig. 1B). At 48 h, competitive infection with UTI89 and UTI89 kpsF::EP185 yielded an index of −1.57 ± 0.73 (n = 5; P < 0.05), suggesting a significant attenuation of the K1 mutant strain. We found no difference in growth kinetics between the kpsF::EP185 mutant and WT strains in vitro that would account for the in vivo differences in fitness (data not shown). Both strains had equivalent mannose-sensitive hemagglutination (data not shown), indicating that differences in type 1 piliation [important for adherence, invasion, and IBC development (28, 40, 72)] did not likely account for the fitness differences in vivo.
FIG. 1.

Unencapsulated UPEC has neutrophil-dependent and -independent fitness defects. (A) Organization of the capsule gene locus. Region I genes encode capsule assembly and export factors. Region II encodes synthesis factors. Region III products are involved in capsule export. Region I interruption (strain UTI89 kpsF::EP185) is designated by the upward-pointing arrow. Deletions of region I (Δkps) and region II (Δneu) used in later experiments are indicated by lines below the diagram. (B and C) CFU counts of isolates UTI89 (WT; closed squares) and UTI89 kpsF:: EP185 (open triangles) in infected bladders at time points between 6 h and 2 weeks in female C3H/HeN (TLR4+) and C3H/HeJ (TLR4−) mice, respectively. Horizontal lines indicate the median values for the groups. P values were calculated using the Mann-Whitney nonparametric test.
TLR4 has a central role in pathogen recognition during lower UTI, with its stimulation resulting in induction of innate immunity and a proinflammatory response (27, 32, 54, 56, 57, 60). An example of a major TLR4-regulated response is neutrophil recruitment, a primary host defense during cystitis. In the murine model of cystitis, abundant neutrophil infiltration generally occurs 12 to 16 h after initiation of the infection in the mouse strain used in these experiments (31). In this temporal window, the capsule mutant had a significant attenuation in the TLR4-responsive C3H/HEN mouse (Fig. 1B).
We hypothesized that K1 capsule would be important independently of the TLR4-induced innate immune response. To test this hypothesis, we infected C3H/HeJ mice that are functionally TLR4 nonresponsive. A major deficit in this mouse strain is abrogated lipopolysaccharide-mediated induction of interleukin-6 signaling, which is important for neutrophil recruitment in mice (50, 63, 64). Thus, these mice have abrogated neutrophil recruitment into the bladder during infection (24). As depicted in Fig. 1C, the kpsF::EP185 mutant was still attenuated in the C3H/HeJ mice, although the timing of the attenuation was delayed relative to the infection in C3H/HeN mice (P = 0.008 at 48 h and 2 weeks p.i.). These data suggest that the K1 capsule has a role in protecting E. coli from the action of the TLR4-dependent innate immune response and makes another contribution independent of protection from the TLR4-dependent innate immune response.
To demonstrate that the observed loss of virulence in the kpsF::EP185 mutant strain was not specific to the nature of the genetic defect (a polar interruption in the capsule assembly genes) or from an unlinked mutation, we performed additional in vivo experiments with a strain bearing a complete deletion of the region I capsule assembly genes, UTI89 Δkps. Competitive experiments were performed with an antibiotic resistance-marked parent strain, UTI89 attλ::PSSH10-1, carrying a spectinomycin resistance marker at the benign lambda integration site, and a kanamycin-resistant derivative of the kps deletion mutant, UTI89 Δkps attHK022::COM-GFP, which has antibiotic resistance and gfp expression cassettes at the innocuous HK phage integration site. Growth studies in Luria broth revealed no significant differences between the two (data not shown). Competitive infections were performed in vivo with each strain bearing control vector pBAC-LacZ. At 48 h postinfection, the kps deletion mutant had a significant defect in virulence relative to the coinfected parental reference strain (Fig. 2A; P = 0.032). Full complementation of the virulence defect for the kps mutant was observed when the region I kps genes were provided in trans in UTI89 Δkps attHK022::COM-GFP (Fig. 2B; P = 0.76), demonstrating that the loss of virulence in the kps mutant is directly attributable to the capsule defect and the interruption or deletion of the region I genes results in a loss of virulence in vivo.
FIG. 2.

Complementation of a region I kps deletion restores virulence. Competitive infections were performed with WT strain UTI89 attλ::PSSH10-1 (spectinomycin resistant) and the isogenic region I capsule deletion mutant UTI89 Δkps attHK::COM-GFP (kanamycin resistant; GFP expressing) for 48 h in C3H/HEN (TLR4+) mice. (A) Each strain carried the control vector pBAC-LacZ (designated VC). (B) UTI89 attλ::PSSH10-1 (WT) carries control vector pBAC-LacZ, and UTI89 Δkps attHK::COM-GFP carries complementing vector pBAC-LAC RegI kps, containing the entire region I capsule gene cluster under the control of its native promoter (designated C). A total of 10 mice were infected. Horizontal bars represent the geometric means. Statistical comparisons were performed using the Mann-Whitney nonparametric test.
The K1 assembly mutant has a modest intracellular proliferation defect during early cystitis.
We hypothesized that one mechanism for the loss of fitness by the capsule assembly mutant may be due to altered adherence and/or invasion. To determine if the loss of capsule modified acute invasion and intracellular proliferation, resulting in an early loss of fitness in vivo, we quantified the intracellular UPEC population produced by the WT or unencapsulated bacteria at 1 and 6 h p.i. in C3H/HeN mice. We approached our hypothesis by comparing two different capsule mutants: a region I assembly gene deletion mutant (Δkps) and a region II synthesis gene deletion mutant (Δneu). Both mutants lacked whole-cell agglutination with K1 antiserum and were resistant to killing by the K1-specific bacteriophage K1F, indicating that they lacked surface presentation of capsule (Fig. 3A). However, unlike the synthesis mutant, the assembly mutant accumulates intracellular polysaccharide, as shown by whole-cell radial immunodiffusion (Fig. 3B), which may affect adherence, invasion, or intracellular proliferation, independently of the loss of extracellular capsule, possibly due to cellular stress or alterations in monosaccharide metabolism. Mice were transurethrally infected with the WT strain, the Δkps mutant strain, or the Δneu mutant strain, each carrying control vector pBR329 in trans. An additional group of mice was infected with the Δkps mutant strain complemented with kpsFEDUCS on plasmid pSX50. At each endpoint, the infected bladders were procured, washed with PBS to enumerate the extracellular bacteria, and then treated with gentamicin ex vivo to specifically eliminate any remaining extracellular bacteria. The remaining intracellular bacteria were subsequently enumerated by plating a gentamicin-free homogenate of the bladder tissue. Within 1 h of infection, there were no significant differences in the extracellular and intracellular numbers of bacteria of any of the strains (Fig. 4A), suggesting that initial adherence and invasion were not significantly altered by the loss of capsule, regardless of the underlying mutation. At 6 h of infection, a significant decrease in intracellular proliferation was observed in the Δkps mutant strain that was not present in the Δneu mutant strain (Fig. 4B). These data suggest that extracellular capsule per se does not alter intracellular proliferation over the first 6 h of infection but that disruption of capsule assembly produces a modest independent effect on bacterial replication in the intracellular compartment of the bladder epithelium. Control experiments in vitro did not reveal differences in the growth of the Δkps or Δneu mutant in Luria broth or in the production of type 1 pili that would account for the observed in vivo differences in intracellular proliferation. One additional observation is that at 6 h of infection, the total number of bacteria of all of the strains per bladder in the ex vivo gentamicin assays (intracellular plus extracellular) was lower than the total bacterial counts enumerated in the mouse infections described in Fig. 1. This may reflect slightly delayed growth kinetics due to the presence of plasmid vectors in the ex vivo gentamicin experiments.
FIG. 3.
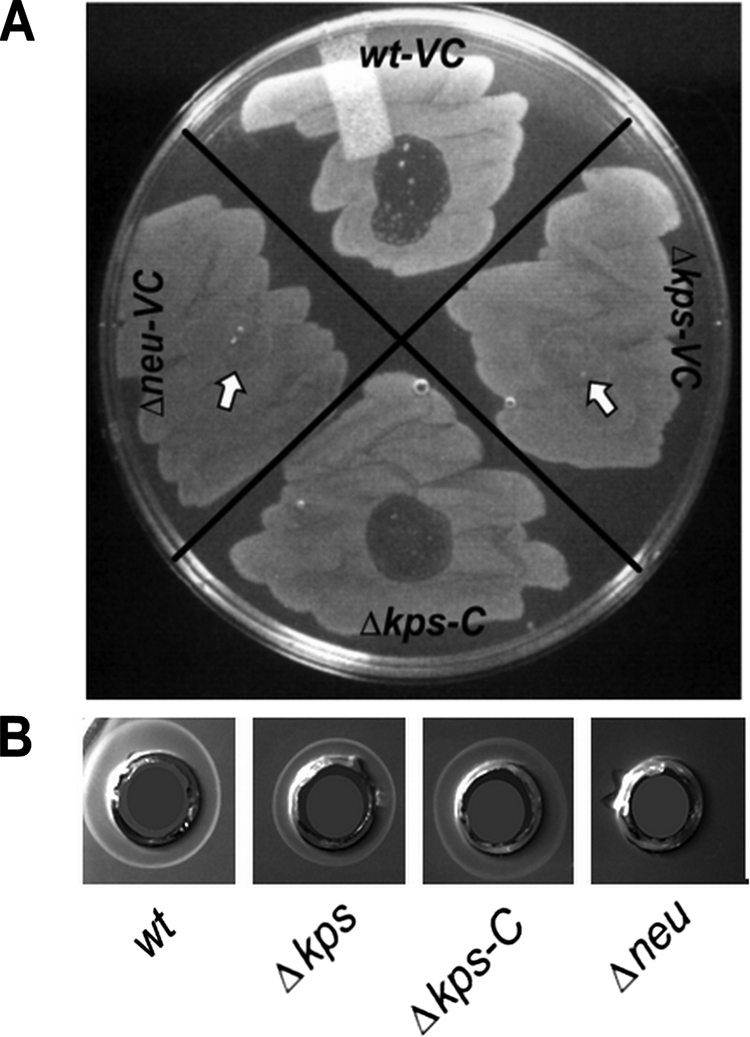
Phage sensitivity and radial immunodiffusion of capsule synthesis and assembly mutants. (A) Lawns of each strain grown on L agar were exposed to a spot of fresh K1F phage lysate and incubated for ∼6 h at 37°C. Phage sensitivity, indicating fully surface-assembled K1 capsule, is evident by lytic zones. Arrows indicate where phage was spotted on nonlysed bacterial lawns. VC = vector control, pBR329. C = complement, pSX50. Identical results were obtained for VC = pBAC-LacZ and C = pBAC-LacZ RegI kps (data not shown). (B) Radial immunodiffusion plates containing K1-reactive polyclonal horse serum. Whole-cell lysates of each strain indicated were distributed into the respective wells. VC = vector control, pBR329. C = complement, pSX50.
FIG. 4.

Adherence, invasion, and early intracellular replication by K1 capsule mutants. Gentamicin protection assays were performed ex vivo on previously infected bladders of C3H/HeN mice at 1 and 6 h (panels A and B, respectively). Δkps-C designates UTI89 Δkps carrying the region I genes on pSX50. The remaining strains carried the control vector pBR329. Bars indicate the median values for the group, and P values were calculated by the Mann-Whitney nonparametric test. P > 0.1 for all comparisons at 6 h (panel B), except where indicated otherwise.
Intracellular collections of the K1 capsule assembly mutant are readily penetrated by neutrophils.
The fitness experiments over 2 weeks of infection (Fig. 1) suggested that K1 capsule confers an advantage on UTI89 from TLR4-dependent and TLR4-independent host responses. To explore these effects further, we first performed histological analyses of mouse bladders infected with the WT and Δkps mutant strains. We have previously proposed that IBC formation protects UPEC in the urinary tract, shielding the community from host defenses and environmental stressors (2, 31). The intracellular location of the bacteria is a first-line defense against neutrophil killing, but it has been shown that neutrophils can selectively migrate to infected cells and surround the IBC (31). Histological examination of bladder sections from C3H/HeN mice infected for 16 h with the WT strain showed numerous stereotypic IBC in the bladder epithelium (Fig. 5A). In these bladders, neutrophils were found closely associated with the periphery of the IBC between the superficial and transitional epithelium, uplifting the associated IBC. These observations support our previous report showing that normal IBC maturation produces a biomass that resists neutrophil infiltration and penetration (31). In contrast, only disorganized collections of the Δkps mutant strain were identified that had neutrophils located throughout the bacterial population (Fig. 5B). The disorganized collections of the Δkps mutant strain were difficult to identify; however, in three bladders infected with the Δkps mutant strain, five independent nonadjacent sections were found with neutrophils penetrating the bacterial mass while in three bladders infected with WT bacteria, 12 IBC had neutrophils only at the periphery. Thus, the collections of bacteria formed by the Δkps mutant strain were more likely to be penetrated by neutrophils. We also observed that the Δkps mutant strain occasionally became filamentous during this neutrophil penetration, although this appears to be a general effect of neutrophil contact with UPEC rather than a result of loss of capsule (31, 32).
FIG. 5.
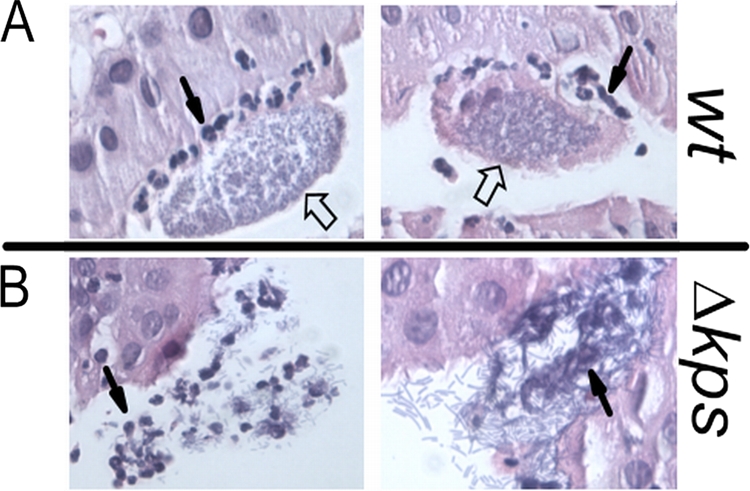
Neutrophil infiltration of IBC formed by WT and isogenic acapsular UPEC. Two representative sections of IBC formed by the WT or unencapsulated UTI89 Δkps at 12 h postinfection in C3H/HeN mice. Each is visualized under ×40 magnification and by hematoxylin-eosin staining. Closed arrows indicate representative neutrophils. Open arrows indicate characteristic UPEC-containing IBC.
The K1 capsule assembly mutant fails to produce multiple generations of IBC-like structures.
Based on the observation that neutrophils more readily penetrated the intracellular collections of the Δkps mutant, we hypothesized that the Δkps mutant forms aberrant community structures compared to WT IBC. As mentioned earlier, WT IBC are biofilm-like structures known to contain bacteria producing surface-associated fibers such as type 1 pili, aggregation factors such as antigen 43, and previously uncharacterized polysaccharides (2). In vitro studies on artificial surfaces have demonstrated roles for multiple different proteins and sugar-based molecules in biofilm formation (reviewed in references 25, 55, and 62). Therefore, we surmised that the polysaccharide capsule of UPEC might provide matrix constituents necessary for the appropriate development and maintenance of IBC.
We postulated that the failure of UPEC to produce K1 capsule would result in decreased aggregation of the intracellular bacteria. We first sought to determine if capsule expression occurred during WT IBC formation. As a surrogate measure of expression, we used a single-copy chromosomal transcriptional fusion of the UTI89 K1 capsule region I promoter to gfp. This fusion was integrated into the att λ site as described in Materials and Methods. We found that the capsule genes were expressed throughout IBC formation by examining 6- and 16-h samples (Fig. 6A). Fusion expression was demonstrated throughout the IBC structures, but the absolute expression from the region I promoter appeared variable on a cell-to-cell basis.
FIG. 6.

Expression of capsule during IBC formation and contribution to IBC morphogenesis. (A) kps-gfp expression during IBC formation in WT UPEC. Infected bladders from female C3H/HEN mice infected for 6 h were counterstained with TO-PRO-3 (red) and imaged in whole mount by confocal microscopy. Representative images are shown. Scale bar = 50 μm. (B) IBC morphologies of UTI89 (WT) and UTI89 kpsF:: EP185 during bladder infection (C3H/HEN) from 6 h to 2 weeks. Bladders were counterstained with wheat germ agglutinin-Alexa 594. Arrows indicate microclusters of bacteria. Scale bar = 50 μm. (C) Magnified view of bacterial grouping (C3H/HEN mice) and morphology within intracellular collections of WT and kpsF:: EP185 mutant strains at 6 and 16 h p.i. Two representative images are shown for each strain at each time point. Scale bar = 10 μm. (D) Coinfection of bladders of female C3H/HEN mice with the GFP-expressing WT and the non-GFP expressing Δkps mutant at 6 h p.i. Representative images were taken in whole mount by confocal microscopy following counterstaining with TO-PRO-3. IBC formed by the WT is yellow (open arrows), and Δkps mutant bacteria are red (closed arrows). (E) Mixed IBC in the bladders during WT and Δkps mutant coinfection of female C3H/HEN mice at 6 h p.i. The Δkps mutant is green; the WT is red. Three representative examples are shown.
We next examined the morphologies of IBC generated by the WT strain or acapsular derivatives over the first 48 h of infection. To improve tracking and visualization of the strains, each carried a plasmid producing constitutive GFP expression. For the WT strain, tightly aggregative bacterial formations consistent with IBC were the predominant organizations of bacteria observed over the first 48 h in C3H/HeN (TLR4+) mice (Fig. 6B and C). In contrast, the kpsF::EP185 strain produced dispersed, irregular intracytoplasmic collections of bacteria (Fig. 6B and C). The density and volumes of the WT and kpsF::EP185 mutant bacterial collections were quantified by capturing complete Z stacks of each strain within infected murine bladder epithelium and analyzing the geometries of each using Volocity software as described in Materials and Methods. The relative mean volumes of the WT and kpsF::EP185 mutant bacterial collections were 2,370 ± 837 μm3GFP (n = 9) and 420 ± 343 μm3GFP (n = 12), respectively (P < 0.0001 by unpaired two-tailed t test). The relative mean density of WT IBC at 6 h p.i. was calculated as 0.24 ± 0.05 (n = 4) versus 0.13 ± 0.07 (μm3GFP/μm3TOTAL) for kpsF::EP185 (P = 0.04 by unpaired two-tailed t test).
Several characteristics of IBC have been described that are consistent with a biofilm-like community. IBC contain extracellular matrix and the autoaggregative factor Ag43 (2). Furthermore, the bacteria within IBC undergo characteristic morphological changes seen in biofilm communities on abiotic surfaces (31). Microscopic studies of the WT and assembly mutant strains revealed significant differences in the morphological changes among bacteria proliferating within the bladder epithelium. WT bacteria within IBC underwent stereotypical changes from bacillar to coccoid morphologies (31), while the capsule mutant consistently appeared as bacilli, even among large collections of cytoplasmic bacteria (Fig. 6C). These observations provide additional evidence that interruption of capsule assembly, in turn, results in a failure to develop the biofilm-like state found within IBC.
UPEC proliferates rapidly during acute cystitis in IBC formations, and failure to produce IBC, disperse from IBC, or undergo multiple rounds of IBC formation results in attenuation in the associated infections (32, 72). By 24 h postinfection, kpsF::EP185 mutant bacteria appeared mostly in microclusters, defined as discrete collections of two to six closely associated bacteria contained within an epithelial cell (Fig. 6B). In contrast, IBC contained >104 bacteria with >250 bacteria in the central Z-stack slice of the formation, as imaged by confocal microscopy. Furthermore, bacteria in the microcluster formations were predominantly bacilli, while the bacteria at the core of the IBC appeared as cocci, reflecting the developmental changes that occur during IBC formation. The intracellular location of the microclusters, like that of IBC, was determined by localizing the bacterial formations beneath the apical surface of the superficial bladder epithelium stained with fluorescent wheat germ agglutinin. The differences in numbers of microclusters observed for the WT and kpsF::EP185 mutant strains were quantified at 24 h and 48 h p.i. in a blinded study (Table 3). These data represent the percentage of bacteria organized in microclusters versus total intracellular bacterial organizations (IBC, dispersed intracellular bacteria, and microclusters). The high percentage of microclusters produced by the kpsF::EP185 mutant strain between 24 h and 48 h was significantly different from that produced by the WT (P < 0.0001) and correlated with the time frame in which this mutant strain, relative to the WT parent strain, had a loss of fitness in the C3H/HeJ (TLR4−) mouse (previously shown in Fig. 1C). Thus, during 24 to 48 h of infection, the formation of low-density microclusters by the K1 assembly mutant relative to the high-density IBC formations made by WT bacteria likely contributes to the overall loss of bacterial burden, even in the TLR4 mutant mouse.
TABLE 3.
Quantitation of microcluster formation during UTI
| Time (h) and strain | Microclusters as % of totala (no. counted) |
|---|---|
| 24 | |
| WT | 0 (100) |
| kpsF::EP185 | 72 (89) |
| 48 | |
| WT | 35 (66) |
| kpsF::EP185 | 79 (34) |
Total = microclusters plus IBC. P < 0.0001 between the WT and the kpsF::EP185 mutant at 24 and 48 h by Fisher's exact test.
The intracellular phenotype of the K1 capsule mutant was also confirmed using the Δkps region I deletion strain. As a direct comparison of the intracellular phenotype of the Δkps mutant strain to that of the WT, a coinfection experiment was performed with equal proportions of the two strains. To differentiate the strains, the WT strain produced GFP constitutively, while the Δkps mutant bacteria were unmarked. Bladder tissue from infected mice was recovered at 6 h p.i. and counterstained with the fluorescent DNA-intercalating dye TO-PRO-3, thus labeling the WT bacteria yellow and the Δkps mutant red. A representative three-dimensional reconstruction of a confocal Z stack is shown in Fig. 6D. We found that while the WT strain formed IBC, the Δkps mutant strain was dispersed in the cytoplasm, lacking the aggregation typical of IBC.
During the coinfection experiments, we also observed examples of interbacterial complementation. Bladders from mice infected for 6 h with an equal mixture of the WT and GFP-expressing Δkps mutant strains produced unified IBC (Fig. 6E). In epithelial cells infected with Δkps mutant bacteria alone, the dispersed cytoplasmic phenotype was still evident, as was the aggregative IBC phenotype of the WT strain when it solely infected target epithelial cells (data not shown). However, some rare IBC were identified with adjoined WT and Δkps mutant bacteria and appeared to have a core of WT bacteria with Δkps mutant bacteria peripherally localized (n = 5 in three bladders; Fig. 6C). All Δkps mutant bacteria within epithelial cells coinfected with the WT strain appeared to be incorporated into the adjoined community, rather than dispersed in the cytoplasm, suggesting that the mixed community may have arisen following an early intersection between the coinfecting strains. Thus, WT bacteria appear to be able to complement the defect of the Δkps mutant within an individual IBC.
One hypothesis to explain the lack of tight intracellular aggregations for the Δkps mutant is that the loss of the K1 capsule results in the exposure of an antiaggregation factor. While additional candidates may be postulated, we hypothesized that CdiA, a known contact-dependent inhibition factor that inhibits biofilm formation (4), may be unmasked by the loss of capsule. Using a genetic approach, we examined if CdiA, exposed by the loss of encapsulation, inhibited IBC formation and promoted intracellular dispersal of Δkps. Contrary to our hypothesis, Δkps ΔcdiA mutant bacteria did not form IBC at 6 h postinfection (Fig. 7A). However, we did note that the ΔcdiA strain formed large IBC, suggesting that CdiA may be expressed within WT IBC, restricting community growth and development (Fig. 7B).
FIG. 7.

CdiA is not responsible for failure of capsule assembly mutants to form IBC. (A) Comparative IBC formation at 6 h postinfection (C3H/HEN mice) by capsule mutants and cdiA mutants in C3H/HeN mice. IBC were enumerated by Lac staining (see Materials and Methods). The horizontal bars represent the median values. P value was determined by the nonparametric Mann-Whitney test. (B) Representative images of Lac-stained IBC derived from UTI89 (WT) and the UTI89 ΔcdiA mutant at 6 h postinfection. Arrows indicate IBC present in the representative images. Each image was taken at the same magnification.
K1 capsule is a central determinant of IBC formation, and NanR-sialic acid dysregulation exacerbates the IBC defect of the K1 assembly-deficient Δkps mutant.
As mentioned earlier, loss of the K1 region I capsule assembly genes (such as in the Δkps mutant strain) results in intracellular accumulation of polysialic acid (68). In contrast, K1 region II synthesis mutants (such as the Δneu mutant strain) do not synthesize sialic acid or accumulate intracellular polysaccharide. Thus, the two types of mutants share the acapsular phenotype, while the assembly mutant has the additional phenotype of intracellular accumulation of polysaccharide and the potential for altered intracellular sialic acid pools. We hypothesized that the assembly (Δkps) and synthesis (Δneu) mutants would produce significantly fewer IBC than the encapsulated WT strain due to the loss of extracellular polysaccharide production. However, we also hypothesized that the assembly mutant (Δkps) would have a more profound IBC defect due to the accumulation of intracellular sialic acid, resulting in altered regulation by the sialic acid-sensitive regulator NanR (34).
As predicted, both the Δkps and Δneu mutants had severe defects in IBC formation (Fig. 8). C3H/HEN mice were inoculated with the respective strains by transurethral catheterization, and after 6 h of infection, the resected bladders were fixed and Lac stained to identify IBC formations. IBC were not identified in bladders infected with the Δkps mutant (Fig. 8A). To ensure that the loss of IBC formation by the Δkps mutant was attributable to the loss of capsule assembly, we complemented the Δkps mutant strain with pBAC-LacZ RegI kps, containing the K1 capsule region I gene cluster. Complementation produced a complete restoration of IBC formation (Fig. 8A; P = 0.25, WT versus Δkps/kps mutant). Transverse histochemical sections demonstrated that the complemented Δkps mutant strain produced IBC like the WT strain and notably different than the dispersed intracellular organization of the uncomplemented strain (Fig. 8B). In contrast to the Δkps mutant strain, which accumulates intracellular sialic acid, we found that the Δneu mutant strain, which does not synthesize sialic acid and makes no K1 capsule, produced rare IBC, although the number of IBC of the Δneu mutant was not found to be significantly different from the number of IBC of the Δkps mutant. The rare IBC formed by Δneu mutant bacteria appeared grossly similar to those produced by WT bacteria (Fig. 8B). Microscopy did not successfully localize the Δneu mutant strain to other parts of the epithelium, although the 6-h ex vivo gentamicin assays suggest that Δneu mutant bacteria are likely widely distributed within the bladder epithelium at 6 h p.i. (Fig. 4B).
FIG. 8.

Mutation of nanR partially restores IBC formation in the capsule assembly mutant. (A) IBC formation was determined for each UTI89 derivative by whole-mount Lac staining at 6 h postinfection in female C3H/HeN mice. Horizontal bars indicate median values. P values are shown as calculated by the Mann-Whitney test in comparison to UTI89 Δkps. Complementation in trans was performed with pBAC-LAC RegI kps and pBAC-LAC nanR. (B) Hematoxylin-eosin-stained 8-μm sections of infected C3H/HEN bladders demonstrating IBC formation. Images were captured at ×400 magnification. Large filled arrows indicate IBC. Open arrows indicate apical epithelial cell nuclei. Thin black arrows indicate dispersed bacteria. A capital T indicates the transitional epithelium layer.
We next addressed our hypothesis that accumulation of intracellular sialic acid as a consequence of interrupting capsule assembly in the Δkps mutant strain resulted in NanR dysregulation and exacerbation of the IBC defect. NanR is a sialic acid-sensitive regulator in E. coli (34). When it is not engaged with sialic acid, NanR represses the nanATEK-yjhK operon, which encodes the sialic acid lyase, inner membrane sialic acid transporter, and other sialic acid metabolism accessory factors (34). NanR similarly represses the expression of nanC, which encodes a major sialic acid-importing outer membrane pore (14). In UPEC K5 grown in the absence of sialic acid, NanR was shown to activate fimB expression, which in turn promotes phase ON variation of type 1 pilus expression (59). The addition of sialic acid resulted in fimB repression, most likely by binding and inducing NanR to release the fimB promoter. Thus, NanR has been shown to inhibit and activate gene expression in response to sialic acid, depending upon the specific target. However, we found that the intracellular defect of the capsule mutant was not due to dysregulation of type 1 pili, as we did not find a difference in type 1 pilus expression during in vitro growth when comparing the Δkps and Δneu mutant strains to the isogenic parental strain and measuring phase PCR, anti-type 1 pilus immunoblots, and mannose-sensitive yeast agglutination (data not shown).
Considering the intracellular accumulation of sialic acid in the Δkps mutant strain (Fig. 3B), we anticipated that deleting nanR in the Δkps mutant strain might relieve the regulatory consequences of imbalances in sialic acid pools and partially restore IBC development. We constructed a Δkps ΔnanR double-mutant strain and compared its capacity to form IBC relative to those of the WT and the other capsule mutants. The Δkps ΔnanR double mutant had a partially restored IBC phenotype (P = 0.023 for the Δkps mutant strain versus the Δkps ΔnanR double-mutant strain), suggesting that altered sialic acid-dependent regulation contributed to the Δkps IBC defect (Fig. 8A). Furthermore, the introduction of nanR in trans into the Δkps ΔnanR double-mutant strain abrogated the detection of IBC in whole-mount bladders by Lac staining (Fig. 8A). We did not anticipate full restoration of IBC formation in the Δkps ΔnanR double-mutant strain due to the absence of K1 capsule. Together, these data demonstrate that K1 polysaccharide capsule is important in IBC formation and that imbalances in sialic acid trafficking and metabolism may disrupt stereotypic IBC formation. It will be interesting to further investigate the molecular details of NanR-mediated capsule regulation in future experiments.
DISCUSSION
Encapsulation of bacterial pathogens with polysaccharide capsules is a well-established theme in bacterial virulence. The assembled capsules have many recognized functions, including masking of surface-associated antigens, inhibition of complement-mediated killing, and molecular mimicry (29, 52, 71). K1 capsule has a known role in evading neutrophil-related killing through inhibition of phagocytosis (70). Because neutrophil function is considered a central inflammatory defense in UTIs, K1 encapsulation would be expected to serve as a primary protection for UPEC. Molecular epidemiology and serologic studies show that there is a high prevalence of group 2 capsules (29), and the assembly and export mechanisms conserved among group 2 encapsulated UPEC strains may be the target for chemical inhibitors, thus representing a clinically important therapeutic opportunity. The K1 serotype is particularly prevalent among UPEC strains, and its importance in UTI pathogenesis is evident from the dramatic attenuation in numbers that unencapsulated strains suffer in vivo. Neutrophil recruitment occurs at 12 to 16 h postinfection in our murine model of cystitis, and unencapsulated strains experienced a significant loss of fitness during this time period and at times thereafter (Fig. 1). Complementation of the kps mutant confirmed that the defect is due to the loss of capsule (Fig. 2), and it is possible that capsule shields bacterial membrane molecules from components of the host innate immune response. One further observation is that the coinfection of the WT strain with the capsule assembly mutant resulted in lower CFU counts for the WT strain than when the WT strain was coinfected with the complemented capsule assembly mutant (Fig. 2). This may reflect greater induction of the innate immune response by the unencapsulated strain, which in turn predisposes the WT strain to heightened immune killing. Surprisingly, a marked loss of fitness was observed in TLR4-deficient mice (C3H/HEJ) that fail to significantly recruit neutrophils into the bladder (31); the region I capsule assembly mutants retained a virulence defect compared to the WT strain (Fig. 1), suggesting alternative roles for encapsulation or capsule-dependent carbohydrate metabolism in UPEC virulence.
It is also possible that loss of fitness of the K1 capsule mutant during cystitis is the result of greater susceptibility to the complement system. However, while complement-mediated clearance appears to be important in the upper urinary tract (39, 42, 65), the extent to which complement contributes to bacterial clearance in the bladder remains questionable (38). While we cannot exclude a role for the complement system in eliminating the unencapsulated strain during cystitis, the TLR4-independent loss of fitness of the UPEC K1 capsule mutants may be more attributable to other defects, such as a failure to undergo repeated rounds of intracellular replication in IBC.
We hypothesized that polysaccharide would be involved in the aggregative phenotype of UPEC in IBC formation and that the loss of aggregation would impact the ability of UPEC to proliferate intracellularly. Interruption of capsule assembly (Δkps) resulted in a significant but small loss of intracellular proliferation during the first 6 h of infection. The defect was specific to interrupted capsule assembly and not necessarily the presence or absence of capsule, since the capsule synthesis mutant (Δneu) did not demonstrate the same defect (Fig. 4). However, the two most striking alterations in the in vivo capsule phenotype over the first 48 h of infection were (i) the failure to form IBC following the first cycle of adherence and invasion of the apical bladder epithelium and (ii) the failure to undergo high-density intracellular replication during later stages of infection when the WT strain continued to form serial generations of IBC. In contrast to the aggregative intracellular IBC phenotype of the WT strain, the capsule mutant had intracellular proliferation in unorganized collections that did not proceed through the IBC developmental stages (Fig. 6). This intracellular defect is strikingly different than the phenotype we previously described for intracellular UPEC unable to express type 1 pili (72). In that previous study, intracellular collections of bacteria were rarely observed but the phenotype of the rare intracellular bacterial collections that were identified resembled that of the capsular mutant in that they were unable to aggregate into IBC. Thus, type 1 pili may be necessary both in the early nucleation phase of the community and during IBC development, while the polysaccharide capsule has a greater role in IBC development, promoting the aggregative phenotype of the biomasses. Despite the ability of the capsule assembly mutant to invade and replicate efficiently in the bladder epithelium early in cystitis, the failure to organize into IBC was accompanied by neutrophil infiltration of the biomasses (Fig. 5). Thus, these data suggest that while some K capsule types may have a one-on-one role in defending individual bacteria from phagocytosis by neutrophils, the polysaccharide also has a role in facilitating biofilms that, in turn, preclude phagocyte penetration.
The loss of capsule resulted in significantly decreased bacterial numbers at later time points (Fig. 1 and 2). A prior study using real-time video microscopy showed that after invasion, individual WT bacteria replicate rapidly into protected communities of bacteria that disperse and reenter neighboring cells through apical membrane invasion (31). The bacteria form additional IBC in these neighboring cells, resulting in high densities of organisms during the first several days of infection. The later generations of high-density intracellular infection appeared to be absent in the capsular mutant strains, replaced by the formation of microclusters, i.e., low-density groupings of intracellular bacteria (Fig. 6). In our present work, we cannot specifically define the point of this defect. One possibility is that the Δkps mutant invades neighboring cells during second-round IBC formation through a different pathway than the WT strain or that the mutant remains contained within endosomes, while the WT organism continues to escape into the cytoplasm. Kim et al. have previously shown that K1 capsule prevents lysosomal fusion in human brain microvascular cells (35). In the transitional cells of the bladder epithelium, the capsule mutant might remain trapped, in a viable state, within endosomes. Given that over 105 bacteria may reside in each fully matured IBC, relatively subtle defects in UPEC in endosome escape or IBC nucleation may be expected to have significant effects on decreased numbers of IBC and thus the overall burden of infection.
A recent study showed that the K2 capsule is also a virulence determinant in the pathogenesis of UTI (10). The effect of the K2 synthesis gene deletion on the persistence of pyelonephritis isolate CFT073 in the urinary tract was the most pronounced in the urine and kidneys. However, the capsule mutant had reduced bladder CFU counts, approaching statistical significance, while complementation of the capsule mutant with a low-copy-number plasmid resulted in a significant advantage during a competitive infection with an uncomplemented isogenic strain, indicating that the K2 capsule conferred a gain in virulence. The observed differences in the relative importance of the K2 and K1 capsules to the respective clinical isolates CFT073 and UTI89 during lower UTI may arise from one of several differences. Since the study by Buckles et al. does not describe tissue pathology, we cannot determine if IBC were present or absent or, if they were present, the morphology of the bacterial structures in the absence of the K2 capsule. A prior study demonstrated that CFT073 does form aggregative IBC structures (23). One plausible explanation is that CFT073 expresses alternative matrix components to capsular polysaccharides during IBC formation. Indeed, genetic comparisons of CFT073 and UTI89 reveal a great deal of diversity (12). Alternatively, the two strains may have different requirements for the subversion of the innate immune response. The study by Buckles et al. used only a fully immunocompetent host and evaluated fitness at a single time point of infection (48 h). Thus, conclusions cannot be drawn about how well the K2 mutant derivative of CFT073 would survive in a host with a compromised innate response or what events occurred early or late in disease.
In the case of UTI89, the K1 cystitis isolate, the striking disorganization of the intracellular bacteria that resulted from interrupting capsule assembly suggested that loss of capsule results from altered expression of a surface factor involved in bacterial aggregation. Intriguingly, mutation of cdiA, which codes for an antiaggregation factor (4), resulted in large IBC (Fig. 7B), suggesting that CdiA may be normally expressed to some degree during WT IBC formation and may inhibit or restrain IBC growth. However, mutation of cdiA in the Δkps mutant background provided no restoration of IBC formation (Fig. 7), suggesting that interruption of capsule biogenesis results in a more fundamental defect and not simply an unmasking of CdiA.
A further hypothesis to explain the aberrant morphology of the Δkps mutant strain is that the disruption of capsule assembly created an accumulation of intracellular sialic acid polymer and monomer. Indeed, we observed the presence of sialic acid intermediates in the Δkps mutant strain even though this material was not displayed on the bacterial surface (Fig. 3). This accumulation might lead to altered signaling through the sialic acid-dependent regulator NanR. In prior work using E. coli K-12 derivatives, Ringenberg et al. suggested that without coexistent mutations in nanA, which codes for a sialate aldolase that converts sialic acid to N-acetyl-d-mannosamine, free sialic acid does not significantly accumulate in the cytoplasm, even with disruption of K1 capsule assembly (51). However, this is contingent on free sialic acid inactivating NanR, thus derepressing the nanATEK operon and expressing NanA. We postulated that, prior to the induction of nanA expression, small amounts of free sialic acid could interact with NanR and have significant consequences on the NanR-sialic acid-dependent regulon, perturbing biofilm formation through as-yet-undescribed components of the regulon. A nanR deletion was able to partially restore IBC formation by the Δkps mutant and complementation of the nanR deletion in the Δkps background abrogated IBC formation, which supports our hypothesis of sialic acid-dependent NanR regulation during IBC formation (Fig. 8). Recently, Alteri et al. demonstrated that NanA is significantly upregulated in human urine, but a NanA mutant of isolate CFT073 did not have a fitness defect in vivo during UTI (1). These experiments may suggest that catabolism of sialic acid is not essential during UTI but leave open questions about the independent roles of sialic acid-dependent sensing and regulation. Further studies are required to address the different phenotypes of clinical UPEC isolates in response to sialic acid. Regardless, given that the unencapsulated K1 strain has defects in both acute and chronic stages of persistence, disrupting sialic acid acquisition, synthesis, or regulation may have significant effects on virulence for high-prevalence strain types such as K1 and might be a target of therapeutic design.
In summary, we have demonstrated a novel role for the K1 polysaccharide capsule of UPEC during acute cystitis and chronic persistence in the bladder. Because IBC formation has been observed in human samples (53), it is likely that our results reported herein hold great clinical relevance. By elucidating factors that contribute to the success of intracellular persistence by UPEC during UTI, new therapeutic targets will come to light. Given the multiple ways in which capsule is important during uropathogenesis and the conservation of group 2 assembly and export, it may be an attractive target for antivirulence chemotherapeutics.
Acknowledgments
We thank Joseph St. Geme for his insightful and critical reading of the manuscript. pSX50 was the kind gift of Eric Vimr. Richard Silver kindly provided the horse group B meningococcal antiserum (H46).
S.J.H. is supported by R01DK51406 and ORWH SCOR P50DK64540. P.C.S. received support from K08DK074443 and ORWH SCOR P50DK064540.
Editor: S. M. Payne
Footnotes
Published ahead of print on 19 January 2010.
REFERENCES
- 1.Alteri, C. J., S. N. Smith, and H. L. Mobley. 2009. Fitness of Escherichia coli during urinary tract infection requires gluconeogenesis and the TCA cycle. PLoS Pathog. 5:e1000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, G. G., J. J. Palermo, J. D. Schilling, R. Roth, J. Heuser, and S. J. Hultgren. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105-107. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, J. B., F. Parivar, G. Lee, T. B. Wallington, A. G. MacIver, R. A. Bradbrook, and J. C. Gingell. 1989. The enigma of interstitial cystitis—an autoimmune disease? Br. J. Urol. 63:58-63. [DOI] [PubMed] [Google Scholar]
- 4.Aoki, S. K., R. Pamma, A. D. Hernday, J. E. Bickham, B. A. Braaten, and D. A. Low. 2005. Contact-dependent inhibition of growth in Escherichia coli. Science 309:1245-1248. [DOI] [PubMed] [Google Scholar]
- 5.Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bahrani-Mougeot, F. K., E. L. Buckles, C. V. Lockatell, J. R. Hebel, D. E. Johnson, C. M. Tang, and M. S. Donnenberg. 2002. Type 1 fimbriae and extracellular polysaccharides are preeminent uropathogenic Escherichia coli virulence determinants in the murine urinary tract. Mol. Microbiol. 45:1079-1093. [DOI] [PubMed] [Google Scholar]
- 7.Bishop, B. L., M. J. Duncan, J. Song, G. Li, D. Zaas, and S. N. Abraham. 2007. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat. Med. 13:625-630. [DOI] [PubMed] [Google Scholar]
- 8.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 9.Boulnois, G. J., I. S. Roberts, R. Hodge, K. R. Hardy, K. B. Jann, and K. N. Timmis. 1987. Analysis of the K1 capsule biosynthesis genes of Escherichia coli: definition of three functional regions for capsule production. Mol. Gen. Genet 208:242-246. [DOI] [PubMed] [Google Scholar]
- 10.Buckles, E. L., X. Wang, M. C. Lane, C. V. Lockatell, D. E. Johnson, D. A. Rasko, H. L. Mobley, and M. S. Donnenberg. 2009. Role of the K2 capsule in Escherichia coli urinary tract infection and serum resistance. J. Infect. Dis. 199:1689-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burman, W. J., P. E. Breese, B. E. Murray, K. V. Singh, H. A. Batal, T. D. MacKenzie, J. W. Ogle, M. L. Wilson, R. R. Reves, and P. S. Mehler. 2003. Conventional and molecular epidemiology of trimethoprim-sulfamethoxazole resistance among urinary Escherichia coli isolates. Am. J. Med. 115:358-364. [DOI] [PubMed] [Google Scholar]
- 12.Chen, S. L., C. S. Hung, J. Xu, C. S. Reigstad, V. Magrini, A. Sabo, D. Blasiar, T. Bieri, R. R. Meyer, P. Ozersky, J. R. Armstrong, R. S. Fulton, J. P. Latreille, J. Spieth, T. M. Hooton, E. R. Mardis, S. J. Hultgren, and J. I. Gordon. 2006. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc. Natl. Acad. Sci. U. S. A. 103:5977-5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9-14. [DOI] [PubMed] [Google Scholar]
- 14.Condemine, G., C. Berrier, J. Plumbridge, and A. Ghazi. 2005. Function and expression of an N-acetylneuraminic acid-inducible outer membrane channel in Escherichia coli. J. Bacteriol. 187:1959-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Covarrubias, L., and F. Bolivar. 1982. Construction and characterization of new cloning vehicles. VI. Plasmid pBR329, a new derivative of pBR328 lacking the 482-base-pair inverted duplication. Gene 17:79-89. [DOI] [PubMed] [Google Scholar]
- 16.Danese, P. N., L. A. Pratt, and R. Kolter. 2000. Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J. Bacteriol. 182:3593-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Da Re, S., and J. M. Ghigo. 2006. A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J. Bacteriol. 188:3073-3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duncan, M. J., G. Li, J. S. Shin, J. L. Carson, and S. N. Abraham. 2004. Bacterial penetration of bladder epithelium through lipid rafts. J. Biol. Chem. 279:18944-18951. [DOI] [PubMed] [Google Scholar]
- 21.Eto, D. S., T. A. Jones, J. L. Sundsbak, and M. A. Mulvey. 2007. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog. 3:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foxman, B., R. Barlow, H. D'Arcy, B. Gillespie, and J. D. Sobel. 2000. Urinary tract infection: self-reported incidence and associated costs. Ann. Epidemiol. 10:509-515. [DOI] [PubMed] [Google Scholar]
- 23.Garofalo, C. K., T. M. Hooton, S. M. Martin, W. E. Stamm, J. J. Palermo, J. I. Gordon, and S. J. Hultgren. 2007. Escherichia coli from urine of female patients with urinary tract infections is competent for intracellular bacterial community formation. Infect. Immun. 75:52-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagberg, L., R. Hull, S. Hull, J. R. McGhee, S. M. Michalek, and C. Svanborg Eden. 1984. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect. Immun. 46:839-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall-Stoodley, L., J. W. Costerton, and P. Stoodley. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2:95-108. [DOI] [PubMed] [Google Scholar]
- 26.Hannan, T. J., I. U. Mysorekar, S. L. Chen, J. N. Walker, J. M. Jones, J. S. Pinkner, S. J. Hultgren, and P. C. Seed. 2008. LeuX tRNA-dependent and -independent mechanisms of Escherichia coli pathogenesis in acute cystitis. Mol. Microbiol. 67:116-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hedlund, M., B. Frendeus, C. Wachtler, L. Hang, H. Fischer, and C. Svanborg. 2001. Type 1 fimbriae deliver an LPS- and TLR4-dependent activation signal to CD14-negative cells. Mol. Microbiol. 39:542-552. [DOI] [PubMed] [Google Scholar]
- 28.Iwahi, T., Y. Abe, M. Nakao, A. Imada, and K. Tsuchiya. 1983. Role of type 1 fimbriae in the pathogenesis of ascending urinary tract infection induced by Escherichia coli in mice. Infect. Immun. 39:1307-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson, J. R. 1991. Virulence factors in Escherichia coli urinary tract infection. Clin. Microbiol. Rev. 4:80-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson, L., A. Sabel, W. J. Burman, R. M. Everhart, M. Rome, T. D. MacKenzie, J. Rozwadowski, P. S. Mehler, and C. S. Price. 2008. Emergence of fluoroquinolone resistance in outpatient urinary Escherichia coli isolates. Am. J. Med. 121:876-884. [DOI] [PubMed] [Google Scholar]
- 31.Justice, S. S., C. Hung, J. A. Theriot, D. A. Fletcher, G. G. Anderson, M. J. Footer, and S. J. Hultgren. 2004. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 101:1333-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Justice, S. S., D. A. Hunstad, P. C. Seed, and S. J. Hultgren. 2006. Filamentation by Escherichia coli subverts innate defenses during urinary tract infection. Proc. Natl. Acad. Sci. U. S. A. 103:19884-19889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Justice, S. S., S. R. Lauer, S. J. Hultgren, and D. A. Hunstad. 2006. Maturation of intracellular Escherichia coli communities requires SurA. Infect. Immun. 74:4793-4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalivoda, K. A., S. M. Steenbergen, E. R. Vimr, and J. Plumbridge. 2003. Regulation of sialic acid catabolism by the DNA binding protein NanR in Escherichia coli. J. Bacteriol. 185:4806-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim, K. J., S. J. Elliott, F. Di Cello, M. F. Stins, and K. S. Kim. 2003. The K1 capsule modulates trafficking of E. coli-containing vacuoles and enhances intracellular bacterial survival in human brain microvascular endothelial cells. Cell Microbiol. 5:245-252. [DOI] [PubMed] [Google Scholar]
- 36.Kim, K. S., H. Itabashi, P. Gemski, J. Sadoff, R. L. Warren, and A. S. Cross. 1992. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Invest. 90:897-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinder, S. A., J. L. Badger, G. O. Bryant, J. C. Pepe, and V. L. Miller. 1993. Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O8 and construction of a transformable R-M+ mutant. Gene 136:271-275. [DOI] [PubMed] [Google Scholar]
- 38.Lannergard, A., G. Friman, and A. Larsson. 2003. Serum amyloid A: a novel serum marker for the detection of systemic inflammatory response in cystitis. J. Urol. 170:804-806. [DOI] [PubMed] [Google Scholar]
- 39.Li, K., S. H. Sacks, and N. S. Sheerin. 2008. The classical complement pathway plays a critical role in the opsonisation of uropathogenic Escherichia coli. Mol. Immunol. 45:954-962. [DOI] [PubMed] [Google Scholar]
- 40.Martinez, J. J., M. A. Mulvey, J. D. Schilling, J. S. Pinkner, and S. J. Hultgren. 2000. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19:2803-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller, V. L., and J. J. Mekalanos. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moffitt, M. C., and M. M. Frank. 1994. Complement resistance in microbes. Springer Semin. Immunopathol. 15:327-344. [DOI] [PubMed] [Google Scholar]
- 43.Mulvey, M. A., Y. S. Lopez-Boado, C. L. Wilson, R. Roth, W. C. Parks, J. Heuser, and S. J. Hultgren. 1998. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282:1494-1497. [DOI] [PubMed] [Google Scholar]
- 44.Mulvey, M. A., J. D. Schilling, and S. J. Hultgren. 2001. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69:4572-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy, K. C., and K. G. Campellone. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagy, G., U. Dobrindt, G. Schneider, A. S. Khan, J. Hacker, and L. Emody. 2002. Loss of regulatory protein RfaH attenuates virulence of uropathogenic Escherichia coli. Infect. Immun. 70:4406-4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Opal, S., A. Cross, and P. Gemski. 1982. K antigen and serum sensitivity of rough Escherichia coli. Infect. Immun. 37:956-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otto, K., and M. Hermansson. 2004. Inactivation of ompX causes increased interactions of type 1 fimbriated Escherichia coli with abiotic surfaces. J. Bacteriol. 186:226-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Platt, R., C. Drescher, S. K. Park, and G. J. Phillips. 2000. Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid 43:12-23. [DOI] [PubMed] [Google Scholar]
- 50.Poltorak, A., I. Smirnova, X. He, M. Y. Liu, C. Van Huffel, O. McNally, D. Birdwell, E. Alejos, M. Silva, X. Du, P. Thompson, E. K. Chan, J. Ledesma, B. Roe, S. Clifton, S. N. Vogel, and B. Beutler. 1998. Genetic and physical mapping of the Lps locus: identification of the Toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol. Dis. 24:340-355. [DOI] [PubMed] [Google Scholar]
- 51.Ringenberg, M., C. Lichtensteiger, and E. Vimr. 2001. Redirection of sialic acid metabolism in genetically engineered Escherichia coli. Glycobiology 11:533-539. [DOI] [PubMed] [Google Scholar]
- 52.Roberts, I. S. 1996. The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 50:285-315. [DOI] [PubMed] [Google Scholar]
- 53.Rosen, D. A., T. M. Hooton, W. E. Stamm, P. A. Humphrey, and S. J. Hultgren. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4:e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuelsson, P., L. Hang, B. Wullt, H. Irjala, and C. Svanborg. 2004. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 72:3179-3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schembri, M. A., M. Givskov, and P. Klemm. 2002. An attractive surface: gram-negative bacterial biofilms. Sci. STKE 2002:re6. [DOI] [PubMed] [Google Scholar]
- 56.Schilling, J. D., S. M. Martin, C. S. Hung, R. G. Lorenz, and S. J. Hultgren. 2003. Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 100:4203-4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schilling, J. D., M. A. Mulvey, C. D. Vincent, R. G. Lorenz, and S. J. Hultgren. 2001. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J. Immunol. 166:1148-1155. [DOI] [PubMed] [Google Scholar]
- 58.Silver, R. P., W. Aaronson, and W. F. Vann. 1988. The K1 capsular polysaccharide of Escherichia coli. Rev. Infect. Dis. 10(Suppl. 2):S282-S286. [DOI] [PubMed] [Google Scholar]
- 59.Sohanpal, B. K., S. El-Labany, M. Lahooti, J. A. Plumbridge, and I. C. Blomfield. 2004. Integrated regulatory responses of fimB to N-acetylneuraminic (sialic) acid and GlcNAc in Escherichia coli K-12. Proc. Natl. Acad. Sci. U. S. A. 101:16322-16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song, J., M. J. Duncan, G. Li, C. Chan, R. Grady, A. Stapleton, and S. N. Abraham. 2007. A novel TLR4-mediated signaling pathway leading to IL-6 responses in human bladder epithelial cells. PLoS Pathog. 3:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steenbergen, S. M., and E. R. Vimr. 1990. Mechanism of polysialic acid chain elongation in Escherichia coli K1. Mol. Microbiol. 4:603-611. [DOI] [PubMed] [Google Scholar]
- 62.Stoodley, P., K. Sauer, D. G. Davies, and J. W. Costerton. 2002. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56:187-209. [DOI] [PubMed] [Google Scholar]
- 63.Sultzer, B. M. 1968. Genetic control of leucocyte responses to endotoxin. Nature 219:1253-1254. [DOI] [PubMed] [Google Scholar]
- 64.Sultzer, B. M., R. Castagna, J. Bandekar, and P. Wong. 1993. Lipopolysaccharide nonresponder cells: the C3H/HeJ defect. Immunobiology 187:257-271. [DOI] [PubMed] [Google Scholar]
- 65.Tang, S., W. Zhou, N. S. Sheerin, R. W. Vaughan, and S. H. Sacks. 1999. Contribution of renal secreted complement C3 to the circulating pool in humans. J. Immunol. 162:4336-4341. [PubMed] [Google Scholar]
- 66.Valdivia, R. H., A. E. Hromockyj, D. Monack, L. Ramakrishnan, and S. Falkow. 1996. Applications for green fluorescent protein (GFP) in the study of host-pathogen interactions. Gene 173:47-52. [DOI] [PubMed] [Google Scholar]
- 67.Valle, J., S. Da Re, N. Henry, T. Fontaine, D. Balestrino, P. Latour-Lambert, and J. M. Ghigo. 2006. Broad-spectrum biofilm inhibition by a secreted bacterial polysaccharide. Proc. Natl. Acad. Sci. U. S. A. 103:12558-12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vimr, E. R., W. Aaronson, and R. P. Silver. 1989. Genetic analysis of chromosomal mutations in the polysialic acid gene cluster of Escherichia coli K1. J. Bacteriol. 171:1106-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang, X., J. F. Preston III, and T. Romeo. 2004. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 186:2724-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weiss, J., M. Victor, A. S. Cross, and P. Elsbach. 1982. Sensitivity of K1-encapsulated Escherichia coli to killing by the bactericidal/permeability-increasing protein of rabbit and human neutrophils. Infect. Immun. 38:1149-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whitfield, C., and I. S. Roberts. 1999. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 31:1307-1319. [DOI] [PubMed] [Google Scholar]
- 72.Wright, K. J., P. C. Seed, and S. J. Hultgren. 2007. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell. Microbiol. 9:2230-2241. [DOI] [PubMed] [Google Scholar]
- 73.Wright, K. J., P. C. Seed, and S. J. Hultgren. 2005. Uropathogenic Escherichia coli flagella aid in efficient urinary tract colonization. Infect. Immun. 73:7657-7668. [DOI] [PMC free article] [PubMed] [Google Scholar]