

Abstract
As an etiological agent of bacterial sepsis and wound infections, Vibrio vulnificus is unique among the Vibrionaceae. Its continued environmental persistence and transmission are bolstered by its ability to colonize shellfish, form biofilms on various marine biotic surfaces, and generate a morphologically and physiologically distinct rugose (R) variant that yields profuse biofilms. Here, we identify a c-di-GMP-regulated locus (brp, for biofilm and rugose polysaccharide) and two transcription factors (BrpR and BrpT) that regulate these physiological responses. Disruption of glycosyltransferases within the locus or either regulator abated the inducing effect of c-di-GMP on biofilm formation, rugosity, and stress resistance. The same lesions, or depletion of intracellular c-di-GMP levels, abrogated these phenotypes in the R variant. The parental and brp mutant strains formed only scant monolayers on glass surfaces and oyster shells, and although the R variant formed expansive biofilms, these were of limited depth. Dramatic vertical expansion of the biofilm structure was observed in the parental strain and R variant, but not the brp mutants, when intracellular c-di-GMP levels were elevated. Hence, the brp-encoded polysaccharide is important for surface colonization and stress resistance in V. vulnificus, and its expression may control how the bacteria switch from a planktonic lifestyle to colonizing shellfish to invading human tissue.
In nature, the majority of microorganisms live in biofilm communities that are attached to a surface (49, 58, 71). Biofilms provide a stable, protective milieu for growth and a means to facilitate transmission by acting as a source for the dissemination of large numbers of microorganisms (30, 43, 84). Compared with their planktonic counterparts, biofilm consortia are extraordinarily resistant to antibiotics, biocides, and the immune defense responses of the host.
Biofilm formation has been described as a stepwise process (74). Flagellar motility is crucial for approaching the surface (58), whereas pili are required for surface colonization and microcolony formation (40, 65, 70). Once formed, these microcolonies can develop distinct three-dimensional architecture, consisting of tower- and mushroom-shaped microcolonies sheathed in a hydrated matrix of exopolymeric substances that are composed of polysaccharides, nucleic acids, and proteins that are produced by the resident microorganisms (20, 77).
The polysaccharide components of the biofilm matrix have been the subject of intense study. Polysaccharides are composed of monosaccharides joined by glycosidic linkages (86). In general, they are synthesized and assembled by a series of proteins (glycosyltransferases [GT]) that are encoded by genes clustered in specific biosynthesis loci on the chromosome. Polysaccharides often constitute the outermost layer of the bacterial cell. As such, they can mediate direct interactions between the bacterium and its environment. Capsular polysaccharide (CPS) and exopolysaccharide (EPS) are important virulence and adhesion factors for many pathogens. CPS mediates resistance of bacteria to complement-mediated bacteriolysis and phagocytosis (31, 34). EPS possesses both adsorptive and adhesive properties (6, 42, 89). EPS has also been shown to impart a wrinkled, raised look to the normally flat, featureless appearance of wild-type cells. This phenotype, known as rugose (R) colony development, is associated with increased biofilm formation and bacterial survival under various stress conditions (2, 52, 66).
Vibrio species are ubiquitous in aquatic ecosystems, and several species can form pathogenic or symbiotic relationships with eukaryotic hosts (12, 79). The adaptation of Vibrio species to the changing environments of aquatic ecosystems and the colonization of their respective hosts are critical for their long-term survival. As such, biofilm formation and rugose colony formation are expected to play key roles in the ecology and transmission of Vibrio species; however, the genetics and physiological impact of rugosity are not well understood. Vibrio vulnificus is an environmentally successful and potent opportunistic human and animal pathogen (45, 73, 76). The most common routes for V. vulnificus infection are through wounds or the ingestion of contaminated water or food. Unlike other members of the Vibrionaceae, which cause gastroenteritis (8, 12, 62), V. vulnificus is infamous for causing septicemia. The fatality rate of susceptible patients with primary septicemia is greater than 50% (33, 41, 45, 85), and it carries the highest death rate of any food-borne disease agent (82). In marine and estuarine environments, the bacterium associates asymptomatically with filter feeders, such as oysters (32, 88), and has been found to form biofilms on the surface of plankton, algae, fish, and eels (7, 46, 51). Three morphologically distinct phase variants, called opaque (O), translucent (T), and rugose (R), arise in response to environmental stresses (83). O variants produce copious amounts of CPS, whereas T variants produce little or no CPS. The rugose phenotype was shown to enhance biofilm formation, and the R variant was more resistant to serum killing than the parental strain. However, the phenotype did not correlate with resistance to any other environmental stresses (24). The CPS of V. vulnificus is essential for virulence (25, 26, 37, 45, 53, 76) but is not required for biofilm or rugose colony formation (25, 26, 37, 45, 53, 76). Therefore, other factors participate in these developmental pathways. NtrC was identified as a regulator of biofilm formation in V. vulnificus (38). Comparative proteomics of ntrC mutant and wild-type strains grown under planktonic and biofilm conditions suggested that NtrC exerted its effect, in part, via regulation of an ADP-glycero-manno-heptose-6-epimerase that is normally involved in biosynthesis of the inner core region of lipopolysaccharides (LPS) (14, 75). Recently, reverse transcription-PCR (RT-PCR) was used to identify a polysaccharide locus whose transcription was increased in the R variant of V. vulnificus (24). However, it was not determined if this locus played any role in biofilm formation or the development of the rugose phenotype.
It has recently come to light that the second messenger bis-(3′-5′)-cyclic-di-GMP (c-di-GMP) regulates a variety of cellular processes associated with the transition between planktonic and sessile lifestyles in many bacteria (36, 67, 68, 87). In general, high intracellular concentrations of c-di-GMP promote EPS production, biofilm formation, and rugosity while repressing motility and virulence gene expression. Diguanylate cyclases (DGCs) synthesize c-di-GMP, while phosphodiesterases (PDEs) degrade it. We previously characterized a DGC, DcpA, from V. vulnificus that induced biofilm formation and rugosity when overexpressed (53). We used DcpA to demonstrate that c-di-GMP regulated the production of an EPS that was both structurally and functionally distinct from CPS; however, the c-di-GMP-regulated EPS locus was not identified, nor was it known if the EPS itself played a role in biofilm and rugose colony formation. Here, we identify a c-di-GMP-regulated EPS locus (designated brp, for biofilm and rugose polysaccharide) and two transcriptional regulators (designated BrpR and BrpT) that govern these physiological responses in V. vulnificus.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. All primers used in this study are listed in Table 2. Strains were grown in Luria-Bertani medium (LB) or heart infusion medium containing 2% NaCl (HIN) (25) as indicated. Escherichia coli S17.1λpir (72) was used as the donor strain in conjugations with V. vulnificus. Antibiotics were added at the following concentrations: ampicillin (Ap), 100 μg/ml; kanamycin (Km), 25 μg/ml for E. coli and 160 μg/ml for V. vulnificus; rifampin (Rf), 100 μg/ml; chloramphenicol (Cm), 25 μg/ml for E. coli and 6 μg/ml for V. vulnificus. l-Arabinose (l-ara) and isopropyl-beta-d-thiogalactopyranoside (IPTG) were added to final concentrations of 0.2% and 0.3 mM, respectively, when indicated. Genomic DNA was extracted using DNAzol (Invitrogen) following the manufacturer's instructions. Sequencing was done at the Centre for Applied Genomics (TCAG) at the Hospital for Sick Children (Toronto). The genomes of V. vulnificus strains YJ016 and CMCP6 were obtained from the J. Craig Venter Institute website (http://cmr.jcvi.org). Homology searches were conducted using BLAST (1) analysis, and protein parameters and domain predictions were performed with ProtParam (22) and the PROSITE research tool from the Expert Protein analysis system (35). The nomenclature for genes identified in the genome of strain ATCC 27562 followed that used for strain CMCP6.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| S17.1λpir | Donor strain for conjugation | 72 |
| V. vulnificus | ||
| ATCC 27562 | Type strain; clinical isolate from Florida; Rfr | Institut Pasteur |
| ATCC 27562R | Natural rugose variant of ATCC 27562; Rfr | This study |
| ΔbrpF mutant | Mutant of ATCC 27562 with a disruption of the brpF glycosyltransferase; Cmr | This study |
| ΔbrpI mutant | Mutant of ATCC 27562 with a disruption of the brpI glycosyltransferase; Cmr | This study |
| ΔbrpR::Tn10 | Mutant of ATCC 27562 containing a Tn10 insertion in brpR | This study |
| ΔbrpT::Tn10 | Mutant of ATCC 27562 containing a Tn10 insertion in brpT | This study |
| RΔbrpF | Mutant of ATCC 27562R with a disruption of the brpF glycosyltransferase; Cmr | This study |
| RΔbrpI | Mutant of ATCC 27562R with a disruption of the brpI glycosyltransferase; Cmr | This study |
| RΔbrpR | Mutant of ATCC 27562R with a disruption of the brpR transcription factor; Cmr | This study |
| RΔbrpT | Mutant of ATCC 27562R with a disruption of the brpT transcription factor; Kmr | This study |
| Plasmids | ||
| pBAD24T | Mobilizable derivative of pBAD24; Apr | 29 |
| pBAD24T::dcpA | pBAD24T containing dcpA under the control of PBAD; Apr | 53 |
| pSU38T | Mobilizable derivative of pSU38; Kmr | This study |
| pSU38T::PTAC-brpF | pSU38T containing brpF under the control of PTAC; Kmr | This study |
| pSU38T::PTAC-brpI | pSU38T containing brpI under the control of PTAC; Kmr | This study |
| pSW23T | Suicide vector; Cmr | 15 |
| pSW23T::ΔbrpF | pSW23T containing 400-bp internal fragment of brpF; Cmr | This study |
| pSW23T::ΔbrpI | pSW23T containing 400-bp internal fragment of brpI; Cmr | This study |
| pSW23T::ΔbrpR | pSW23T containing 400-bp internal fragment of brpR; Cmr | This study |
| pNKTXI-SceI | Mini-Tn10 mutagenesis plasmid; Kmr Apr | 54 |
TABLE 2.
Primers used in this study
| Primer | Sequencea |
|---|---|
| PCR | |
| wcrFfwd | CCGCAATTGACCATGAGAATTTTACATATTATCAATG |
| wcrFrev | CCGTCTAGAAGCTCAATACAGGCAACCCT |
| wcrIfwd | CCGGAATTCATGAAAAAAGTACTGCATATTACA |
| wcrIrev | CCGTCTAGATCAAATATAGAGGCTGCTGAG |
| ΔwcrFfwd | CCGGGATCCGCTGGCCAGACATTGTTCAT |
| ΔwcrFrev | CCGTCTAGAAGCTCAATACAGGCAACCCT |
| ΔwcrIfwd | CCGGAATTCCAAAACTTATCTATACCCCACA |
| ΔwcrIrev | CCGTCTAGACTTTGTCACGCAACTGCTGTT |
| ptacKpnI | CCGGGTACCGTATAAGTGTGGAATTGTGAGC |
| ptacEcoRI | CCGGAATTCGTATAATGTGTGGAATTGTGAGC |
| ptac-wcrFfwd | TTCACCAACAAGGACCATAGCATATGAGAATTTTACATATTATCAATG |
| ptac-wcrFrev | CATTGATAATATGTAAAATTCTCATATGCTATGGTCCTTGTTGGTGAA |
| ptac-wcrIfwd | TTCACCAACAAGGACCATAGCATATGAAAAAAGTACTGCATATTACA |
| ptac-wcrIrev | TGTAATATGCAGTACTTTTTTCATATGCTATGGTCCTTGTTGGTGAA |
| RT-PCR | |
| vvuL20RT1 | GCTCGTGCACGTCATAAGAA |
| vvuL20RT2 | GACGGTCACGGTAAGCGTAT |
| dcpART1 | CAAGCTTTATGCCCTCGATG |
| dcpART2 | CATCCACAAGCCATTGACAGT |
| wcrART1 | CGTGGCGTACCAGTTCAATTCGA |
| wcrART2 | GTTTAATCCCAAGCGACGGAGGT |
| wcrDRT1 | TGGCTGGACAGTAATGCCAGACA |
| wcrDRT2 | GGTTTTGTGCGGATTTTCGCAGT |
| wcrFRT1 | GAAGGTGCAGGAAGAGTTGA |
| wcrFRT2 | TAGATAACCAGCGTTGCCGA |
| wcrIRT1 | AGAAGCATTTGGCGGTGGTGTAC |
| wcrIRT1 | TCACATCCGGTTGGAGTTGCTTG |
Restriction sites are in bold.
Subculture assay for isolation of the R variant.
An isolated colony of each indicated strain was inoculated in HIN containing the appropriate antibiotics and incubated overnight at 30°C with shaking. Cultures were passaged daily by diluting 1:100 in fresh media, and the development of the rugose phenotype was monitored by plating aliquots on HIN plus antibiotic until a rugose (R) variant was obtained (typically between 10 and 14 days for wild-type cells).
RNA isolation and RT-PCR.
For isolation of total RNA, strains were grown in HIN. Cells were isolated from the biofilm fraction during mid-exponential and stationary phases. RNA was isolated using an RNeasy kit (Qiagen) and treated with DNase I (Ambion). The integrity of the isolated RNA was verified by formaldehyde agarose gel electrophoresis, and its concentration/purity was determined from the A260/A280 values on a NanoDrop 1000 (Thermo Scientific). Primer sets were optimized for PCR using genomic DNA from V. vulnificus strain ATCC 27562, and PCR was performed on RNA samples to verify that the samples did not contain residual contaminating DNA. The Qiagen OneStep RT-PCR kit was used to amplify regions from RNA targets according to the manufacturer's protocol. The primer pairs and target genes are listed in Table 2. RT-PCR was carried out using an MBS multiblock thermocycler system (Thermo Hybaid). Reverse transcription reactions were performed at 50°C for 30 min. cDNA was amplified using an initial hot start of 95°C for 15 min followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and elongation at 72°C for 1 min. PCR products were visualized by agarose gel electrophoresis and quantified by densitometry using the GeneTools gel analysis software package (Syngene). The transcript levels for the respective target genes were normalized relative to the level of rplT (L20) transcript in the same sample that was run on the same gel.
Construction and complementation of the ΔbrpF and ΔbrpI mutant strains.
Primers brpF KO-F/-R and brpI KO-F/-R were used to amplify internal 400-bp fragments of brpF and brpI, respectively. PCR products were cloned in the suicide plasmid pSW23T (15). The resulting plasmids, pSW23T::ΔbrpF and pSW23T::ΔbrpI, were conjugated to V. vulnificus ATCC 27562 and the R variant. Cointegrants were selected on LB or HIN plates containing Rf and Cm, and correct integration was confirmed by PCR. To complement the mutant strains, the RP4 origin of conjugative transfer was amplified from pSW23T (15) with primers oriT1NotI and oriT2NotI, ligated to NotI-digested pSU38 (4), and transformed into E. coli S17.1λpir to create pSU38T. The brpF and brpI genes were fused to the PTAC promoter by superprimer PCR as follows: the primers ptacKpnI/ptac-brpFrev and ptac-EcoRI/ptac-brpIrev were used in separate reactions to amplify the region from −40 to the start ATG of the PTAC promoter. The primer pairs ptac-brpFfwd/brpFXbaIrev and ptac-brpIfwd/brpIXbaIrev were used to amplify brpF and brpI, respectively, from strain ATCC 27562 genomic DNA. Equal amounts (20 ng) of the ptac-brpFfwd/brpFXbaIrev and ptacKpnI/ptac-brpFrev or the ptac-EcoRI/ptac-brpIrev and ptac-brpIfwd/brpIXbaIrev PCR products were mixed in separate reactions that contained all the necessary PCR reagents but lacked primers. Five PCR cycles were run at 94°C for 30 s, 60°C for 30 s, and 72°C for 60 s. The ptacKpnI/brpFXbaIrev and ptac-EcoRI/brpIXbaIrev primers were added to the appropriate tubes, and 25 additional cycles were run. The brpF and brpI promoter fusions were digested with XbaI and KpnI and XbaI and EcoRI, respectively, and cloned into the same sites of pSU38T. The resulting plasmids, pSU38T::PTAC-brpF and pSU38T:: PTAC-brpI, were conjugated to the ΔbrpF and ΔbrpI mutant strains for complementation studies.
Antiserum production and slide agglutination assays.
The production of antisera to formalin-killed whole cells of V. vulnificus strain ATCC 27562 and subsequent slide agglutination assays were performed as previously described (54).
Biofilm and aggregate formation in culture tubes.
Strains were grown in HIN at 30°C with shaking to an optical density at 600 nm (OD600) of 0.4 to 0.6 and induced with l-ara and IPTG where indicated. Incubation with shaking was continued. Biofilm formation was observed at 3 h postinduction, while aggregate formation was examined following overnight growth. Images of the biofilm at the air-liquid interface and the cell aggregate at the bottom of the tube were taken with a Kodak DX6490 digital camera.
Biofilm assays in 96-well microtiter plates.
Strains were grown in HIN at 30°C with shaking until they reached an OD600 of 1; the growth rates for parental and mutant strains were similar. Cultures were adjusted to 1 × 106 CFU ml−1 in fresh media, and 150 μl was inoculated into 96-well microtiter plates (two wells per strain). Microtiter plates were statically incubated for 20 h at 30°C. Following measurement of the planktonic cell density at OD600 with a BioTek PowerWave 340 microplate spectrophotometer, media were carefully removed and the wells were stained with 150 μl of 0.1% crystal violet (CV) solution for 30 min. The stained wells were then washed three times with phosphate-buffered saline (PBS) (130 mM NaCl, 5 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4), and biofilms were resolubilized in 150 μl of isopropanol-acetone (4:1). The OD595 of each well was then measured. The ratio of ODcv/ODcells was calculated to determine the relative level of biofilm formation.
Colony morphology assays.
Strains were grown overnight in HIN containing l-ara and IPTG where indicated at 30°C with shaking. Cultures were then diluted 1:200 in HIN, and 2 μl was spotted onto HIN plates containing l-ara and IPTG when necessary. Plates were incubated at 30°C for 48 h and photographed with an AxioCam MRc5 (Zeiss) digital camera attached to a dissecting microscope.
Construction and screening of a transposon (Tn) library.
The pNKTXI-SceI plasmid, which carries a mini-Tn10 transposon (54), was conjugated to strain ATCC 27562, and mutants with Tn insertions were selected on LB Rf Km plates. These clones were pooled and used as recipients in conjugations with E. coli S17.1λpir carrying pBAD24T::dcpA. Exconjugants were selected on LB Rf Km Ap plates containing l-ara. These clones were screened for their ability to form a biofilm in 96-well microtiter plates. Attenuated clones were expected to contain Tn insertions in regulatory and structural genes of the c-di-GMP/EPS signaling pathway. Genomic DNA was extracted from translucent colonies, and Tn insertion sites were identified by arbitrarily primed PCR mapping (59).
Construction and complementation of the ΔbrpR and ΔbrpT mutants in an R variant.
Primers brpRintEcoRI and brpRintXbaI were used to amplify an internal 400-bp fragment of brpR for cloning into the suicide plasmid pSW23T as described above for conjugation to an R variant of V. vulnificus ATCC 27562. Correct integration was confirmed by PCR. Since brpT is only 633 bp in length, we reasoned that recovering a bona fide knockout using the pSW23T suicide plasmid might be difficult. We instead elected to use chitin-induced natural transformation (9, 27, 48) to transfer the brpT::Tn10 lesion into an R variant. Genomic DNA from a brpT::Tn10 mutant that we identified in our library screen was used as the donor DNA for the transformation experiment. Transformants were selected on HIN Rf Km plates, and transfer of the brpT::Tn10 lesion was confirmed by PCR.
Scanning electron microscopy (SEM).
Strains were grown in HIN at 30°C with shaking, induced with 0.2% l-ara when the OD600 reached 0.4 to 0.6, and grown to a final OD600 of 1.0. Samples were diluted 1:10 in fresh media, 1 ml of diluted culture was added to either glass coverslips or oyster shells in 12-well plates, and the plates were incubated statically at 30°C overnight. Biofilms were rinsed briefly in 0.1 M cacodylate buffer, pH 7.4, and fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, for 2 h at room temperature. Biofilms were washed three times for 5 min with 0.1 M cacodylate buffer, pH 7.4, and dehydrated in a graded ethanol series (25%, 50%, 70%, 95%, and 100% two times for 10 min). Samples were critical-point dried in CO2, gold coated, and viewed with a Hitachi 3400 scanning electron microscope at 5 to 10 kV of accelerating potential at the Microscopy Imaging Lab (University of Toronto).
Resistance assays.
Strains were analyzed to determine their resistance to chlorine, oxidative, and osmotic stress as previously described for V. cholerae (83, 89). Survival was calculated as CFU of treated cultures/CFU of untreated cultures, where treated cultures were exposed to 3 ppm NaOCl (chlorine stress), 5 mM H2O2 (oxidative stress), or 1 to 2.5 M NaCl (osmotic stress) diluted in PBS. Untreated cultures were exposed to PBS alone, and survival in PBS was defined as 100%. To monitor bacterial growth in the presence of increasing NaCl concentrations, 107 cells of the indicated strains were grown statically in LB containing NaCl at 0, 2, 10, or 30 ppt (0/00) for 20 h at 30°C before the OD600 was measured with a BioTek PowerWave 340 microplate spectrophotometer.
Deposition of nucleic acid sequences and accession numbers.
The nucleic acid sequence for the brp locus of strain 27562 has been deposited at the National Centre for Biotechnology Information (NCBI) GenBank database under accession number G4384922.
RESULTS
Identification of a c-di-GMP-regulated EPS locus in V. vulnificus.
We utilized comparative genomics to identify six putative polysaccharide loci containing at least three genes in the genomes of V. vulnificus strains YJ016 and CMCP6. Two of these loci coded for known CPS and putative LPS biosynthesis genes, respectively. We focused our attention on one of the four remaining loci (vv21574 to vv21582), as its expression was recently reported to be associated with the R variant (24). However, its regulation and impact, if any, on biofilm or rugose colony development were not known.
The locus, found on chromosome II, is a nine-gene cluster organized in two operons and encodes a number of glycosyltransferases, polysaccharide biosynthesis, and export protein homologs (Fig. 1). To verify that expression of the locus was associated with the R variant, we monitored the expression pattern of vv21574, vv21577, vv21578, and vv21580 in strain 27562 by RT-PCR analysis. The transcript levels for the respective target genes were normalized relative to the level of the rplT (L20) transcript in the same sample that was run on the same gel. Transcription of these genes was difficult to detect in stationary-phase cells of the parental strain and the R variant (Fig. 2, bottom panel, compare lanes 1 and 3). Conversely, transcription of these target genes was elevated 2- to 7-fold in the R variant relative to the parental strain during mid-exponential growth (Fig. 2, top panel, compare lanes 1 and 3). This suggested that the expression of this locus was increased in the R variant. Expression of the locus also appeared to be greater during mid-exponential growth and decreased during stationary-phase growth.
FIG. 1.

The vv21574-vv21582 (brp) polysaccharide gene cluster in the genome of V. vulnificus CMCP6. Open arrows represent the locations and directions of transcription of the respective open reading frames (ORFs). Small angled arrows indicate the positions of putative promoters, including a conserved ops (operon polarity suppressor) sequence (55), while the black hairpin structure following vv21582 denotes a putative Rho-independent transcriptional terminator. The open circles above vv21578 and vv21580 indicate the positions of targeted disruptions using the pSW23T suicide plasmid (15). Red, gray, green, and blue arrows denote glycosyltransferase, hypothetical proteins, export proteins, and EPS biosynthesis genes, respectively.
FIG. 2.

In vivo expression of the vv21574-vv21582 (brp) locus. The indicated strains were grown in HIN. RNA was isolated from mid-exponential (ME)- and stationary (S)-phase cells of the parental ATCC 27562 strain carrying pBAD24T (lane 1) or pBAD24T::dcpA (lane 2) and from an R variant, ATCC 27562R, with pBAD24T (lane 3). RT-PCR was used to determine if vv21574, vv21577, vv21578, and vv21580 were being transcribed. Expressions of the dcpA and the ribosomal rplT genes were used as positive controls. Primer sets were optimized for PCR using genomic DNA from V. vulnificus strain ATCC 27562, and PCR was performed on RNA samples to verify that they did not contain residual contaminating DNA. Target genes are indicated above the gels. M, 100-bp marker.
Since we previously demonstrated that rugose colony formation in V. vulnificus was regulated by c-di-GMP, we speculated that expression of the locus might be subject to c-di-GMP regulation as well. Transcription of vv21574, vv21577, vv21578, and vv21580 in the parental strain carrying pBAD24T::dcpA, which expresses the V. vulnificus DGC DcpA (53), was elevated relative to that found in the same strain carrying the empty vector during mid-exponential growth (Fig. 2, compare lanes 1 and 2). This suggested that expression of the locus was subject to regulation by c-di-GMP.
The locus is required for c-di-GMP-induced biofilm, aggregate, and rugose colony formation.
We reasoned that the effect of c-di-GMP on biofilm, aggregate, and rugose colony formation was linked to its regulation of the vv21574 to vv21582 cluster. To investigate this, glycosyltransferases from each operon (vv21578 and vv21580) were disrupted, and biofilm and aggregate formation by strains carrying either pBAD24T::dcpA or the empty vector was assessed relative to that of the parental control strains. The parental, Δvv21578, and Δvv21580 strains carrying the vector control remained opaque on solid media (Fig. 3) and continued to agglutinate in the presence of antisera to wild-type ATCC 27562 cells. This suggested that the locus was not required for production of the CPS. Biofilm formation by the Δvv21578 and Δvv21580 strains carrying pBAD24T was lower than that for the wild-type strain carrying the same vector (P < 0.001). This suggested that the locus was being expressed in wild-type bacteria but not at a level sufficient to promote biofilm formation under the conditions tested here. When we elevated the intracellular c-di-GMP levels by inducing dcpA expression, biofilm formation was induced 4.8-fold (P < 0.001) in the parental strain, whereas only 2.5-fold and 2.1-fold increases were observed for the Δvv21578 and Δvv21580 strains, respectively (P < 0.001 for both; Fig. 4A, top panel, and 4B). Furthermore, a cell aggregate was clearly visible for the parental strain, while no such aggregate was observed for either of the mutants (Fig. 4A, middle panel). Complementation of both mutant strains with the appropriate wild-type gene restored biofilm and aggregate formation. It is noteworthy that the vv21578 and vv21580 lesions in wild-type cells carrying pBAD24T::dcpA did not reduce biofilm levels to those of the same mutants carrying the vector control. One explanation is that mutation of more than one gene within the locus is required to completely prevent production of the encoded polysaccharide. Alternatively, c-di-GMP could also regulate the expression of additional factors, such as adhesins or other polysaccharides, that participate in biofilm formation (50).
FIG. 3.
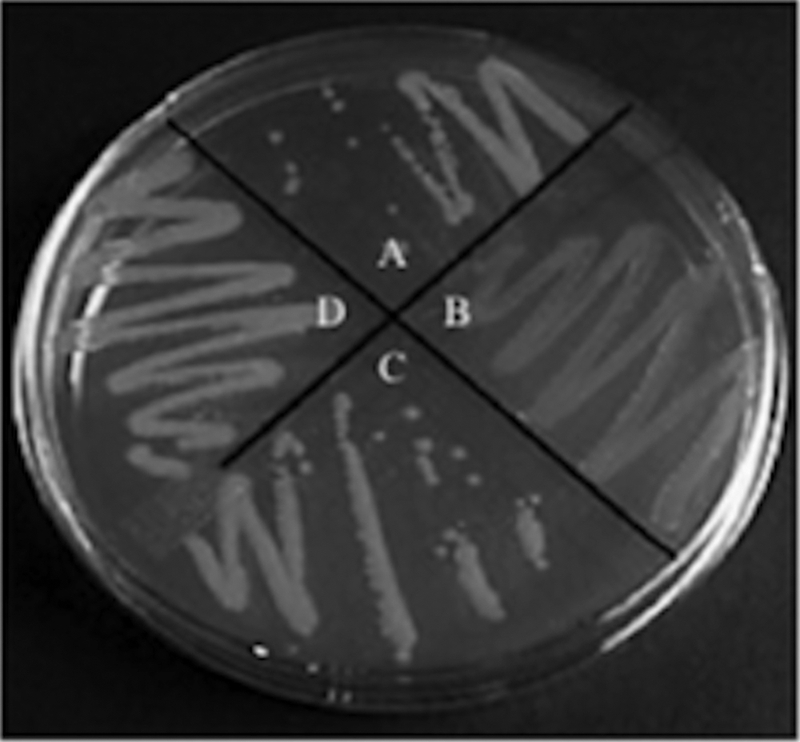
Lesions within the brp locus do not affect CPS production. The parental ATCC 27562 strain (A) appears opaque on solid medium, while the CPS biosynthesis mutant, Δwzy (B), appears translucent. The ΔbrpF (C) and ΔbrpI (D) mutant strains appear opaque. The strains were grown in HIN.
FIG. 4.

c-di-GMP-induced biofilm and aggregate formation is dependent on the vv21574-vv21582 (brp) locus. The indicated strains were grown in HIN. (A) Biofilm (top panel) and aggregate (middle panel) formation in culture tubes by the parental, mutant (Δvv21574 and Δvv21580), and complemented (Δvv21574/pSU38T::vv21574 and Δvv21580/pSU38T::vv21580) strains carrying the pBAD24T control vector or pBAD24T::dcpA. Control strains for complementation studies carried the empty pSU38T plasmid. Colony morphologies for the same strains are shown in the bottom panel. (B) Quantitative measurement of biofilm formation of the corresponding strains in panel A by CV staining. Results shown are representative of at least three independent experiments. Error bars represent standard deviations.
Colony morphology assays supported the involvement of the locus in rugose colony formation. Strains carrying the empty pBAD24T vector were flat and featureless in appearance (Fig. 4A, bottom panel). The parental strain carrying pBAD24T::dcpA developed the raised, wrinkled phenotype that typifies the R variant, while the mutant strains carrying the same vector did not. Complementation of both mutant strains with the appropriate wild-type gene restored c-di-GMP-induced rugose colony formation. These results suggested that in the signaling pathway leading to biofilm and rugose colony development, the input of c-di-GMP culminated in the production of the polysaccharide encoded by this locus.
This locus was previously designated wcr, so named because polysaccharide nomenclature traditionally designates “w” for any putative polysaccharide gene (64) and because it was preferentially expressed in the rugose (“r”) variant (24). The “c” designation stemmed from the observation that a translucent (CPS−) mutant with a Tn insertion in vv21580 failed to generate opaque colonies in a reversion assay that would be indicative of restored CPS production (24). However, the mutant was never complemented to verify that vv21580 was required for CPS production; hence the role of the wcr locus in CPS production was not definitively demonstrated. Furthermore, several known and unknown mechanisms exist by which a CPS− phenotype can arise in V. vulnificus (13), including spontaneous deletions in the wza-wzb-wzc region. Thus, background mutations leading to a CPS− phenotype in the Δvv21580 mutant could not be excluded. The Δvv21578 and Δvv21580 mutants characterized here had an opaque phenotype and agglutinated with antisera to formalin-killed whole cells of the parental strain, whereas a bona fide CPS− strain we created, wzy::Tn10 (54), was translucent and did not agglutinate. This suggested that the locus was not required for CPS production. To accurately reflect its role, we have renamed the locus brp, for biofilm and rugose polysaccharide.
The brp locus is required for the development, maintenance, and resistance of the R variant.
We sought to address the role of the brp locus in the emergence, maintenance, and resistance of the R phenotype. Prolonged growth of V. vulnificus in HIN has been reported to support the development of the R phenotype (25). The parental and brpF (Δvv21578) and brpI (Δvv21578) mutant strains were subcultured in HIN for up to 9 weeks. The parental strain typically gave rise to R variants (ATCC 27562R) within 14 days, while R variants could not be recovered from either mutant even after 62 days of growth. To definitively demonstrate that brpF and brpI were required for development of the R phenotype, we disrupted these genes in the ATCC 27562R strain. Lesions in either brpF or brpI in an R variant (strains ATCC 27562RΔbrpF and ATCC 27562RΔbrpI) resulted in a shift to the smooth phenotype (Fig. 5A, bottom panel), with a concomitant loss of the copious biofilm characteristics of the ATCC 27562R strain (Fig. 5A, top panel, and 5B); biofilm formation by the ATCC 27562RΔbrpF and ATCC 27562RΔbrpI mutants decreased significantly (8-fold, P < 0.001). Complementation of the ATCC 27562RΔbrpF mutant with the respective wild-type gene led to partial restoration (62%) of biofilm formation (P < 0.05) and rugosity. Complementation of the ATCC 27562RΔbrpI mutant with the wild-type gene restored biofilm formation and rugosity to levels similar to that of the parental strain (P, not significant).
FIG. 5.

Expression of the brp locus is required for biofilm and rugose colony formation by the R variant. The indicated strains were grown in HIN. (A) Biofilm formation in culture tubes (top panel) and colony morphology (bottom panel) of the parental and ATCC 27562R strains and the ATCC 27562RΔbrpF and ATCC 27562RΔbrpI mutant strains carrying the pSU38T vector control. Complemented strains carry pSU38T::brpF or pSU38T::brpI, respectively. (B) Quantitative measurement of biofilm formation of the corresponding strains in panel A by CV staining. Results shown are representative of at least three independent experiments. Error bars represent standard deviations.
The resistance of the ATCC 27562R, ATCC 27562RΔbrpF, and ATCC 27562RΔbrpI strains to NaOCl, NaCl, and H2O2 relative to that of the parental strain carrying pBAD24T::dcpA or the vector control was evaluated. Wild-type bacteria harboring pBAD24T::dcpA and ATCC 27562R showed a 2.7-fold increased tolerance (P < 0.005) toward the killing effects of chlorine relative to that for the parental control (Fig. 6A). Disruption of either brpF or brpI in ATCC 27562R decreased chlorine tolerance (P < 0.001), albeit not to the baseline level observed for the wild-type strain; chlorine resistance was 1.7-fold higher in the mutants than in the parental control. These results suggested that the brp locus participated in the development of chlorine resistance, but additional factors also contributed to this phenotype. To assess the tolerance of the strains to osmotic stress, bacteria grown in Miller's LB (0.17 M NaCl) were exposed to 1, 2, or 2.5 M NaCl and direct plate counting was used to monitor bacterial survival. There was little difference between the strains in response to osmotic shock regardless of the NaCl concentration used, and bacterial recovery was relatively low, with a 90% drop in CFU being recorded for each strain (data not shown). We then opted to monitor growth of the strains in LB containing different concentrations of NaCl: 0, 2, 10, and 30 ppt (0/00), which approximates the salinity range of estuarine environments (21). While none of the strains grew in LB containing 00/00 NaCl (Fig. 6B), they all grew to some extent, albeit poorly (1.3- to 1.8-fold increase, P < 0.05), in 20/00 NaCl. Growth of all the strains was significantly better (P < 0.001) at 100/00 NaCl (4.7- to 7-fold increase). This trend of poor growth at low NaCl concentrations reflects the halophilic nature of the species. Growth of the parental control, ATCC 27562RΔbrpF, and ATCC 27562RΔbrpF strains was significantly lower at 300/00 NaCl than at 100/00 NaCl (P < 0.05). The growth of wild-type bacteria harboring pBAD24T::dcpA at 300/00 NaCl was similar to the growth at 100/00 NaCl (P, not significantly different). Notably, growth of the R variant continued to increase significantly (P < 0.001) at 300/00 NaCl. These results suggested that the brp-encoded EPS promoted growth of the bacteria at NaCl levels that approach the salinity of natural seawater, which is approximately 350/00 (21). All of the strains were equally sensitive to 5 mM hydrogen peroxide (>99.5% killing for each), suggesting that neither increased c-di-GMP levels nor the brp-encoded EPS conferred resistance to oxidative stress (data not shown).
FIG. 6.

Sensitivity of the parental, ATCC 27562R, ATCC 27562RΔbrpF, and ATCC 27562RΔbrpI strains to chlorine and increasing salinity. The indicated strains were grown in LB containing 100/00 NaCl. (A) The strains were then incubated in PBS containing 3 ppm NaOCl for 5 min. The surviving bacteria were enumerated by plate counts. The number of viable bacteria relative to that obtained following treatment with PBS alone is plotted. (B) Strains were grown in LB containing 00/00 (white bars), 20/00 (dark gray bars), 100/00 (light gray bars), and 300/00 (black bars) NaCl. Bacterial growth (OD595) is plotted. Results shown are representative of at least three independent experiments. Error bars represent standard deviations.
The brp locus is required for the development of robust biofilm architecture.
The development of a substantive, stable three-dimensional biofilm has been difficult to document for V. vulnificus. Our data suggested that this was likely because the brp locus was poorly expressed, if at all, in the absence of an inducing signal. Since expression of the locus appeared to be regulated by c-di-GMP, we carried out SEM analysis of the biofilm structures formed on biotic (oyster shells) and abiotic (glass) surfaces by the parental, ΔbrpF mutant, ΔbrpI mutant, and ATCC 27562R strains carrying pBAD24T::dcpA or the empty vector. An extensive network of cell aggregates separated by water channels was evident on both surfaces for wild-type cells carrying pBAD24T::dcpA, and the cells appeared to be enrobed in a smooth matrix (Fig. 7, second row). Wild-type cells harboring the vector control were devoid of this matrix, and only dispersed individual cells adhered to either surface type (Fig. 7, first row); similar results were obtained for the brpF and brpI mutant strains, regardless of whether they were carrying pBAD24T::dcpA or the vector control (data not shown). The ATCC 27562R strain carrying the vector control exhibited far greater surface coverage on both glass and oyster shells than the parental strain, and the matrix encapsulating the cells was even more apparent (Fig. 7, third row). Interestingly, as can be seen at low magnification, vertical expansion of the biofilm macrostructure appeared to be curtailed. This was particularly evident for biofilms formed on glass coverslips, where the thickness of the biofilm did not exceed more than 12 to 15 cells. Alternatively, the vertically developing structure may have been fragile and detached during preparation of the sample for SEM. This limitation was overcome in ATCC 27562R cells carrying pBAD24T::dcpA (Fig. 7, fourth row). An extensive, vertically developed biofilm macrostructure with an interlacing network of deep fluid channels could be clearly seen at lower magnification, and a mesh-like sheath covering and connecting the cells was evident at higher magnification. The dramatic increase in biofilm and aggregate formation by ATCC 27562 cells that contained pBAD24T::dcpA relative to that observed for cells carrying the vector control was also evident when the strains were grown in culture tubes (data not shown). These results suggested that biofilm formation and matrix production required an intact brp cluster and that the continued synthesis of c-di-GMP promoted the vertical expansion and/or stability of the developing biofilm, either through continued brp expression or perhaps through the expression of additional factors that lead to the development of more complex three-dimensional structures.
FIG. 7.

Scanning electron micrographs of biofilms formed by V. vulnificus. Biofilm formation by the parental and ATCC 27562R strains carrying pBAD24T::dcpA or the vector control on glass coverslips or oyster shells is shown. The indicated strains were grown in HIN. Images are in pairs of low (L) and high (H) magnification. Scale bars are indicated at the bottom of each image.
Depleting intracellular c-di-GMP levels prevents biofilm formation and development of the rugose phenotype.
To determine if lowering the intracellular level of c-di-GMP could reverse the effect of c-di-GMP on biofilm and rugose colony formation, we expressed YhjH, an E. coli PDE (80), in the R variant. A biofilm was typically observed for ATCC 27562R within 6 h of growth in culture tubes (Fig. 8, top panel). The expression of yhjH in ATCC 27562R during the 6-h growth period prevented biofilm formation. Maintenance of the rugose phenotype was also inhibited when the intracellular c-di-GMP level decreased (Fig. 8, bottom panel). The expression of yhjH in ATCC 27562R led to loss of the R phenotype, while the same strain carrying the vector control remained rugose. Together, these results support the notion that controlled augmentation or depletion of the intracellular level of c-di-GMP can regulate the switch between the planktonic/biofilm and smooth/rugose states of the bacteria.
FIG. 8.

Depletion of intracellular c-di-GMP levels in the R variant prevents biofilm and rugose colony formation. The indicated strains were grown in HIN. (Top) ATCC 27562R carrying pME6041T or pME6041T::yhjH was grown in the presence (+) or absence (−) of l-arabinose. The arrow marks the position of the biofilm ring. (Bottom) Colony morphology of the strains above. Images shown are representative of at least three independent experiments.
brpR and brpT encode regulators of brp expression.
To identify additional genes in the c-di-GMP/brp signaling pathway, we screened an ATCC 27562 Tn10 mutant library for clones that failed to form a biofilm in response to elevated c-di-GMP levels. As expected, Tn insertions in several of the brp structural genes were recovered. In addition, we identified lesions in two genes (vv10525 and vv21570), of 1,335 and 633 bp, that we designated brpR and brpT, respectively. brpR was situated on chromosome I, while brpT was adjacent to the brp locus. Unlike the parental strain, neither the brpR::Tn10 nor the brpT::Tn10 mutant harboring pBAD24T::dcpA formed a biofilm or exhibited a rugose phenotype (data not shown). Accordingly, lesions in brpR and brpT also led to a significant decrease (P < 0.001) in biofilm formation and loss of the rugose phenotype in the R variant (Fig. 9). Complementation of the ATCC 27562RΔbrpR and ATCC 27562RΔbrpT mutants with the respective wild-type gene restored biofilm formation and rugosity (12- to 14-fold increase for each after complementation; P < 0.001). BrpR contained a helix-turn-helix (HTH) DNA binding domain and shared 79% and 84% amino acid identity with VpsR of V. cholerae and CpsR of V. parahaemolyticus. BrpT also harbored an HTH DNA binding domain and shared 55% amino acid identity with VpsT of V. cholerae. These results suggested that BrpR and BrpT were transcription factors that regulated biofilm and rugose colony formation in V. vulnificus.
FIG. 9.

BrpR and BrpT are required for biofilm and rugose colony formation by the R variant. The indicated strains were grown in HIN. (A) Biofilm formation in culture tubes (top panel) and colony morphology (bottom panel) of the parental strain, ATCC 27562R, and the ATCC 27562RΔbrpR and ATCC 27562RΔbrpT mutant strains carrying the pBAD24T vector control. Complemented strains carry pBAD24T::brpR or pBAD24T::brpT, respectively. (B) Quantitative measurement of biofilm formation of the corresponding strains in panel A by CV staining. Results shown are representative of at least three independent experiments. Error bars represent standard deviations.
DISCUSSION
The global distribution of V. vulnificus in coastal and estuarine waters (16, 56, 57), coupled with its extremely invasive pathology (76), has spawned many investigations into the physical properties that contribute to its environmental persistence and its attachment to, and colonization of, marine biotic surfaces. Biofilm and rugose colony formation is predicted to be important for the pathogen's survival in aquatic ecosystems. Biofilm formation promotes surface colonization, and biofilms are refractory to host defenses and antibiotic treatment. The R variant exhibits an enhanced capacity to form stress-resistant biofilms. Studies of the impact of bacterial surface structures on biofilm and rugose colony formation have implicated the involvement of CPS, EPS, pili, and flagella in these processes, yet many of the structural genes and the genetic regulators responsible for their expression are unknown. In this work we identify a locus, brp, coding for an EPS that mediates biofilm and rugose colony formation in V. vulnificus. Expression of the brp locus was low in wild-type cells but was induced by increasing c-di-GMP levels. Increased brp expression correlated with increased biofilm and rugose colony formation and the development of stress-resistant characteristics that typify the R variant. Disruption of putative glycosyltransferases within the locus resulted in the loss of these phenotypes. Furthermore, we show that biofilm and rugose colony formation is linked to at least two genes, brpR and brpT, that encode transcriptional regulators. Both BrpR and BrpT contain a helix-turn-helix DNA-binding motif near their C terminus. It is therefore possible that both regulators bind to the promoter regions of the brp cluster and directly activate transcription of the brp biosynthetic genes. Alternatively, BrpR and BrpT could mediate their effect on brp expression indirectly, by controlling the expression of another positive regulator of brp expression. Since elevated c-di-GMP levels could not bypass lesions in brpR or brpT to enhance biofilm and rugose colony formation, this suggested that the input of c-di-GMP in the signaling pathway either precedes or occurs concomitantly with that of these transcription factors.
The brp locus is homologous to the vps locus of V. cholerae and the cps locus of V. parahaemolyticus (Fig. 10). The respective polysaccharide loci are organized as two operons, and each is subject to regulation by c-di-GMP (10, 39, 44, 81). BLAST analysis suggested that BrpR was a transcriptional regulator homologous to VpsR of V. cholerae (5, 63) and CpsR of V. parahaemolyticus (28), while BrpT was homologous to the VpsT transcription factor of V. cholerae (5, 11). The brp cluster is also conserved in numerous Vibrio species, including V. fischeri, V. splendidus, and V. alginolyticus (24). This degree of evolutionary conservation leads us to speculate that this polysaccharide cluster may be a common mechanism used by many Vibrio species to promote environmental persistence and bacterium-host interactions. If so, then comparative studies of these loci should advance our understanding of such interactions and bacterial survival. However, significant differences between the systems exist. The V. cholerae vps and V. parahaemolyticus cps loci contain 17 and 11 genes, respectively. Putative biosynthetic genes within these loci are absent from V. vulnificus, resulting in a reduced set of nine genes for the brp locus. This raises the question of whether these “extra” genes in the vps and cps loci actually contribute to VPS and CPS production in V. cholerae and V. parahaemolyticus. The rbmABCDEF locus of V. cholerae, which is situated in the vpsI/II intergenic region and modulates biofilm and rugose colony formation (17, 18), is not present in V. parahaemolyticus or V. vulnificus. Likewise, the scrABC locus of V. parahaemolyticus, which affects cps expression (10), is absent from V. cholerae and V. vulnificus. These differences likely reflect variations in the structures of the respective polysaccharides, their expression in response to different environmental stimuli, and the host ranges of the bacterial species.
FIG. 10.

Genomic comparison of the vps, cps, and brp loci. (A) Comparison of genes encoding the V. cholerae (Vch) vps, V. parahaemolyticus (Vpa) cps, and V. vulnificus (Vvu) brp loci. Open arrows represent the locations and directions of transcription of the respective genes. Genes depicted in gray are dissimilar. Homologous genes are shown in the same color and are connected by dotted lines. The rbmABCDEF genes, which modulate vps expression, are located between vpsI and vpsII. (B) Simplified schematic of the regulatory pathway of biofilm formation in V. cholerae, V. parahaemolyticus, and V. vulnificus. Activators are shown as circles, and repressors are shown as rectangles. Homologs are shown in the same color.
Estuarine environments, in which Vibrio species are commonly found, are characterized by having constantly changing chlorine and salt gradients due to the mixing of sea and freshwater, and these gradients can range from 0.5 to 30 ppt (21). Chlorine resistance in the V. cholerae R variant is due to the capacity of the VPS to inactivate chlorine (89). It was previously reported that the R variant of V. vulnificus produced copious biofilms and was more resistant to serum killing than the parental strain; however, the phenotype did not correlate with an increased resistance to chlorine (24), and resistance to osmotic and oxidative stress was not examined. We observed that the R variant of V. vulnificus indeed produced substantial biofilms and, like V. cholerae, exhibited increased resistance to chlorine. The V. cholerae R variant was 20 times more resistant to the killing effects of osmotic shock than the nonrugose parental strain (83). Although there was no difference in the survival of the parental strain and that of the R variant following osmotic stress, V. vulnificus was less susceptible to its killing effects than V. cholerae (10% survival for V. vulnificus versus 0.5% survival for V. cholerae [83]). We also noted that the V. vulnificus R variant grew better than the parental strain at NaCl concentrations that approached the salinity of seawater. This may reflect an increased tolerance of the R variant for fluctuations in osmotic conditions that occur in a salt- and freshwater mix. The UV irradiation of water generates reactive oxygen species such as hydrogen peroxide that damage cellular lipids, proteins, and nucleic acids (3, 19). This effect increases with increasing salinity in a synergistic, rather than simply additive, manner (23). Thus, bacteria that may be exposed to the photic zone of the water column must evolve protective mechanisms against phototoxicity. The increased resistance of the V. cholerae R variant to oxidative stress is due to the increased production of catalases (89). Hydrogen peroxide sensitivity remained unchanged between the V. vulnificus parental strain and the R variant, suggesting that the R phenotype did not correlate with an increased resistance to photoxic attack. Interestingly, attempts to use UV light and microfiltration to depurate V. vulnificus from oyster stocks failed at temperatures above 21°C, the approximate temperature of seawater during the summer months (78). It was proposed that the tight association of V. vulnificus with oysters, fish, crustaceans, mollusks, and plankton likely provided the bacteria with protective environments where it could freely reproduce. In such closed systems, bacterial replication may be enhanced and, as a result, large numbers of bacteria can be released into the surrounding seawater at rates exceeding the bactericidal effects of H2O2 that is generated by UV light, obviating the need to employ additional phototoxic resistance mechanisms. Collectively, these characteristics likely contribute to the organism's environmental persistence and colonization of host surfaces.
Despite being capable of attaching to abiotic surfaces (37, 53), eel skin (46), and HEp-2 cells (60, 61), there exist few structural data on the macrostructure of V. vulnificus biofilms. Studies of this kind have been hampered by the difficulty in obtaining stable biofilms under standard laboratory conditions, and this is one of the few studies to observe the biofilm morphology of V. vulnificus in detail. Microscopic examination of the wild-type strain revealed that the cells adhered poorly to biotic and abiotic surfaces. In contrast, development of the R phenotype or increased c-di-GMP production via expression of the DGC DcpA led to the formation of a compact, well-developed biofilm with prominent pillar-like structures and water channels. Cells within the biofilm were enrobed in a matrix that was not observed when DcpA was overexpressed in the brpF or brpI mutant, supporting our hypothesis that the product of the brp locus functions to modify the cell surface.
Although V. vulnificus is capable of forming extensive biofilms, biofilm development varies considerably among V. vulnificus isolates (47). Our data suggest that this may be due, in part, to differences in the levels of brp expression in these isolates. Since the bacteria must be able to alternate between sessile and planktonic states, they must be able to regulate their ability to form and disperse the biofilm structure. We have shown that increased c-di-GMP levels induced the production of a cell surface EPS encoded by the brp locus that conferred biofilm formation, rugosity, chlorine resistance, and osmotic stress resistance to V. vulnificus. Depletion of intracellular c-di-GMP levels halted and even reversed these phenotypes. Tight regulation of brp expression could form the basis for rapid bacterial release from a developed biofilm, either as clusters or as individual cells, when intracellular c-di-GMP levels drop. In this scenario, c-di-GMP would act as a key intracellular regulator for controlling biofilm stability by shifting the state of biofilm cells between attached and detached in a concentration-dependent manner. Hence, the brp-encoded EPS likely plays an important role in surface colonization by, and the persistence of, V. vulnificus, and its regulated expression may control how the bacteria switch from a planktonic lifestyle to colonizing shellfish to invading human tissue.
Acknowledgments
We are indebted to Steven Doyle and Battista Calvieri for their expertise with SEM analysis. We also thank Alfred Spormann (Stanford University) for the pARA-yhjH plasmid.
This work was supported by funding from the Canadian Institutes of Health Research (CIHR), the Natural Sciences and Engineering Research Council of Canada (NSERC), and a Premier's Research Excellence award (PREA) to D.A.R.-M. Y.G. is a recipient of a CIHR Master's award.
Editor: A. Camilli
Footnotes
Published ahead of print on 11 January 2010.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anriany, Y. A., R. M. Weiner, J. A. Johnson, C. E. De Rezende, and S. W. Joseph. 2001. Salmonella enterica serovar Typhimurium DT104 displays a rugose phenotype. Appl. Environ. Microbiol. 67:4048-4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arana, I., A. Muela, J. Iriberri, L. Egea, and I. Barcina. 1992. Role of hydrogen peroxide in loss of culturability mediated by visible light in Escherichia coli in a freshwater ecosystem. Appl. Environ. Microbiol. 58:3903-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartolome, B., Y. Jubete, E. Martinez, and F. de la Cruz. 1991. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102:75-78. [DOI] [PubMed] [Google Scholar]
- 5.Beyhan, S., K. Bilecen, S. R. Salama, C. Casper-Lindley, and F. H. Yildiz. 2007. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J. Bacteriol. 189:388-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhaskar, P. V., and N. B. Bhosle. 2006. Bacterial extracellular polymeric substance (EPS): a carrier of heavy metals in the marine food-chain. Environ. Int. 32:191-198. [DOI] [PubMed] [Google Scholar]
- 7.Bisharat, N., V. Agmon, R. Finkelstein, R. Raz, G. Ben-Dror, L. Lerner, S. Soboh, R. Colodner, D. N. Cameron, D. L. Wykstra, D. L. Swerdlow, and J. J. Farmer III. 1999. Clinical, epidemiological, and microbiological features of Vibrio vulnificus biogroup 3 causing outbreaks of wound infection and bacteraemia in Israel. Lancet 354:1421-1424. [DOI] [PubMed] [Google Scholar]
- 8.Blake, P. A., R. E. Weaver, and D. G. Hollis. 1980. Diseases of humans other than cholera caused by vibrios. Annu. Rev. Microbiol. 34:341-367. [DOI] [PubMed] [Google Scholar]
- 9.Blokesch, M., and G. K. Schoolnik. 2007. Serogroup conversion of Vibrio cholerae in aquatic reservoirs. PLoS Pathog. 3:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boles, B. R., and L. L. McCarter. 2002. Vibrio parahaemolyticus scrABC, a novel operon affecting swarming and capsular polysaccharide regulation. J. Bacteriol. 184:5946-5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casper-Lindley, C., and F. H. Yildiz. 2004. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J. Bacteriol. 186:1574-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty, S., G. B. Nair, and S. Shinoda. 1997. Pathogenic vibrios in the natural aquatic environment. Rev. Environ. Health 12:63-80. [DOI] [PubMed] [Google Scholar]
- 13.Chatzidaki-Livanis, M., M. K. Jones, and A. C. Wright. 2006. Genetic variation in the Vibrio vulnificus group 1 capsular polysaccharide operon. J. Bacteriol. 188:1987-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coleman, W. G., Jr. 1983. The rfaD gene codes for ADP-L-glycero-D-mannoheptose-6-epimerase. An enzyme required for lipopolysaccharide core biosynthesis. J. Biol. Chem. 258:1985-1990. [PubMed] [Google Scholar]
- 15.Demarre, G., A. M. Guerout, C. Matsumoto-Mashimo, D. A. Rowe-Magnus, P. Marliere, and D. Mazel. 2005. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res. Microbiol. 156:245-255. [DOI] [PubMed] [Google Scholar]
- 16.DePaola, A., G. M. Capers, and D. Alexander. 1994. Densities of Vibrio vulnificus in the intestines of fish from the U.S. Gulf Coast. Appl. Environ. Microbiol. 60:984-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fong, J. C., K. Karplus, G. K. Schoolnik, and F. H. Yildiz. 2006. Identification and characterization of RbmA, a novel protein required for the development of rugose colony morphology and biofilm structure in Vibrio cholerae. J. Bacteriol. 188:1049-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fong, J. C., and F. H. Yildiz. 2007. The rbmBCDEF gene cluster modulates development of rugose colony morphology and biofilm formation in Vibrio cholerae. J. Bacteriol. 189:2319-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foote, C. S. 1976. Photosensitized oxidation and singlet oxygen: consequences in biological systems, p. 85-134. In W. A. Pryor (ed.), Free radicals in biology. Academic Press, New York, NY.
- 20.Frolund, B., T. Griebe, and P. H. Nielsen. 1995. Enzymatic activity in the activated-sludge floc matrix. Appl. Microbiol. Biotechnol. 43:755-761. [DOI] [PubMed] [Google Scholar]
- 21.Garrison, T. 1995. Essentials of oceanography. Wadsworth Publishing Company, Belmont, CA.
- 22.Gasteiger, E., C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel, and A. Bairoch. 2005. Protein identification and analysis tools on the ExPASy server, p. 571-607. In J. M. Walker (ed.), The proteomics protocols handbook. Humana Press, Totowa, NJ.
- 23.Gourmelon, M., D. Touati, M. Pommepuy, and M. Cormier. 1997. Survival of Escherichia coli exposed to visible light in seawater: analysis of rpoS-dependent effects. Can. J. Microbiol. 43:1036-1043. [DOI] [PubMed] [Google Scholar]
- 24.Grau, B. L., M. C. Henk, K. L. Garrison, B. J. Olivier, R. M. Schulz, K. L. O'Reilly, and G. S. Pettis. 2008. Further characterization of Vibrio vulnificus rugose variants and identification of a capsular and rugose exopolysaccharide gene cluster. Infect. Immun. 76:1485-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grau, B. L., M. C. Henk, and G. S. Pettis. 2005. High-frequency phase variation of Vibrio vulnificus 1003: isolation and characterization of a rugose phenotypic variant. J. Bacteriol. 187:2519-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gulig, P. A., K. L. Bourdage, and A. M. Starks. 2005. Molecular pathogenesis of Vibrio vulnificus. J. Microbiol. 43:118-131. [PubMed] [Google Scholar]
- 27.Gulig, P. A., M. S. Tucker, P. C. Thiaville, J. L. Joseph, and R. N. Brown. 2009. USER friendly cloning coupled with chitin-based natural transformation enables rapid mutagenesis of Vibrio vulnificus. Appl. Environ. Microbiol. 75:4936-4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guvener, Z. T., and L. L. McCarter. 2003. Multiple regulators control capsular polysaccharide production in Vibrio parahaemolyticus. J. Bacteriol. 185:5431-5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall-Stoodley, L., and P. Stoodley. 2005. Biofilm formation and dispersal and the transmission of human pathogens. Trends Microbiol. 13:7-10. [DOI] [PubMed] [Google Scholar]
- 31.Harris-Young, L., M. L. Tamplin, J. W. Mason, H. C. Aldrich, and J. K. Jackson. 1995. Viability of Vibrio vulnificus in association with hemocytes of the American oyster (Crassostrea virginica). Appl. Environ. Microbiol. 61:52-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harwood, V. J., J. P. Gandhi, and A. C. Wright. 2004. Methods for isolation and confirmation of Vibrio vulnificus from oysters and environmental sources: a review. J. Microbiol. Methods 59:301-316. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann, T. J., B. Nelson, R. Darouiche, and T. Rosen. 1988. Vibrio vulnificus septicemia. Arch. Intern. Med. 148:1825-1827. [PubMed] [Google Scholar]
- 34.Hor, L. I., T. T. Chang, and S. T. Wang. 1999. Survival of Vibrio vulnificus in whole blood from patients with chronic liver diseases: association with phagocytosis by neutrophils and serum ferritin levels. J. Infect. Dis. 179:275-278. [DOI] [PubMed] [Google Scholar]
- 35.Hulo, N., A. Bairoch, V. Bulliard, L. Cerutti, E. De Castro, P. S. Langendijk-Genevaux, M. Pagni, and C. J. A. Sigrist. 2006. The PROSITE database. Nucleic Acids Res. 34:D227-D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenal, U., and J. Malone. 2006. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 40:385-407. [DOI] [PubMed] [Google Scholar]
- 37.Joseph, L. A., and A. C. Wright. 2004. Expression of Vibrio vulnificus capsular polysaccharide inhibits biofilm formation. J. Bacteriol. 186:889-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim, H. S., M. A. Lee, S. J. Chun, S. J. Park, and K. H. Lee. 2007. Role of NtrC in biofilm formation via controlling expression of the gene encoding an ADP-glycero-manno-heptose-6-epimerase in the pathogenic bacterium, Vibrio vulnificus. Mol. Microbiol. 63:559-574. [DOI] [PubMed] [Google Scholar]
- 39.Kim, Y. K., and L. L. McCarter. 2007. ScrG, a GGDEF-EAL protein, participates in regulating swarming and sticking in Vibrio parahaemolyticus. J. Bacteriol. 189:4094-4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knight, S. D., J. Berglund, and D. Choudhury. 2000. Bacterial adhesins: structural studies reveal chaperone function and pilus biogenesis. Curr. Opin. Chem. Biol. 4:653-660. [DOI] [PubMed] [Google Scholar]
- 41.Kumamoto, K. S., and D. J. Vukich. 1998. Clinical infections of Vibrio vulnificus: a case report and review of the literature. J. Emerg. Med. 16:61-66. [DOI] [PubMed] [Google Scholar]
- 42.Kumar, A. S., K. Mody, and B. Jha. 2007. Bacterial exopolysaccharides-a perception. J. Basic Microbiol. 47:103-117. [DOI] [PubMed] [Google Scholar]
- 43.Leriche, V., R. Briandet, and B. Carpentier. 2003. Ecology of mixed biofilms subjected daily to a chlorinated alkaline solution: spatial distribution of bacterial species suggests a protective effect of one species to another. Environ. Microbiol. 5:64-71. [DOI] [PubMed] [Google Scholar]
- 44.Lim, B., S. Beyhan, and F. H. Yildiz. 2007. Regulation of Vibrio polysaccharide synthesis and virulence factor production by CdgC, a GGDEF-EAL domain protein, in Vibrio cholerae. J. Bacteriol. 189:717-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linkous, D. A., and J. D. Oliver. 1999. Pathogenesis of Vibrio vulnificus. FEMS Microbiol. Lett. 174:207-214. [DOI] [PubMed] [Google Scholar]
- 46.Marco-Noales, E., M. Milan, B. Fouz, E. Sanjuan, and C. Amaro. 2001. Transmission to eels, portals of entry, and putative reservoirs of Vibrio vulnificus serovar E (biotype 2). Appl. Environ. Microbiol. 67:4717-4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDougald, D., W. H. Lin, S. A. Rice, and S. Kjelleberg. 2006. The role of quorum sensing and the effect of environmental conditions on biofilm formation by strains of Vibrio vulnificus. Biofouling 22:133-144. [DOI] [PubMed] [Google Scholar]
- 48.Meibom, K. L., M. Blokesch, N. A. Dolganov, C. Y. Wu, and G. K. Schoolnik. 2005. Chitin induces natural competence in Vibrio cholerae. Science 310:1824-1827. [DOI] [PubMed] [Google Scholar]
- 49.Miller, M. B., and B. L. Bassler. 2001. Quorum sensing in bacteria. Annu. Rev. Microbiol. 55:165-199. [DOI] [PubMed] [Google Scholar]
- 50.Monds, R. D., P. D. Newell, R. H. Gross, and G. A. O'Toole. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol. Microbiol. 63:656-679. [DOI] [PubMed] [Google Scholar]
- 51.Morris, J. G., Jr. 2003. Cholera and other types of vibriosis: a story of human pandemics and oysters on the half shell. Clin. Infect. Dis. 37:272-280. [DOI] [PubMed] [Google Scholar]
- 52.Morris, J. G., Jr., M. B. Sztein, E. W. Rice, J. P. Nataro, G. A. Losonsky, P. Panigrahi, C. O. Tacket, and J. A. Johnson. 1996. Vibrio cholerae O1 can assume a chlorine-resistant rugose survival form that is virulent for humans. J. Infect. Dis. 174:1364-1368. [DOI] [PubMed] [Google Scholar]
- 53.Nakhamchik, A., C. Wilde, and D. A. Rowe-Magnus. 2008. Cyclic-di-GMP regulates extracellular polysaccharide production, biofilm formation, and rugose colony development by Vibrio vulnificus. Appl. Environ. Microbiol. 74:4199-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakhamchik, A., C. Wilde, and D. A. Rowe-Magnus. 2007. Identification of a Wzy polymerase required for group IV capsular polysaccharide and lipopolysaccharide biosynthesis in Vibrio vulnificus. Infect. Immun. 75:5550-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nieto, J. M., M. J. Bailey, C. Hughes, and V. Koronakis. 1996. Suppression of transcription polarity in the Escherichia coli haemolysin operon by a short upstream element shared by polysaccharide and DNA transfer determinants. Mol. Microbiol. 19:705-713. [DOI] [PubMed] [Google Scholar]
- 56.Oliver, J. D., R. A. Warner, and D. R. Cleland. 1982. Distribution and ecology of Vibrio vulnificus and other lactose-fermenting marine vibrios in coastal waters of the southeastern United States. Appl. Environ. Microbiol. 44:1404-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliver, J. D., R. A. Warner, and D. R. Cleland. 1983. Distribution of Vibrio vulnificus and other lactose-fermenting vibrios in the marine environment. Appl. Environ. Microbiol. 45:985-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O'Toole, G., H. B. Kaplan, and R. Kolter. 2000. Biofilm formation as microbial development. Annu. Rev. Microbiol. 54:49-79. [DOI] [PubMed] [Google Scholar]
- 59.O'Toole, G. A., and R. Kolter. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449-461. [DOI] [PubMed] [Google Scholar]
- 60.Paranjpye, R. N., J. C. Lara, J. C. Pepe, C. M. Pepe, and M. S. Strom. 1998. The type IV leader peptidase/N-methyltransferase of Vibrio vulnificus controls factors required for adherence to HEp-2 cells and virulence in iron-overloaded mice. Infect. Immun. 66:5659-5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paranjpye, R. N., and M. S. Strom. 2005. A Vibrio vulnificus type iv pilin contributes to biofilm formation, adherence to epithelial cells, and virulence. Infect. Immun. 73:1411-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powell, J. L. 1999. Vibrio species. Clin. Lab. Med. 19:537-552, vi. [PubMed] [Google Scholar]
- 63.Rashid, M. H., C. Rajanna, D. Zhang, V. Pasquale, L. S. Magder, A. Ali, S. Dumontet, and D. K. Karaolis. 2004. Role of exopolysaccharide, the rugose phenotype and VpsR in the pathogenesis of epidemic Vibrio cholerae. FEMS Microbiol. Lett. 230:105-113. [DOI] [PubMed] [Google Scholar]
- 64.Reeves, P. R., M. Hobbs, M. A. Valvano, M. Skurnik, C. Whitfield, D. Coplin, N. Kido, J. Klena, D. Maskell, C. R. Raetz, and P. D. Rick. 1996. Bacterial polysaccharide synthesis and gene nomenclature. Trends Microbiol. 4:495-503. [DOI] [PubMed] [Google Scholar]
- 65.Reisner, A., J. A. Haagensen, M. A. Schembri, E. L. Zechner, and S. Molin. 2003. Development and maturation of Escherichia coli K-12 biofilms. Mol. Microbiol. 48:933-946. [DOI] [PubMed] [Google Scholar]
- 66.Rice, E. W., C. J. Johnson, R. M. Clark, K. R. Fox, D. J. Reasoner, M. E. Dunnigan, P. Panigrahi, J. A. Johnson, and J. G. Morris, Jr. 1992. Chlorine and survival of “rugose” Vibrio cholerae. Lancet 340:740. [DOI] [PubMed] [Google Scholar]
- 67.Romling, U., and D. Amikam. 2006. Cyclic di-GMP as a second messenger. Curr. Opin. Microbiol. 9:218-228. [DOI] [PubMed] [Google Scholar]
- 68.Romling, U., M. Gomelsky, and M. Y. Galperin. 2005. C-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57:629-639. [DOI] [PubMed] [Google Scholar]
- 69.Reference deleted.
- 70.Sauer, F. G., M. A. Mulvey, J. D. Schilling, J. J. Martinez, and S. J. Hultgren. 2000. Bacterial pili: molecular mechanisms of pathogenesis. Curr. Opin. Microbiol. 3:65-72. [DOI] [PubMed] [Google Scholar]
- 71.Sauer, K. 2003. The genomics and proteomics of biofilm formation. Genome Biol. 4:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simon, R., U. B. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1:784-791. [Google Scholar]
- 73.Starks, A. M., T. R. Schoeb, M. L. Tamplin, S. Parveen, T. J. Doyle, P. E. Bomeisl, G. M. Escudero, and P. A. Gulig. 2000. Pathogenesis of infection by clinical and environmental strains of Vibrio vulnificus in iron-dextran-treated mice. Infect. Immun. 68:5785-5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stoodley, P., K. Sauer, D. G. Davies, and J. W. Costerton. 2002. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56:187-209. [DOI] [PubMed] [Google Scholar]
- 75.Stroeher, U. H., K. E. Jedani, B. K. Dredge, R. Morona, M. H. Brown, L. E. Karageorgos, M. J. Albert, and P. A. Manning. 1995. Genetic rearrangements in the rfb regions of Vibrio cholerae O1 and O139. Proc. Natl. Acad. Sci. U. S. A. 92:10374-10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strom, M. S., and R. N. Paranjpye. 2000. Epidemiology and pathogenesis of Vibrio vulnificus. Microb. Infect. 2:177-188. [DOI] [PubMed] [Google Scholar]
- 77.Sutherland, I. W. 2001. The biofilm matrix—an immobilized but dynamic microbial environment. Trends Microbiol. 9:222-227. [DOI] [PubMed] [Google Scholar]
- 78.Tamplin, M. L., and G. M. Capers. 1992. Persistence of Vibrio vulnificus in tissues of Gulf Coast oysters, Crassostrea virginica, exposed to seawater disinfected with UV light. Appl. Environ. Microbiol. 58:1506-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thompson, F. L., T. Iida, and J. Swings. 2004. Biodiversity of vibrios. Microbiol. Mol. Biol. Rev. 68:403-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thormann, K. M., S. Duttler, R. M. Saville, M. Hyodo, S. Shukla, Y. Hayakawa, and A. M. Spormann. 2006. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J. Bacteriol. 188:2681-2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tischler, A. D., and A. Camilli. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 53:857-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Todd, E. C. 1989. Costs of acute bacterial foodborne disease in Canada and the United States. Int. J. Food Microbiol. 9:313-326. [DOI] [PubMed] [Google Scholar]
- 83.Wai, S. N., Y. Mizunoe, A. Takade, S. I. Kawabata, and S. I. Yoshida. 1998. Vibrio cholerae O1 strain TSI-4 produces the exopolysaccharide materials that determine colony morphology, stress resistance, and biofilm formation. Appl. Environ. Microbiol. 64:3648-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wai, S. N., Y. Mizunoe, and S. Yoshida. 1999. How Vibrio cholerae survive during starvation. FEMS Microbiol. Lett. 180:123-131. [DOI] [PubMed] [Google Scholar]
- 85.Warnock, E. W., III, and T. L. MacMath. 1993. Primary Vibrio vulnificus septicemia. J. Emerg. Med. 11:153-156. [DOI] [PubMed] [Google Scholar]
- 86.Whitfield, C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75:39-68. [DOI] [PubMed] [Google Scholar]
- 87.Wolfe, A. J., and K. L. Visick. 2008. Get the message out: cyclic-di-GMP regulates multiple levels of flagellum-based motility. J. Bacteriol. 190:463-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wright, A. C., R. T. Hill, J. A. Johnson, M. C. Roghman, R. R. Colwell, and J. G. Morris, Jr. 1996. Distribution of Vibrio vulnificus in the Chesapeake Bay. Appl. Environ. Microbiol. 62:717-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yildiz, F. H., and G. K. Schoolnik. 1999. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 96:4028-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]