

Abstract
Neisseria gonorrhoeae is an obligate human pathogen that causes gonorrhea. We have shown previously that complement receptor 3 and Akt kinase play important roles in mediating cervical infection. At present, there are limited data to indicate how hormonally induced changes to the mucosal epithelia of the female genital tract mediate the course of gonococcal disease. Hence, I have expanded upon previous work to investigate the interaction of gonococci with primary human cervical epithelial (pex) cells under the variable estrogen and progesterone concentrations likely to be encountered in vivo throughout the female menstrual cycle. My data indicated that the ability of gonococci to survive and to replicate within pex cells was increased under progesterone-predominant conditions. Using bacterial survival, immunological, and kinase assays, I show that progesterone functioned in an additive manner with gonococcal phospholipase D to augment Akt kinase activity. This, in turn, resulted in a parallel increase in nitric oxide synthase expression. Nitric oxide production by pex cells was dependent upon Akt activity and was increased under progesterone-predominant conditions. Whereas both inducible and endothelial nitric oxide synthase contributed to nitric oxide production, only inducible nitric oxide synthase activity promoted gonococcal survival within pex cells. Collectively, these data provide the first clues as to how steroid hormones potentially modulate the course of gonococcal disease in women. In addition, these data demonstrate that host-derived nitric oxide likely is not protective against gonococci, in vivo; rather, nitric oxide may be required to sustain cervical bacterial disease.
Neisseria gonorrhoeae is an exclusive human pathogen that causes the sexually transmitted disease, gonorrhea. The gonococcus is highly human adapted and thus has developed variable mechanisms of pathogenesis that are, in part, dependent upon the site of infection (reviewed in reference 6). We have shown previously that complement receptor type 3 (CR3) serves as the key receptor by which gonococci initiate cervical infection in vivo, as well as ex vivo in primary human cervical epithelial (pex) cells (8). Gonococci exposed to pex cells release a group of proteins, including a phospholipase D (NgPLD), that facilitate cervical adherence and invasion (9).
Invasion of certain epithelial cells by gonococci triggers (host) Akt activity (7, 31). Akt is a serine-threonine kinase implicated in diverse cellular functions, and several bacterial and viral pathogens have developed mechanisms by which to subvert Akt signaling to promote disease (7, 22, 26, 28, 53, 56). In contrast to vulvular epidermal carcinoma (A-431) cells, in which gonococcus-induced Akt activity requires phosphatidylinositol 3-kinase (PI3K) (31), Akt activation in pex cells occurs independently of PI3K and results from the direct interaction of NgPLD with cervical Akt (7).
Despite the many advances made in our understanding of gonococcal pathogenesis, the factors governing the divergent presentation of gonococcal disease observed between men and women remain unclear. That is, whereas infection of the male urethra typically results in an acute inflammatory response, cervical gonorrhea is predominantly asymptomatic. This asymptomatic state has an extreme impact on the prevalence of gonorrhea in the general population and in the chronic disease sequelae observed, disproportionately, among women. Infections in women are compounded by a number of (host) physiological factors that cooperatively complicate disease. For example, clinical data indicate that gonococcal disease in females may be governed, in part, by steroid hormones (23, 24, 29, 33, 37). Notwithstanding, there are limited data addressing how steroid hormones modulate gonococcal pathogenesis.
Estradiol (E2) and progesterone (Pg) represent the major form of estrogens and progestagens, respectively, found in women. Although other estrogens and progestagens (e.g., estrone and 17-α-hydroxyprogesterone) exist in vivo, these hormone derivatives represent minor hormone species and thus are only present in minute quantities in women. With regard to asymptomatic cervical infection, it is noteworthy that steroid hormones (e.g., E2 and Pg) can exhibit anti-inflammatory, as well as immunomodulatory, properties. Although not shown for cervical epithelial cells, there are data to indicate that, under some conditions, hormone-dependent Akt activation can induce nitric oxide synthase (NOS) expression and, consequently, nitric oxide (NO) generation (14, 17, 18, 20). Although little is known about the expression or the subcellular location of NOS isoforms within the cervix, an increase in NO intermediates (i.e., nitrate, nitrite, and NO [referred to as NOX collectively]) within the female genital tract is believed to play a role in cervical ripening and in other processes associated with normal and preterm labor (51). Thus, the ability of gonococci to induce NO production by cervical epithelial cells, in an Akt-dependent manner, could have important consequences with regard to women's health. Hence, I initiated studies to examine the effect(s) of physiological levels of E2 and Pg on N. gonorrhoeae infection of primary cervical cells. My data suggest that the CR3-NgPLD-Akt pathway plays a role in promoting N. gonorrhoeae infection through Pg-enhanced NOS activity with subsequent NO production. My data further demonstrate that host-derived NO does not exert a bactericidal effect on gonococci but instead plays an important role in promoting the intracellular survival of these bacteria during pex cell challenge.
MATERIALS AND METHODS
Cell culture, bacteria, and infection studies.
Primary cervical epithelial (pex) cells were procured from surgical cervical tissue and maintained as described previously (11). Cervical tissue was obtained from premenopausal women undergoing hysterectomy at The Ohio State University Medical Center for medically indicated reasons not related to our study and was provided by the Cooperative Human Tissue Network (The Research Institute at Nationwide Children's Hospital, Columbus, OH). In accordance with National Institutes of Health (NIH) guidelines, these tissues do not constitute human subjects. Pex cell monolayers were incubated in the presence of various concentrations (as noted) of E2 and/or Pg for at least 24 h before their use, as indicated in the experiments described below. Hormones were maintained in the culture medium throughout the course of each assay. E2 (Sigma, St. Louis, MO) and Pg (Sigma) were selected for use in infection studies because these hormones represent the major forms of estrogens and of progestagens (respectively) found in women. To avoid potential complicating factors associated with phenol red (35), antibiotic- and phenol red-free medium was used in assays designed to assess the affect(s) of steroid hormones on gonococcal infection. E2 and Pg were solubilized in 2-butanol (Sigma) as our previous studies demonstrated that this reagent does not interfere with the ability of gonococci to associate with or to invade pex cells (9). Pharmacological agents used in these studies were not cytotoxic (viability greater than 95%) at the indicated concentrations as determined by trypan blue exclusion (pex cells) or by counting CFU of gonococci incubated in the presence of, compared to the absence of, each reagent.
N. gonorrhoeae strains 1291 (1) and 1291ΔPLD (9) were used in the infection studies described herein. Strain 1291 was originally isolated from a male with gonococcal urethritis and is commonly used to study gonococcal pathogenesis. Infection studies were performed as previously described using a multiplicity of infection of 100 (11); gonococci were enumerated spectrophotometrically. The pex cells were challenged with gonococci for variable time periods (as noted). Uninfected, control cell monolayers were simultaneously processed with challenged cell monolayers, as described previously and as outlined below. Where indicated, pharmacological agents were included in or excluded from infection studies, and these included 1 mM (±)-S-nitroso-N-acetylpenicillamine (SNAP; NO donor), 1 mM SIN-1 HCl (SIN; NO donor), 500 μM S-nitrosoglutathione (GSNO; NO donor/low-molecular-mass thiol), 300 nM S-methyl-l-thiocitrulline (MTC; NOS1 inhibitor), 10 μM S-methylisothiourea (SMT; NOS2 inhibitor), 500 nM l-N5-(1-iminoethyl)ornithine (NIO; NOS3 inhibitor), 50 μM 1-(2-trifluoromethylphenyl)imidazole (TRIM; inhibitor of NOS1 and NOS2), and 500 nM NG-nitro-l-arginine methyl ester (NAME; inhibitor of NOS1 and NOS3) (all obtained from Calbiochem, La Jolla, CA) and 150 nM N-(4S)-(4-amino-5-[aminoethyl]aminopentyl)-N′-nitroguanidine tris (trifluoroacetate) (NOS1inh), 700 nM NG-monomethyl-l-arginine (NMMA; inhibitor of NOS1 and NOS3), 10 nM 1400W (NOS2 inhibitor), 10 mM 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO; NO-specific scavenger), and 20 mM NaNO2 (nitrite) (all from Sigma). Akt inhibitor VII (1 μM) was also included in select experiments. This inhibitor molecule prevents Akt activity by specifically binding to the Akt pleckstrin homology (PH) domain (19), thereby preventing phosphoinositide (as well as NgPLD) from binding to this kinase (7). Cervical cell monolayers were preincubated (1 h, 37°C) with each respective pharmacological agent before the addition of gonococci.
N. gonorrhoeae association, invasion, and survival assays.
The pex cell monolayers were infected with wild-type 1291 or 1291ΔPLD mutant gonococci as outlined above. Where indicated, pex cells were treated with E2, Pg, or E2 plus Pg (E2/Pg) also as described above and in which hormones were maintained in the culture medium over the time course assayed. Quantitative association, invasion, and survival assays were performed as we have described previously (55) and in which chemical agents were included in or excluded from the assay, as noted. Briefly, pex cells were challenged with gonococci at a multiplicity of infection of 100 for 90 min. Gentamicin was then omitted from (association assays) or added to (invasion and intracellular survival assays) infected pex cell monolayers to kill extracellular cell-associated bacteria. The pex cell monolayers were subsequently lysed, or they were subjected to a second incubation in antibiotic-free medium before cell lysis (intracellular survival assays). For each set of experiments, gonococci and pex cells were cultured under standard laboratory conditions (i.e., 5% CO2) or under microaerobic conditions before the onset of pex cell challenge. Mitsubishi (Pouch-MicroAero) gas-generating pouch systems were used to generate microaerobic conditions, because they rapidly (under 30 min) generate the desired oxygen-limited (6% O2, 5 to 8% CO2) environment. For assays performed under microaerobic conditions, microaerobically cultured gonococci were used to challenge microaerobically incubated pex cell monolayers to which an NO scavenger, nitrite, and/or NOS inhibitors were added or omitted. After the addition of gonococci, infected pex cells were returned to a microaerobic environment. Separate gas pouches were used for each assay time point and care was exercised throughout the assay to minimize exposure to air. In this regard, air was vacuum-evacuated from pouches before the 30 min incubation with gentamicin. For all assays, serial dilutions of the cell lysates were plated to enumerate viable CFU. The percent association, invasion, or survival was determined as a function of the original inoculum and the number of colonies formed with subsequent plating of the cellular lysate. Each assay was performed in triplicate on at least three separate occasions. A Kruskal-Wallis analysis of variance was used to determine the statistical significance of the calculated percent association, invasion, or survival for each assay.
Immunoprecipitation, Western blotting, and enzyme-linked immunosorbent assay (ELISA).
Confluent pex cell monolayers in 35-mm tissue culture-treated dishes were challenged for various periods of time, as noted, with wild-type or mutant N. gonorrhoeae as described above, or they were left uninfected. Pex cell challenge was subsequently terminated by immediately transferring the dishes to ice, removing the infection supernatant, and rinsing the cell monolayer thrice with ice-cold phosphate-buffered saline (PBS) before cell lysis in immunoprecipitation (IP) assay buffer. IP then was performed as we described previously (52) using anti-NOS2 (H-174), anti-NOS3 (H-159), or anti-Akt (N-19) antibodies (all from Santa Cruz Biotechnology, Santa Cruz, CA) to capture immune complexes. Captured immune complexes were separated on 4 to 12% denaturing polyacrylamide gradient gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Western blotting was performed according to standard protocols using anti-phospho-Akt antibody (p-Akt1/2/3; Ser 473), anti-NOS2 antibody (C-11), or the anti-phospho-NOS3 antibody (NOS3-P; Ser1177) (all from Santa Cruz). The anti-Akt antibody, p-Akt1/2/3 (Ser 473), recognizes Akt only when phosphorylated on a critical serine residue (Ser473 for Akt1) when Akt is in its active (phosphorylated) state. Serine 1177 of NOS3 serves as a site for Akt-mediated phosphorylation. To ensure equal loading, membranes were stripped and reprobed with the anti-Akt antibody B-1, which recognizes Akt1, Akt2, and Akt3 of human origin. After Western blotting, antibody-labeled NOS2, NOS3, and Akt were detected with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL). The assays were repeated on at least two separate occasions.
As a correlate to and to confirm data obtained by Western blot analyses, ELISAs were performed using whole pex cell lysates obtained from uninfected cells or cells that were challenged in parallel to those used for IP with the Western blot analysis described above. To this end, 100 μl (1/10 vol) of each pex cell lysate was used to coat select wells within microtiter plates, after which wells were rinsed, and nonspecific binding sites were blocked (30 min, room temperature) with PBS-0.25% bovine serum albumin-0.05% Tween 20. NOS2 and actin were then quantitated by using standard ELISA protocols. The anti-NOS2 and anti-actin primary antibodies, C-19 and H-300 (Santa Cruz), respectively, were used, followed by the appropriate fluorescein-conjugated secondary antibodies (Jackson Immunoresearch), to increase assay sensitivity. Arbitrary fluorescence units were recorded (485-nm excitation and 530-nm emission) by using a Synergy HT Multi-Mode microplate reader (BioTek). Blank wells, as well as the omission of the primary or the secondary antibody, served as controls for nonspecific binding and for autofluorescence. Each assay was performed in triplicate on three separate occasions. Statistical significance of data obtained was determined by using Student's t test.
Akt kinase assay.
Confluent pex cell monolayers were left untreated or they were pretreated (37°C, 1 h) with 1 μM Akt inhibitor VII (Calbiochem). Cell monolayers were then left uninfected, or they were challenged with 1291 wild-type bacteria for 3 h. Infections were terminated by immediately transferring the dishes to ice, removing the infection medium, and rinsing the cells thrice with ice-cold PBS before cell lysis in IP assay buffer. Total cellular Akt was captured as outlined above using the anti-Akt C-20AC agarose-conjugated antibody (Santa Cruz). The Akt kinase reaction was carried out using GSK-3α peptide substrate (37 kDa; BioVision, Mountain View, CA) as we have described previously (7). Western blotting was performed using the phospho-GSK-3α/β (Ser21/9) antibody (Cell Signaling Technology, Danvers, MA). Negative controls for each assay included the omission of the immobilized Akt antibody or the GSK-3α peptide substrate. To ensure equal loading, membranes were stripped and reprobed using anti-Akt antibody B-1, as described above.
Quantitation of NOX production by pex cells.
Pex cell monolayers were incubated in Krebs buffer (145 mM NaCl, 5.7 mM NaPO4, 4.86 mM KCl, 0.54 mM CaCl2, 1.22 mM MgSO4, 5.5 mM glucose) 24 h before, and during, their use in assays to measure total cervical NOX production. The pex cells were then left uninfected, or they were challenged with wild-type N. gonorrhoeae, as noted. Culture supernatants were then harvested, centrifuged, and passed through a 0.22-μm-pore-size syringe filter (Millipore) to remove bacteria. Total NOX in clarified pex cell culture supernatants was measured after the reduction of nitrate to nitrite using Griess reagents according to the manufacturer's instructions (Cayman Chemical Co., Ann Arbor, MI). The absorbance of the resulting azo chromophore was read at 540 nm using a Synergy HT Multi-Mode microplate reader (BioTek), and the total NOX was quantitated by comparison to a nitrite standard curve.
Direct determination of NO production by pex cells.
Pex cells were passed to 96-well microtiter plates and allowed to grow to confluence. Before the onset of the infection studies, confluent cell monolayers were incubated (37°C, 1 h) in phenol red-free medium containing cell-impermeant 4-amino-5-methylamino-2′,7′-difluorescein (DAF-FM) or cell-permeant DAF-FM diacetate NO reactive probes (Invitrogen Molecular Probes, Carlsbad, CA). After this incubation, wells containing DAF-FM diacetate were rinsed thrice with PBS to remove any remaining probe that had not permeated the pex cell membrane, thereby allowing the analysis of intracellular NO independently of extracellular NO. For assays in which total NO production was measured, pex cells were incubated with both DAF-FM and DAF-FM diacetate, each of which were retained within the microtiter plate wells throughout the course of the assay. The pex cell monolayers then were left unchallenged, or they were challenged with 1219 wild-type or 1291ΔPLD gonococci. Where indicated, E2/Pg was included (as outlined above) or omitted from the culture medium. Similarly, Akt inhibitor VII or NOS inhibitors were included or omitted from each assay, as noted and as described above. Microtiter plates were immediately placed in a Synergy HT Multi-Mode microplate reader (BioTek) after infection, and fluorescence, which is indicative of NO generation, was read at 530 nm (i.e., 485-nm excitation and 530-nm emission). Arbitrary fluorescence units were recorded every 30 min for at least 18 h. Assays were performed in quadruplicate on at least three separate occasions.
RESULTS
Pg augments gonococcal invasion of and/or their survival within pex cells.
In vivo, during the first 14 days of the menstrual cycle (i.e., the follicular phase), E2 levels gradually rise from ∼0.1 to ∼0.7 nM, after which they again decrease (see Table S1 in the supplemental material). During this time, Pg levels remain relatively stable at ∼3 nM. Within the next 14 days (the luteal phase) of the menstrual cycle, E2 levels moderately increase to ∼0.4 nM, whereas the Pg levels rise dramatically to reach ∼30 nM. The concentration of each of these hormones gradually decreases late in the luteal phase to approximately 0.1 nM E2 and 3 nM Pg before the onset of, and during, menses. My investigations of the affect of steroid hormones on cervical gonorrhea lead to the hypothesis that Pg-predominant conditions, such as would occur during the menstrual cycle luteal phase, promote gonococcal invasion of or survival within pex cells. I thereby measured the invasive and survival capabilities of N. gonorrhoeae using pex cells cultured in the presence or absence of a range of E2 or Pg concentrations. In support of our hypothesis, quantitative gentamicin survival (i.e., invasion) assays demonstrated that the invasive capability of wild-type N. gonorrhoeae1291 was not significantly affected by E2 (Fig. 1A). However, increasing concentrations of Pg resulted in a parallel increase in the ability of these organisms to survive gentamicin treatment after pex cell challenge (Fig. 1B). I then repeated these assays using 0.4 nM E2 combined with various concentrations of Pg (0 to 30 nM) to allow us to more closely mimic the Pg-predominant luteal phase of the menstrual cycle. I again found that a parallel increase in gonococcal invasion occurred with increasing concentrations of Pg (Fig. 1C). Collectively, these data supported my hypothesis that Pg augments gonococcal invasion of and/or their intracellular survival within pex cells.
FIG. 1.

Pg promotes the invasion of pex cells by N. gonorrhoeae. Pex cells were challenged for 90 min with N. gonorrhoeae strain 1291 in the presence or absence of various concentrations of E2 (A), Pg (B), or E2 plus Pg (C), as indicated. Invasion (90 min infection plus 30 min of gentamicin treatment) assays were then performed as described in the text. The values given are the mean (variance) in which the percent invasion of pex cells by gonococci was determined as a function of the original inoculum and the number of CFU formed with subsequent plating of the cervical epithelial cell lysates. The data were obtained from three trials performed in triplicate. P values were determined using a Kruskal-Wallis k-sample analysis of variance. *, P ≤ 0.01 for the invasion of pex cells by gonococci in the presence of (compared to the absence of) Pg or E2/Pg.
Progesterone functions upstream of Akt to promote gonococcal pex cell infection.
I next wanted to perform experiments that would allow me to distinguish between Pg-augmented gonococcal invasion of and/or their intracellular survival within pex cells. To this end, I performed comparative intracellular survival assays (Fig. 2). Our previous data demonstrated that pld mutant gonococci are severely impaired in their ability to invade pex cells. In this regard, we recently showed that a direct NgPLD-Akt interaction occurs during infection of pex cells, which results in increased Akt activity and which is critical for successful gonococcal infection and invasion of these cells (7). Although not shown for the cervix, Akt activity can be hormonally modulated in some cell types (14, 15, 40, 48, 57). Thus, I also wanted to determine whether Pg could rescue the impaired invasive capability of pld mutant gonococci, potentially, by contributing to Akt activation. Thereby, pex cells were left untreated or they were treated with a median (15 nM) concentration of Pg before being challenged with wild-type or pld mutant gonococci, which would permit evaluation of the contribution of Pg in regulating Akt activity in pex cells during gonococcal challenge. An Akt inhibitor was included or omitted from these assays.
FIG. 2.

Effect of Pg and Akt on gonococcal invasion of and survival within pex cells. Pex cells were challenged with N. gonorrhoeae strain 1291 (A) or strain 1291ΔPLD (B) in the presence or absence of 15 nM Pg and/or Akt inhibitor VII, as indicated. Invasion and survival assays were performed as described in the text, where invasion refers to a 90-min infection plus gentamicin treatment. Survival refers to the number of viable gonococci that were recovered from within pex cells at 1 h (Inv + 1 h) or 3 h (Inv + 3 h) after invasion. The values given are the mean (variance) in which the percent invasion or survival of gonococci was determined as a function of the original inoculum and the number of CFU formed with subsequent plating of the cervical epithelial cell lysates. The data were obtained from three trials performed in triplicate. *, P ≥ 0.01 for the survival of gonococci at the indicated time point (Inv + 1 h or Inv + 3 h) compared to the previous time point.
In the absence of Pg, I observed no significant difference (P ≥ 0.24) in the ability of wild-type bacteria to survive within pex cells at 1 h postinvasion compared to their ability to invade these cells (Fig. 2A). However, at 3 h postinvasion the number of viable intracellular gonococci had significantly (P ≤ 0.01) increased by ∼3-fold. In the presence of Pg, a significant increase (compared to the absence of Pg, P ≤ 0.01) in gonococcal invasion, as well as the number of viable gonococci at both 1 h and 3 h postinvasion, was observed (Fig. 2A). Similarly, in the presence of Pg a significant (P ≤ 0.01) increase in gonococcal survival occurred over the time course assayed. In this regard, an ∼1.3-fold increase in viable gonococci occurred at the Inv + 1-h time point after invasion, and an ∼2-fold increase occurred between the Inv + 1-h and Inv + 3-h time points. As expected from our previous studies (7, 10), 1291ΔPLD gonococci were severely impaired in their ability to invade pex cells but also were further impaired in their ability to survive within these cells (Fig. 2B). Pg could not rescue the impaired survival of these mutant bacteria (Fig. 2B), suggesting that Pg promotes gonococcal invasion of and/or their intracellular survival within pex cells in a NgPLD-dependent manner. Consistent with our previous studies (7), Akt inhibition severely impaired the ability of wild-type (Fig. 2A) and pld mutant (Fig. 2B) gonococci to invade pex cells. However, my data further demonstrated that, under conditions of impaired Akt activity, the few invasive gonococci that were able to survive gentamicin treatment did not recover and replicate over the time course assayed, suggesting that Akt also contributes to the intracellular survival of invasive bacteria. This survival defect could not be rescued by the presence of Pg, despite the finding that Pg presence did result in a modest, but significant (P ≤ 0.02), increase in the ability of pld mutant gonococci to invade pex cells treated with an Akt inhibitor (Fig. 2). These data potentially indicated that Pg functions upstream, or at the level, of Akt activation to promote the immediate intracellular survival of N. gonorrhoeae after their invasion into cervical epithelial cells.
Pg augments gonococci-induced Akt activity.
The data presented thus far are highly suggestive of a role for Pg in promoting gonococcal invasion of, and their immediate survival within, pex cells in a NgPLD- and Akt-dependent manner. Hence, I sought to determine whether Pg modulated Akt activity within pex cells challenged with N. gonorrhoeae. To address this line of questioning, I used an antibody to capture total cellular Akt from pex cells that had been cultured in the presence or absence of a median concentration of Pg and subsequently challenged with wild-type or pld mutant gonococci. Western blotting, with an antibody to the phosphorylated, active form of Akt (Akt-P, 55 to 60 kDa) revealed that Akt was not phosphorylated in pld mutant-infected cells in the absence of Pg but did become phosphorylated in the presence of Pg (Fig. 3A). Under these conditions, the level of Akt-P remained constant over the time course assayed (Fig. 3A). To ensure equal loading of Akt immunocomplexes, Western blots were stripped and reprobed using a third anti-Akt antibody to visualize total Akt loaded into each well and onto each gel. Akt-P was readily evident in wild-type-infected pex cells in the absence of Pg; however, in the presence of Pg, a further increase in Akt phosphorylation occurred over the time course assayed (Fig. 3B). Consistent with our previous works (7), an additional band of ∼85 kDa was also evident in wild-type-infected pex cells but was only faintly visible in the absence of Pg. Although we have not yet resolved the identity of this peptide band, we previously speculated that this band results from the covalent interaction of Akt with an unknown peptide (e.g., an adaptor protein) (7). Additional peptide bands of a molecular mass lower then that reported for Akt were also visible and most likely represent degradation products. Phosphorylated Akt was not observed in uninfected pex cells in the presence or absence of Pg, nor was it present when the primary or secondary antibodies were omitted from the initial immunocapture step, demonstrating the specificity of the assay. Thus, these data suggested that Pg and NgPLD function in an additive manner to induce Akt activity and thereby promote gonococcal survival during cervical infection.
FIG. 3.

Pg and NgPLD mediate Akt phosphorylation in pex cells. Pex cells were incubated in the presence or absence of 10 nM Pg, as noted, before and during a time course of 1291ΔPLD mutant (A) or 1291 wild-type (B) gonococcal infection. The length of time for which each pex cell infection was allowed to proceed is indicated across the top of each membrane image. Akt immune complexes were captured in pex cells by using the goat anti-Akt antibody, N-19. Phosphorylated Akt (Akt-P) was detected by using the rabbit anti-phospho-Akt antibody, p-Akt1/2/3 (Ser 473) (upper image in panels A and B). Membranes were stripped and then reprobed using the mouse anti-Akt antibody, B-1, to ensure equal loading of total Akt (bottom image in panels A and B). Akt-P, or total Akt, is visible as a 55- to 60-kDa band on each membrane. UI, uninfected control pex cells; No 1°, the primary antibody was omitted from the initial capture step; No 2°, the agarose-conjugated secondary antibody was omitted from the initial capture step.
Although Akt phosphorylation is indicative of Akt activity, the presence of Akt-P is not a direct measure of Akt kinase activity. Therefore, to confirm the data above, as well as to examine Akt activity in terms of the female menstrual cycle, I performed a kinase assay in which Akt activity was indicated by its ability to phosphorylate the glycogen synthase kinase 3α (GSK-3α; 37 kDa) peptide substrate. Under conditions designed to mimic the Pg-predominant luteal phase of the menstrual cycle, a dose-dependent increase in Akt activity was observed at 3 h postinfection of pex cells with 1291 wild-type gonococci (Fig. 4A). Conversely, E2-predominate conditions had a dose-dependent inhibitory effect on Akt activity (Fig. 4B). I could not detect Akt activity in uninfected cells, in wild-type-infected cells in the presence of an Akt inhibitor, or when I omitted the anti-Akt capture antibody from the initial capture step of the assay or the GSK-3α peptide substrate from the kinase reaction (negative controls). Taken together, these data provide evidence that steroid hormones modulate Akt activity during N. gonorrhoeae infection of pex cells with Pg augmenting, and E2 impairing, Akt activity.
FIG. 4.

Increasing Pg promotes Akt kinase activity. Pex cells were incubated in E2/Pg concentrations reflective of the menstrual cycle luteal (A) or follicular (B) phase and then challenged with N. gonorrhoeae strain 1291, as noted, for 3 h. Akt was immunocaptured from pex cell lysates, and the kinase activity was assessed as described in the text by using a GSK-3α peptide substrate. The presence of an ∼37-kDa band, after immunoblotting with the phospho-GSK-3α/β (Ser21/9) (rabbit) antibody, is consistent with phosphorylation of the GSK-3α peptide (GSK-3α-P) and is indicative of Akt activity. The membranes were stripped and reprobed using the mouse anti-Akt antibody B-1 to ensure the presence of comparable amounts of Akt kinase under each assay condition (lower image in panels A and B). The hormone concentrations (0.4 nM E2 plus 0 to 30 nM Pg in panel A; 3 nM Pg plus 0 to 1 nM E2 in panel B) used are noted across the top of each membrane image. UI, uninfected pex cells; AI, Akt inhibitor VII was included in the assay; Ab, the agarose-conjugated anti-Akt capture antibody, C-20AC, was omitted from the initial capture step; GSK, the GSK-3α peptide substrate was omitted from the kinase reaction.
N. gonorrhoeae-induced nitric oxide synthase expression occurs by a Pg-, NgPLD-, and Akt-mediated pathway.
Akt activation in N. gonorrhoeae-infected pex cells occurs independently of PI3K and results from the direct interaction of NgPLD with cervical Akt (7). In some (noncervical) cells and under certain conditions, PI3K-dependent Akt activation can lead to NOS expression and, consequently, NO generation (14, 17, 18, 20). Although NO is generally believe to exert a cytotoxic effect on bacteria, Neisseria spp. possess a NO detoxification pathway (45) and can use nitrite and/or nitric oxide as an alternative respiratory mechanism (27). NOX are present in cervical secretions (51), and thus these fluids are capable of supporting N. gonorrhoeae growth. In this regard, if gonococcus-induced Akt activity also results in NOS activation, this could have important consequences with regard to women's health. Thereby, I wanted to determine the effect of Pg-modulated Akt activity on NOS expression and NO production by gonococcus-infected pex cells. Pilot assays, performed in the absence of E2/Pg, had revealed that a low level of NOS2 (inducible, iNOS) expression occurs by 3 h postinfection of pex cells with wild-type gonococci that thereafter increased to moderate levels by 6 h postinfection (data not shown). Therefore, I initiated these studies by capturing NOS2 in E2/Pg-treated pex cells that were either uninfected or were challenged for 6 h with wild-type or with pld mutant bacteria to allow examination of the potential role of the NgPLD-Akt pathway in NOS2 induction. Western blot analysis with a second, NOS2-specific antibody revealed that NOS2 (130 kDa) was not expressed in uninfected cells under any of the E2/Pg concentrations assayed (Fig. 5A). However, under conditions reflective of the Pg-predominant luteal phase of the menstrual cycle, infection of pex cells with wild-type N. gonorrhoeae resulted in a visible dose-dependent increase in NOS2 expression (Fig. 5A). Low levels of E2 repressed NOS2 expression compared to the absence of this hormone (i.e., with 3 nM Pg alone). However, increasing concentrations of E2 also resulted in a parallel increase in NOS2 expression, although this level of expression was comparatively lower than that observed for Pg alone (i.e., 3 nM Pg + 0 nM E2) or for Pg-predominant conditions (Fig. 5A).
FIG. 5.

Steroid hormones modulate NOS2 expression in gonococcus-infected pex cells. Pex cells were cultured under conditions that mimic the menstrual cycle, as noted across the top of each Western blot image in panel A or along the x axis in panel B. (A) NOS2 was captured by using (rabbit) antibody H-174 from uninfected cells or from a parallel 6 h infection by using wild-type or pld mutant gonococci. Western blotting was performed with the mouse anti-NOS2 antibody C-11. NOS2 is present in gonococcus-infected, but not uninfected, pex cells as a 130-kDa band. (B) Fluorometric ELISA analysis of pex cell lysates was performed as outlined in the text using the rabbit anti-NOS2 primary antibody C-19 and a fluorescein-conjugated secondary antibody to confirm hormonal modulation of NOS2 expression in pex cells, as is observed in panel A. Fluorescence was recorded at 485-nm excitation and 530-nm emission. Ab (in panel A), the anti-NOS antibody was omitted from the initial NOS2 capture step.
The pex cells challenged using 1291ΔPLD mutant gonococci exhibited an overall lower level of NOS2 expression than that observed for wild-type-infected cells (Fig. 5A). In addition, and in further contrast to data obtained with the use of wild-type gonococci, NOS2 expression did not exhibit a Pg dose-dependent increase when pex cells were challenged with the pld mutant. Rather, in pld mutant-infected pex cells, NOS2 was expressed to the greatest level under conditions of low Pg. Similar data were obtained when I performed semiquantitative, fluorometric, ELISAs to confirm the hormonal modulation of NOS2 expression in pex cells (Fig. 5B). In this regard, analysis of (total) pex cell lysates also revealed an E2/Pg dose-dependent increase in NOS2 expression during the strain 1291 wild-type N. gonorrhoeae challenge; this effect was again more pronounced under conditions reflective of the menstrual cycle luteal phase (Fig. 5B). Similarly, low levels of Pg (i.e., 1 nM Pg plus 0.4 nM E2) again resulted in the greatest increase in NOS2 expression in pex cells challenged with 1291ΔPLD mutant gonococci; however, this increase again was modest compared to wild-type N. gonorrhoeae-challenged cells (Fig. 5B). Only background levels of NOS2 were present in uninfected pex cells (Fig. 5B). Parallel analysis of these same pex cell lysates revealed that the amount of total cellular actin remained unaltered under each condition assayed (see Fig. S1 in the supplemental material). Thereby, these data are consistent with a role for Pg and NgPLD in triggering NOS2 expression in pex cells during gonococcal challenge and, additionally, support our previous publications that demonstrate that pex cells generate alternate functional responses when challenged with strain 1291 wild-type (high-level Akt activity, independent of PI3K) or 1291ΔPLD mutant (low-level Akt activity, dependent upon PI3K) gonococci (7, 10).
Although NOS2 is believed to be the primary NOS isoform associated with defense against microbial pathogens, two other NOS isoforms, NOS1 and NOS3, exist and are constitutively expressed by a variety of cell types. The expression and activity regulation of NOS3 (endothelial, eNOS) within cervical epithelia is unclear. However, in noncervical cells, one way in which NOS3 activity can be induced is by Akt-mediated phosphorylation. Thus, NOS3 function within pex cells has the potential to be upregulated after Akt activation during gonococcal infection. Therefore, it was of interest to investigate the potential role of Pg and Akt in mediating the expression and functional activity of NOS3 in pex cells during N. gonorrhoeae challenge. I again performed IP assays under conditions reflective of the (Pg-predominant) luteal phase of the menstrual cycle and in which an Akt inhibitor was included or omitted. As NOS3 is constitutively expressed, I used an anti-NOS3 antibody that specifically recognizes the active, phosphorylated, form of NOS3 (NOS3-P) for Western blot analysis following its immunocapture. Pex cells were challenged with wild-type gonococci for 3 h. This time point was chosen to allow visualization of both Akt-dependent NOS2 expression and potential Akt-mediated phosphorylation of NOS3. In support of the data described above, increasing concentrations of Pg again resulted in a parallel increase in NOS2 expression (Fig. 6). A Pg dose-dependent increase in NOS3 (140 kDa) phosphorylation was also observed; however, this effect was modest and less pronounced than that observed for (inducible) NOS2 expression (Fig. 6). Neither NOS2 nor NOS3-P were present in pex cell lysates in the presence of the Akt inhibitor or when the primary antibody was omitted from the initial NOS capture step (Fig. 6). Collectively, these data strongly suggest that N. gonorrhoeae infection of pex cells triggers a signal transduction cascade resulting in Akt-dependent NOS activity.
FIG. 6.

Akt mediates NOS expression and activity in gonococcus-infected pex cells. NOS2 (130 kDa) and NOS3 (140 kDa) were captured by using the rabbit antibodies H-174 or H-159, respectively, from a 3-h infection of pex cells cultured under conditions that mimic the menstrual cycle luteal phase. Western blotting was performed using the mouse anti-NOS2 antibody C-11 (upper panel) or the goat antibody NOS3-P Ser1177 (lower panel), which recognizes the Akt phosphorylated form of NOS3. An Akt inhibitor was included or omitted from the assays, as noted. The hormone concentrations (0.4 nM E2 plus 0 to 30 nM) are noted across the top of each blot image. UI, uninfected pex cells; No Ab, the anti-NOS antibody was omitted from the initial capture step.
Gonococcus-infected pex cells generate nitric oxide.
Although NO metabolites (nitrate/nitrite; NOX, collectively) are present in cervical secretions to my knowledge, the ability of cervical epithelial cells to generate NO has not been directly examined. That is, NOX presence within the cervical microenvironment has been typically measured by using the Griess reaction, a colorimetric method for nitrate and nitrite (resulting from nitric oxide oxidation) quantitation. Based on data presented above, I similarly quantitated the effect of N. gonorrhoeae infection on the ability of pex cells to produce NOX (see Fig. S2 in the supplemental material). Upon comparison to a nitrite standard curve, Griess reaction analyses revealed that the levels of NOX present in uninfected and in gonococci-infected pex cells were comparable to those reported for cervical fluid specimens obtained from uninfected women (median, 23.8 μM) or women infected with human papillomavirus (median, 45.2 μM) (43). Thus, I then wanted to directly assess the ability of pex cells to generate NO in response to gonococcal infection. To this end, I performed a series of experiments in which NO production was recorded using cell-permeant and cell-impermeant NO-reactive probes, which allowed me to kinetically measure intracellular, as well as extracellular, NO in real-time using a fluorescence-capable microplate reader. Readings were recorded as arbitrary fluorescence units and were collected every 30 min over 18 h. These studies were performed in the presence or absence of an Akt inhibitor or a panel of NOS inhibitors. The graphs shown in Fig. 7 are representative of data obtained from three assays performed in quadruplicate in which there was some interassay variability in the actual fluorescence units measured; however, there was no variability in the course of the plotted data. Although multiple inhibitors of each NOS isoform were used in these studies, only data obtained with the use of SMT (NOS2 inhibitor) and NIO (NOS3 inhibitor) are shown. Comparable data were obtained with the respective use of each NOS2 or NOS3 inhibitor; however, no effect on NO production was observed with the use of NOS1-specific inhibitors during N. gonorrhoeae infection, supporting previous work in which NOS1 could not be detected within cervical biopsies obtained from pregnant and nonpregnant females by immunohistochemical analysis (50). Under all conditions assayed, background levels of NO production occurred in uninfected cells, but NO was greatly increased during N. gonorrhoeae 1291 challenge (Fig. 7 and see Fig. S3 in the supplemental material). In the presence of the Akt inhibitor both intracellular (Fig. 7A) and extracellular (Fig. 7B) NO was reduced to background levels in wild-type-infected pex cells. Of interest is that, whereas NOS2 inhibition decreased intracellular NO (Fig. 7C), it only minimally affected the presence of extracellular NO (Fig. 7D). Conversely, NOS3 inhibition had no effect on the presence of NO within pex cells (Fig. 7E), but it severely decreased extracellular NO (Fig. 7F). Only background levels of NO were detected intracellularly or extracellularly when the activity of all three NOS isoforms was inhibited by the combined use of NOS1inh, 1400W (NOS2 inhibitor), and NIO, demonstrating the specificity of the assay (see Fig. S3 in the supplemental material).
FIG. 7.

Gonococcal challenge induces Akt-dependent NO production by pex cells. NO production was measured over 18 h in uninfected and 1291 wild-type-infected pex cells, using cell-permeable or cell-impermeable NO-specific fluorescent probes. Arbitrary fluorescence units are given on the y axis. Readings (485-nm excitation and 530-nm emission) were taken every 30 min (x axis). Graphs corresponding to intracellular NO are depicted to the left (A, C, and E), and extracellular NO is shown to the right (B, D, and F). Assays were performed in the presence or absence of Akt inhibitor VII (A and B), 1400W (NOS2 inhibitor; C and D), or NIO (NOS3 inhibitor; E and F). The data shown are the means obtained from three separate experiments performed in quadruplicate.
I then proceeded to measure NO production by pex cells that were incubated in the absence of E2/Pg or that were incubated under conditions reflective of the early and late follicular (i.e., 0.5 nM E2 plus 3 nM Pg) or luteal (i.e., 15 nM Pg plus 0.4 nM E2) phases of the menstrual cycle. Total (intracellular and extracellular) NO was measured in real time, as outlined above. The pex cells were left uninfected, or they were challenged with 1291 wild-type or 1291ΔPLD mutant N. gonorrhoeae. An Akt inhibitor was included or omitted from wild-type gonococcal infections. Only background levels of NO production by uninfected pex cells was observed under each assay condition, and this was only moderately increased upon infection with pld mutant gonococci (Fig. 8). Infection with wild-type gonococci resulted in a further increase in NO production by pex cells, which was reduced to background levels in the presence of an Akt inhibitor (Fig. 8). In support of data obtained by Western blot analyses, Pg had the greatest stimulatory effect on NO production. In this regard, compared to the absence of E2/Pg (Fig. 8A) or E2-predominant conditions (Fig. 8B), the fluorescence intensity recorded under conditions reflective of the menstrual cycle luteal phase was increased >2-fold (Fig. 8C). Taken together, these data demonstrated that gonococcus-induced NO production by pex cells occurred in an Akt-dependent manner that was further modulated by Pg and NgPLD. Importantly, these findings also showed that the level of NOX produced by pex cells was comparable to those levels reported for women, in vivo, and also potentially indicate that the spatial distribution of NO during the course of gonococcal infection may result from the activity of different pex cell NOS isoforms.
FIG. 8.
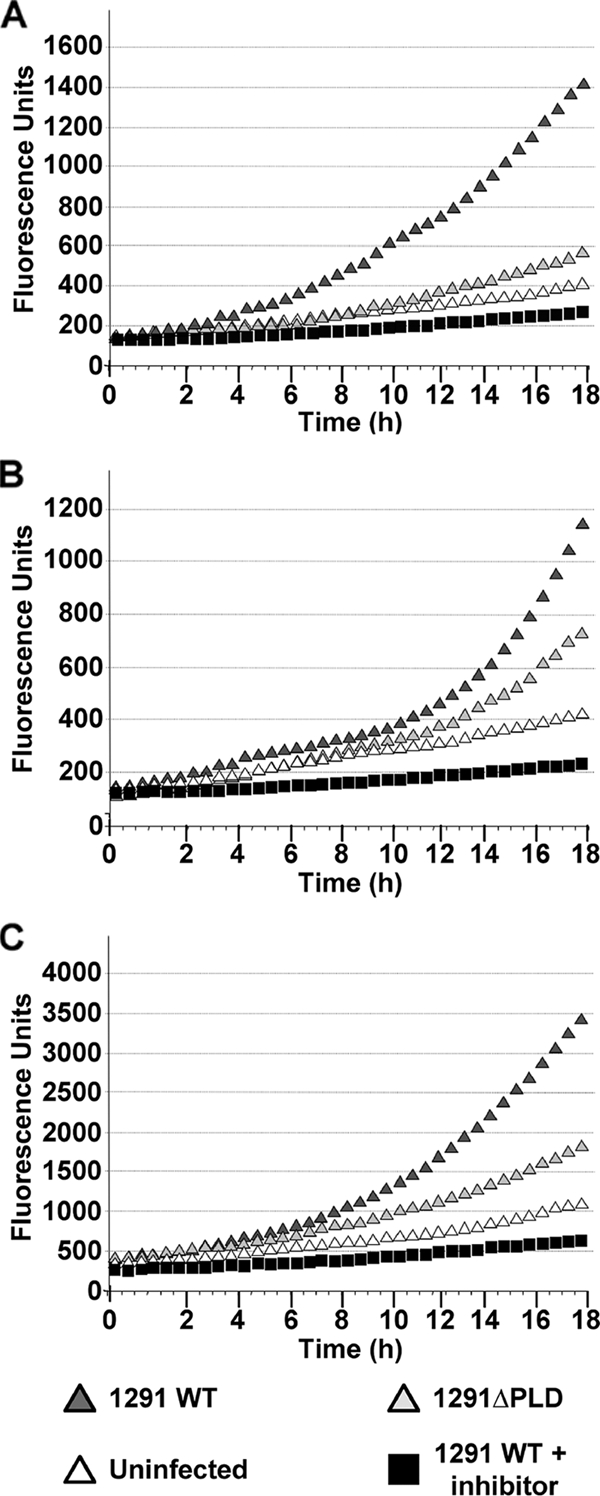
NO production by gonococcus-infected pex cells is enhanced under Pg-predominant conditions. NO production by uninfected pex cells, as well as by1291- and 1291ΔPLD-infected pex cells, was measured as outlined in the text. Akt inhibitor VII was included or omitted from wild-type infections. Pex cells were incubated without E2/Pg (A), with 3 nM Pg plus 0.5 nM E2 (B), or with 0.4 nM E2 plus 15 nM Pg (C) before and during gonococcal challenge. Total (intracellular and extracellular) NO production was measured over 18 h using cell-permeable plus cell-impermeable NO-specific fluorescent probes. Readings (485-nm excitation and 530-nm emission) were recorded every 30 min (x axis). Arbitrary fluorescence units are given on the y axis. The data shown are the means obtained from three separate experiments performed in quadruplicate.
NO promotes the survival of gonococci during pex cell challenge.
My data obtained thus far supported a role for Akt-mediated NO production by pex cells during infection. Therefore, I next wanted to determine whether NO produced by the cervical epithelium exerted a bactericidal effect or, perhaps, promoted the ability of gonococci to survive within pex cells, as we previously show a critical role for Akt activity in N. gonorrhoeae infection and invasion of pex cells. To this end, I performed gonococcus survival assays in the presence or absence of a panel of NOS inhibitors (Fig. 9A). I found that only under conditions when NOS2 was inhibited was the ability of wild-type gonococci to survive within pex cells at 3 h postinvasion significantly impaired, compared to the absence of any inhibitor (Fig. 9A). To confirm these data, I again performed association, invasion, and survival assays with pex cells that were untreated or that were pretreated with L-NAME (NOS1/3 inhibitor) plus TRIM (NOS1/2 inhibitor) to inhibit NO production. To confirm the requirement for host-derived NO in augmenting gonococcal survival during pex cell challenge, nitrite, cPTIO (an NO-specific scavenger), or a panel of NO donors was added to (or omitted from) pex cell monolayers coincident with microaerobically cultured wild-type gonococci. Further, to promote microaerobic respiration by gonococci as well as to more closely mimic ex vivo, the oxygen-limited environment of the human cervix in vivo, infections were allowed to progress under normal laboratory or microaerobic (i.e., 6% O2) conditions, as outlined in the Materials and Methods. These assays showed that gonococcal association with pex cells was moderately, but significantly (P ≤ 0.01), impaired under microaerobic, NO-inhibited, conditions (data not shown). However, association levels could be restored to “normal” levels (i.e., association determined under normal oxygen and NO tensions; i.e., none + O2) by the inclusion of NO donors, but not nitrite, in each assay (data not shown). Among the various conditions assayed, only minor differences were observed in the ability of gonococci to invade pex cells or to survive at 1 h postinvasion of pex cells. Conversely, notable differences in the ability of gonococci to survive within pex cells became evident by 3 h postinvasion (Fig. 9B). In this regard, an ∼3-fold decrease in gonococcal survival was observed at 3 h postinvasion when NO was depleted in pex cells by the addition of cPTIO or by inhibiting NOS activity (compare “none + O2” to “cPTIO +/− O2” or to “none# +/− O2”; P ≤ 0.01 for both comparisons). Nitrite addition to NOS-inhibited pex cells only modestly restored gonococcal intracellular survival in that survival observed under this condition was ∼2-fold less than that observed for “normal” gonococcal survival levels (P ≤ 0.01). Conversely, the addition of the NO donors, SIN and SNAP, to NOS-inhibited pex cells completely restored and modestly enhanced (P ≤ 0.02) the ability of gonococci to survive within these cells (Fig. 9B). Further, the addition of GSNO, a thiol-derived NO donor, resulted in a further significant (P ≤ 0.01) enhancement of the survival capability of intracellular gonococci compared to the use of SIN or SNAP and to “normal” levels of survival (Fig. 9B). Collectively, these data provide strong evidence that host-derived NO promotes the intracellular survival of gonococci during pex cell challenge and, potentially, indicate that NO may be a requirement for sustained cervical disease.
FIG. 9.

NOS2-derived NO promotes the intracellular survival of gonococci within pex cells. (A) Pex cells were treated with a panel of NOS inhibitors. (B) Alternatively, the NO scavenger cPTIO; nitrite; or the NO donors GSNO, SIN, or SNAP were included in the assay, as indicated. Pex cell monolayers were challenged with wild-type bacteria for 90 min, followed by a 30-min gentamicin treatment to kill extracellular bacteria. Invasion and survival assays were performed as described in the text where invasion refers to a 90-min infection plus gentamicin treatment. Survival refers to the number of viable gonococci that were recovered from within pex cells at 1 h (Inv + 1 h) or 3 h (Inv + 3 h) after invasion. Values given are the mean (variance) in which the percent invasion or survival of gonococci was determined as a function of the original inoculum and the number of CFU formed with subsequent plating of the cervical epithelial cell lysates. The data were obtained from at least three trials performed in triplicate. For the “−” column (in panel A), a NOS inhibitor was not included in the assay. Panel A P values: *, P ≤ 0.01 for data obtained in the presence of the indicated NOS inhibitor compared to the absence of any NOS inhibitor. Panel B P values: §, P ≤ 0.01 for data obtained in the presence of GSNO compared to the absence of GSNO or the use of an alternative NO donor; ×, data obtained in the presence of nitrite were not significant compared to the absence of nitrite (i.e., none# +/− O2). The “#” symbol indicates that NO production by pex cells was inhibited by the inclusion of NAME (NOS1/3 inhibitor) and TRIM (NOS1/2 inhibitor).
DISCUSSION
The female reproductive tract is a dynamic environment that is receptive to fluctuating hormone levels. Steroid hormones often have antagonist effects that are not equal in all (mammalian) cell types (4, 5, 25, 49). In this regard, there are at present very limited data describing the response(s) of (primary) cervical epithelial cells to combined, variable physiological concentrations of estrogens and progestagens—such as would occur in vivo throughout the female menstrual cycle—or how these responses potentially might be altered during bacterial infection. Similarly, although clinical data indicate that there is a hormonal component to gonococcal disease in women, how steroid hormones influence the pathology of cervical gonorrhea has remained poorly defined. Our previous works demonstrated that gonococcal infection of primary cervical epithelial (i.e., pex) cells, with CR3 engagement and NgPLD secretion, triggers a signaling pathway in which the serine-threonine kinase, Akt, plays a critical role in promoting the survival of this bacterium. I have now extended these studies to further examine the role of Akt in gonococcal cervicitis under variable E2 and Pg concentrations such as would occur in vivo during the course of the female menstrual cycle.
Clinical studies demonstrate that steroid hormones may modulate the course of gonococcal disease in infected women (23, 24, 29). It is also noteworthy that there are data to indicate that progestin-based contraceptives increase the susceptibility of a woman to develop gonococcal disease (13, 32, 36). I have now similarly shown that Pg, in a dose-dependent manner, augments the ability of gonococci to invade and to survive within pex cells. In this regard, these data suggest that increased Pg mediated the intracellular survival of gonococci by augmenting NgPLD-dependent Akt activity.
Multiple levels of regulation may exist by which Pg augments Akt activity during pex cell challenge. That is, Akt phosphorylation was evident in pld mutant-infected pex cells only when Pg was present. However, infections performed using wild-type gonococci indicated that Pg and NgPLD had an additive effect on Akt phosphorylation (Fig. 3). Although other explanations exist, I suggest that Pg functions indirectly to trigger Akt activity in the absence of NgPLD. It has been documented that in some cell types steroid hormone receptors can trigger Akt activity in a PI3K-dependent manner (17, 18). In the absence of E2/Pg, very low levels of Akt activity are observed in pld mutant-infected pex cells, which is dependent upon the activity of PI3K (7). PI3K can phosphorylate phosphatidylinositol (PtdIns)(4,5)P2 to form PtdIns(3,4,5)P3 that competes with NgPLD for binding to the Akt PH domain (7). Therefore, in the absence of NgPLD, Pg may trigger Akt phosphorylation indirectly through its cognate receptor acting upon the PI3K pathway. However, PI3K-mediated Akt signaling does not appear to promote an intracellular pex cell environment that is favorable for gonococcal survival, because 1291ΔPLD gonococci do not survive within pex cells (Fig. 2), and the ability of wild-type bacteria to invade and/or survive is enhanced when the PI3K activity is chemically inhibited (7).
In the presence of NgPLD (i.e., in wild-type-infected cells), the Pg-dependent additive effect observed in Akt phosphorylation (Fig. 3) may be related to the ability of steroid hormones to modulate the production of proteins comprising the complement system. The sustained release of gonococcal products, including NgPLD, requires complement deposition upon the gonococcus surface, followed by engagement of the CR3 I-domain (10). That complement presence within the female genital tract varies in response to fluctuating hormone levels is demonstrated (16, 38). In this regard, an increase in complement protein C3 occurs during the luteal phase of the menstrual cycle in humans. Although complement production by cervical epithelia in response to steroid hormones has not been examined, we have shown that pex cells produce all of the proteins comprising the alternative pathway of complement (9). Thus, if complement production by pex cells is increased under Pg-predominant conditions, although this is speculative, it might also follow that an increase in NgPLD production and secretion occurs. The increased presence of NgPLD could then result in increased Akt activity and, in this way, account for the observed ability of Pg to augment the survival of gonococci during pex cell challenge.
I have shown that Akt activation triggered the expression and/or the activity of NOS2 and NOS3, which, in turn, resulted in the generation of NO. Although NO is widely believed to mediate bacterial killing, N. gonorrhoeae possesses a NO detoxification pathway (45), which we have previously suggested to play a role in promoting the intracellular survival of gonococci during pex cell challenge (41, 42, 46). In addition, gonococci possess a truncated denitrification pathway comprising AniA, a surface-exposed (30) nitrite reductase (34), and the NO reductase NorB (21), which provides an additional or alternative means of respiration for these bacteria when oxygen becomes limited (27). The cervix is an oxygen-limited environment, with oxygen tensions ranging between 12 and 22 mm Hg (in uninfected, normal cervices) over the menstrual cycle (54); thus, it is not a strictly anaerobic environment. Although the reactivity or toxicity of NO is greatly enhanced in the presence of oxygen, recent data indicate that gonococci are also resistant to the killing action of peroxynitrite (2), a strong oxidant and nitrating agent formed in the presence of NO and superoxide. Cole and coworkers (39) have proposed that host-derived NO induction of N. gonorrhoeae NorB could confer a survival advantage to these bacteria in vivo by serving as an electron acceptor under conditions of oxygen limitation (39). The data shown in Fig. 9 would support the idea that gonococci require host-derived NO during infection under aerobic, as well as microaerobic conditions, although I did not directly assess gonococcal respiration in these studies. However, in the closely related bacterium, Neisseria meningitidis, it has been demonstrated in vitro that oxygen and nitrite reduction both contribute to electron transfer during respiration, i.e., that aerobic denitrification occurs in the presence of and supplements oxygen consumption (44). In addition and consistent with the present data, Read and coworkers (44) demonstrated that a norB mutation in N. meningitidis results in an impaired ability to survive within human macrophages, as well as in nasopharyngeal mucosal explants (47).
Three different eukaryotic NOS isoforms exist that could potentially share different cellular compartments with invasive gonococci during the course of disease. Thus, although NO is readily diffusible, there is great potential for this oxidant to encounter and modify, over a time course of gonococcal challenge, bacterial and host constituents that play a role in NO usage and/or protection from reactive nitrogen or oxidants. My data demonstrate that, during the course of N. gonorrhoeae infection of pex cells, the spatial distribution of NO appears to result from different pex cell NOS isoforms. That is, intracellular NO results primarily from the activity of NOS2, whereas extracellular NO results primarily from NOS3 activity. Under conditions when the activity of NOS2, but not NOS3, was impaired so was the ability of gonococci to survive within pex cells. Whereas NOS2 is an inducible cytosolic protein, NOS3 resides in an inactive state at the plasma membrane. Therefore, it is interesting to speculate that NO resulting from Akt-mediated phosphorylation and activation of NOS3 at the pex cell plasma membrane, where it would be more freely diffusible to the extracellular space, similarly differentially augments the survival of extracellular gonococci. Consistent with this idea are recent data demonstrating a role for the gonococcal NO respiratory pathway in biofilm formation on immortalized cervical cells (12).
In summary, I initiated studies to investigate how steroid hormones potentially modulate the course of gonococcal disease in women, as is demonstrated by clinical data. In this regard, I have presented data to indicate that Pg-predominant conditions promote N. gonorrhoeae infection of pex cells. The ability of Pg to augment gonococcal survival during pex cell challenge corresponds well with clinical data demonstrating the increased susceptibility for acquiring gonococcal disease associated with the use of progestin-based contraceptives. In addition, I have further defined the downstream effects of the CR3-NgPLD-Akt pathway triggered during gonococcal infection of primary cervical cells. My data indicate that Pg functions in an additive manner with NgPLD to induce Akt activity that, in turn, regulates NOS expression and NO production. Importantly, although it has previously been demonstrated, in vitro, that NO is not toxic to gonococci (3), to my knowledge, my data are the first to demonstrate that host-derived NO may be required to sustain cervical bacterial disease. Further, I believe that these data are the first to directly demonstrate NO (versus NOX) production by primary epithelia of the human cervix, as well as to demonstrate that spatial differences in NO production may differentially affect bacterial disease. Thus, the present findings may have broader implications to the study of other mucosal pathogens. However, the contribution of the gonococcal denitrification and detoxification pathways with regard to cervical infection remains to be fully elucidated. These and other unanswered questions are the focus of ongoing investigations in our lab.
Supplementary Material
Acknowledgments
I am grateful to the Cooperative Human Tissue Network (The Research Institute at Nationwide Children's Hospital, Columbus, OH) for providing cervical tissue specimens.
This study was funded by a Young Investigator Award from The Research Institute at Nationwide Children's Hospital and NIAID grant 1R01AI076398.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 4 January 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Apicella, M. A. 1974. Antigenically distinct populations of Neisseria gonorrhoeae: isolation and characterization of the responsible determinants. J. Infect. Dis. 130:619-625. [DOI] [PubMed] [Google Scholar]
- 2.Barth, K. R., V. M. Isabella, L. F. Wright, and V. L. Clark. 2009. Resistance to peroxynitrite in Neisseria gonorrhoeae. Microbiology 155:2532-2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardinale, J. A., and V. L. Clark. 2005. Determinants of nitric oxide steady-state levels during anaerobic respiration by Neisseria gonorrhoeae. Mol. Microbiol. 58:177-188. [DOI] [PubMed] [Google Scholar]
- 4.Curtis, S. W., H. Shi, C. Teng, and K. S. Korach. 1997. Promoter and species specific differential estrogen-mediated gene transcription in the uterus and cultured cells using structurally altered agonists. J. Mol. Endocrinol. 18:203-211. [DOI] [PubMed] [Google Scholar]
- 5.Dana, S. L., P. A. Hoener, D. A. Wheeler, C. B. Lawrence, and D. P. McDonnell. 1994. Novel estrogen response elements identified by genetic selection in yeast are differentially responsive to estrogens and antiestrogens in mammalian cells. Mol. Endocrinol. 8:1193-1207. [DOI] [PubMed] [Google Scholar]
- 6.Edwards, J. L., and M. A. Apicella. 2004. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin. Microbiol. Rev. 17:965-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards, J. L., and M. A. Apicella. 2006. Gonococcal PLD directly interacts with Akt kinase upon infection of primary human cervical epithelial cells. Cell. Microbiol. 8:1253-1271. [DOI] [PubMed] [Google Scholar]
- 8.Edwards, J. L., E. J. Brown, K. A. Ault, and M. A. Apicella. 2001. The role of complement receptor 3 (CR3) in Neisseria gonorrhoeae infection of human cervical epithelia. Cell. Microbiol. 3:611-622. [DOI] [PubMed] [Google Scholar]
- 9.Edwards, J. L., E. J. Brown, S. Uk-Nham, J. G. Cannon, M. S. Blake, and M. A. Apicella. 2002. A co-operative interaction between Neisseria gonorrhoeae and complement receptor 3 mediates infection of primary cervical epithelial cells. Cell. Microbiol. 4:571-584. [DOI] [PubMed] [Google Scholar]
- 10.Edwards, J. L., D. D. Entz, and M. A. Apicella. 2003. Gonococcal phospholipase D modulates the expression and function of complement receptor 3 in primary cervical epithelial cells. Infect. Immun. 71:6381-6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards, J. L., J. Q. Shao, K. A. Ault, and M. A. Apicella. 2000. Neisseria gonorrhoeae elicits membrane ruffling and cytoskeletal rearrangements upon infection of primary human endocervical and ectocervical cells. Infect. Immun. 68:5354-5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falsetta, M. L., T. B. Bair, S. C. Ku, R. N. Vanden Hoven, C. T. Steichen, A. G. McEwan, M. P. Jennings, and M. A. Apicella. 2009. Transcriptional profiling identifies the metabolic phenotype of gonococcal biofilm. Infect. Immun. 77:3522-3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez, R., P. Nelson, J. Delgado, J. Aguilera, R. Massai, L. Velasquez, M. Imarai, H. B. Croxatto, and H. Cardenas. 2001. Increased adhesiveness and internalization of Neisseria gonorrhoeae and changes in the expression of epithelial gonococcal receptors in the fallopian tube of copper T and Norplant users. Hum. Reprod. 16:463-468. [DOI] [PubMed] [Google Scholar]
- 14.Florian, M., Y. Lu, M. Angle, and S. Magder. 2004. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids 69:637-645. [DOI] [PubMed] [Google Scholar]
- 15.Guzeloglu Kayisli, O., U. A. Kayisli, G. Luleci, and A. Arici. 2004. In vivo and in vitro regulation of Akt activation in human endometrial cells is estrogen dependent. Biol. Reprod. 71:714-721. [DOI] [PubMed] [Google Scholar]
- 16.Hasty, L. A., J. D. Lambris, B. A. Lessey, K. Pruksananonda, and C. R. Lyttle. 1994. Hormonal regulation of complement components and receptors throughout the menstrual cycle. Am. J. Obstet. Gynecol. 170:168-175. [DOI] [PubMed] [Google Scholar]
- 17.Haynes, M. P., D. Sinha, K. S. Russell, M. Collinge, D. Fulton, M. Morales-Ruiz, W. C. Sessa, and J. R. Bender. 2000. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ. Res. 87:677-682. [DOI] [PubMed] [Google Scholar]
- 18.Haynes, M. P., L. Li, D. Sinha, K. S. Russell, K. Hisamoto, R. Baron, M. Collinge, W. C. Sessa, and J. R. Bender. 2003. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J. Biol. Chem. 278:2118-2123. [DOI] [PubMed] [Google Scholar]
- 19.Hiromura, M., F. Okada, T. Obata, D. Auguin, T. Shibata, C. Roumestand, and M. Noguchi. 2004. Inhibition of Akt kinase activity by a peptide spanning the betaA strand of the proto-oncogene TCL1. J. Biol. Chem. 279:53407-53418. [DOI] [PubMed] [Google Scholar]
- 20.Hisamoto, K., M. Ohmich, H. Kurachi, J. Hayakawa, Y. Kanda, Y. Nishio, K. Adachi, K. Tasaka, E. Miyoshi, N. Fujiwara, N. Taniguchi, and Y. Murata. 2001. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 276:3459-3467. [DOI] [PubMed] [Google Scholar]
- 21.Householder, T. C., E. M. Fozo, J. A. Cardinale, and V. L. Clark. 2000. Gonococcal nitric oxide reductase is encoded by a single gene, norB, which is required for anaerobic growth and is induced by nitric oxide. Infect. Immun. 68:5241-5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, F. C., Q. Li, and B. J. Cherayil. 2005. A phosphatidyl-inositol-3-kinase-dependent anti-inflammatory pathway activated by Salmonella in epithelial cells. FEMS Microbiol. Lett. 243:265-270. [DOI] [PubMed] [Google Scholar]
- 23.James, J. F., and J. Swanson. 1978. Studies on gonococcus infection. XIII. Occurrence of color/opacity colonial variants in clinical cultures. Infect. Immun. 19:332-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, D. W., K. K. Holmes, P. A. Kvale, C. W. Halverson, and W. P. Hirsch. 1969. An evaluation of gonorrhea case findings in the chronically infected female. Am. J. Epidemiol. 90:438-448. [DOI] [PubMed] [Google Scholar]
- 25.Katzenellenbogen, B. S. 2000. Mechanisms of action and cross-talk between estrogen receptor and progesterone receptor pathways. J. Soc. Gynecol. Invest. 7:S33-S37. [DOI] [PubMed] [Google Scholar]
- 26.Kierbel, A., A. Gassama-Diagne, K. Mostov, and J. N. Engel. 2005. The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol. Biol. Cell 16:2577-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knapp, J. S., and V. L. Clark. 1984. Anaerobic growth of Neisseria gonorrhoeae coupled to nitrite reduction. Infect. Immun. 46:176-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knodler, L. A., B. B. Finlay, and O. Steele-Mortimer. 2005. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J. Biol. Chem. 280:9058-9064. [DOI] [PubMed] [Google Scholar]
- 29.Koch, M. L. 1947. A study of cervical cultures taken in cases of acute gonorrhea with special reference to the phases of the menstrual cycle. Am. J. Obstet. Gynecol. 54:861-866. [DOI] [PubMed] [Google Scholar]
- 30.Ku, S. C., B. L. Schulz, P. M. Power, and M. P. Jennings. 2009. The pilin O glycosylation pathway of pathogenic Neisseria is a general system that glycosylates AniA, an outer membrane nitrite reductase. Biochem. Biophys. Res. Commun. 378:84-89. [DOI] [PubMed] [Google Scholar]
- 31.Lee, S. W., D. L. Higashi, A. Snyder, A. J. Merz, L. Potter, and M. So. 2005. PilT is required for PI(3,4,5)P3-mediated crosstalk between Neisseria gonorrhoeae and epithelial cells. Cell. Microbiol. 7:1271-1284. [DOI] [PubMed] [Google Scholar]
- 32.Louv, W. C., H. Austin, J. Perlman, and W. J. Alexander. 1989. Oral contraceptive use and the risk of chlamydial and gonococcal infections. Am. J. Obstet. Gynecol. 160:396-402. [DOI] [PubMed] [Google Scholar]
- 33.Lysko, P. G., and S. A. Morse. 1980. Effects of steroid hormones on Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 18:281-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellies, J., J. Jose, and T. F. Meyer. 1997. The Neisseria gonorrhoeae gene aniA encodes an inducible nitrite reductase. Mol. Gen. Genet. 256:525-532. [DOI] [PubMed] [Google Scholar]
- 35.Moreno-Cuevas, J. E., and D. A. Sirbasku. 2000. Estrogen mitogenic action. III. Is phenol red a “red herring”? In Vitro Cell Dev. Biol. Anim. 36:447-464. [DOI] [PubMed] [Google Scholar]
- 36.Morrison, C. S., P. Bright, E. L. Wong, C. Kwok, I. Yacobson, C. A. Gaydos, A. Charlotte, H. T. Tucker, and P. D. Blumenthal. 2004. Hormonal contraceptive use, cervical ectopy, and the acquisition of cervical infections. Sex. Transm. Dis. 31:561-567. [DOI] [PubMed] [Google Scholar]
- 37.Morse, S. A., and G. F. Brooks. 1985. Neisseria gonorrhoeae taxonomy, colony phenotypes and disease, p. 3-8. In G. F. Brooks and E. A. Donegan (ed.), Gonococcal infection. Butler and Tanner, Ltd., London, England.
- 38.Oglesby, T. J. 1998. The complement system in reproduction, p. 355-373. In J. E. Volanakis and M. M. Franks (ed.), The human complement system in health and disease. Marcel Dekker, New York, NY.
- 39.Overton, T. W., R. Whitehead, Y. Li, L. A. S. Snyder, N. J. Saunders, H. Smith, and J. A. Cole. 2006. Coordinated regulation of the Neisseria gonorrhoeae truncated denitrification pathway by the nitric oxide-sensitive repressor, NsrR, and nitrite-insensitive NarQ-NarP. J. Biol. Chem. 281:33115-33126. [DOI] [PubMed] [Google Scholar]
- 40.Patten, R. D., I. Pourati, M. J. Aronovitz, J. Baur, F. Celestin, X. Chen, A. Michael, S. Haq, S. Nuedling, C. Grohe, T. Force, M. E. Mendelsohn, and R. H. Karas. 2004. 17β-Estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ. Res. 95:692-699. [DOI] [PubMed] [Google Scholar]
- 41.Potter, A. J., S. P. Kidd, J. L. Edwards, M. L. Falsetta, M. A. Apicella, M. P. Jennings, and A. G. McEwan. 2009. Thioredoxin reductase is essential for protection of Neisseria gonorrhoeae against killing by nitric oxide and for bacterial growth during interaction with cervical epithelial cells. J. Infect. Dis. 199:227-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potter, A. J., S. P. Kidd, J. L. Edwards, M. L. Falsetta, M. A. Apicella, M. P. Jennings, and A. G. McEwan. 2009. Esterase D is essential for protection of Neisseria gonorrhoeae against nitrosative stress and for bacterial growth during interaction with cervical epithelial cells. J. Infect. Dis. 200:273-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahkola, P., T. S. Mikkola, O. Ylikorkala, and M. Väisänen-Tommiska. 2009. Association between high risk papillomavirus and nitric oxide release in the human uterine cervix. Gynecol. Oncol. 114:323-326. [DOI] [PubMed] [Google Scholar]
- 44.Rock, J. D., M. R. Mahnane, M. F. Anjum, J. G. Shaw, R. C. Read, and J. W. Moir. 2005. The pathogen Neisseria meningitidis requires oxygen, but supplements growth by denitrification. nitrite, nitric oxide and oxygen control respiratory flux at genetic and metabolic levels. Mol. Microbiol. 58:800-809. [DOI] [PubMed] [Google Scholar]
- 45.Seib, K. L., H. J. Wu, S. P. Kidd, M. A. Apicella, M. P. Jennings, and A. G. McEwan. 2006. Defenses against oxidative stress in Neisseria gonorrhoeae: a system tailored for a challenging environment. Microbiol. Mol. Biol. Rev. 70:344-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seib, K. L., H. J. Wu, Y. N. Srikhanta, J. L. Edwards, M. L. Falsetta, A. J. Hamilton, T. L. Maguire, S. M. Grimmond, M. A. Apicella, A. G. McEwan, and M. P. Jennings. 2007. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol. Microbiol. 63:54-68. [DOI] [PubMed] [Google Scholar]
- 47.Stevanin, T. M., J. W. Moir, and R. C. Read. 2005. Nitric oxide detoxification systems enhance survival of Neisseria meningitidis in human macrophages and in nasopharyngeal mucosa. Infect. Immun. 73:3322-3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoica, G. E., T. F. Franke, A. Wellstein, F. Czubayko, H. J. List, R. Reiter, E. Morgan, M. B. Martin, and A. Stoica. 2003. Estradiol rapidly activates Akt via the ErbB2 signaling pathway. Mol. Endocrinol. 17:818-830. [DOI] [PubMed] [Google Scholar]
- 49.Teng, C. T., Y. Liu, N. Yang, D. Walmer, and T. Panella. 1992. Differential molecular mechanism of the estrogen action that regulates lactoferrin gene in human and mouse. Mol. Endocrinol. 6:1969-1981. [DOI] [PubMed] [Google Scholar]
- 50.Tschugguel, W., C. Schneeberger, H. Lass, F. Stonek, M. B. Zaghlula, K. Czerwenka, C. Schatten, A. Kaider, P. Husslein, and J. C. Huber. 1999. Human cervical ripening is associated with an increase in cervical inducible nitric oxide synthase expression. Biol. Reprod. 60:1367-1372. [DOI] [PubMed] [Google Scholar]
- 51.Väisänen-Tommiska, M., M. Nuutila, K. Aittomäki, V. Hiilesmaa, and O. Ylikorkala. 2003. Nitric oxide metabolites in cervical fluid during pregnancy: further evidence for the role of cervical nitric oxide in cervical ripening. Am. J. Obstet. Gynecol. 188:779-785. [DOI] [PubMed] [Google Scholar]
- 52.Wen, K.-K., P. C. Giardina, M. S. Blake, J. L. Edwards, M. A. Apicella, and P. A. Rubenstein. 2000. Interaction of the gonococcal porin P.IB with G- and F-actin. Biochem. 39:8638-8647. [DOI] [PubMed] [Google Scholar]
- 53.Wilkowsky, S. E., M. A. Barbieri, P. Stahl, and E. L. Isola. 2001. Trypanosoma cruzi: phosphatidylinositol 3-kinase and protein kinase B activation is associated with parasite invasion. Exp. Cell Res. 264:211-218. [DOI] [PubMed] [Google Scholar]
- 54.Wilson, M. 2005. The reproductive system and its indigenous microbiota: environmental determinants at different regions of the reproductive system, p. 213-219. In M. Wilson (ed.), Microbial inhabitants of humans: their ecology and role in health and disease. Cambridge University Press, New York, NY.
- 55.Wu, H.-J., K. L. Seib, J. L. Edwards, M. A. Apicella, A. G. McEwan, and M. P. Jennings. 2005. Azurin of pathogenic Neisseria is involved in defense against hydrogen peroxide and survival within cervical epithelial cells. Infect. Immun. 73:8444-8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yilmaz, O., T. Jungas, P. Verbeke, and D. M. Ojcius. 2004. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 72:3743-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu, X., R. V. Rajala, J. F. McGinnis, F. Li, R. E. Anderson, X. Yan, S. Li, R. V. Elias, R. R. Knapp, X. Zhou, and W. Cao. 2004. Involvement of insulin/phosphoinositide 3-kinase/Akt signal pathway in 17β-estradiol-mediated neuroprotection. J. Biol. Chem. 279:13086-13094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.