

Abstract
Japanese encephalitis virus (JEV) is a mosquito-borne RNA virus and one of the most important flaviviruses in the medical and veterinary fields. Although cholesterol has been shown to participate in both the entry and replication steps of JEV, the mechanisms of infection, including the cellular receptors of JEV, remain largely unknown. To clarify the infection mechanisms of JEV, we generated pseudotype (JEVpv) and recombinant (JEVrv) vesicular stomatitis viruses bearing the JEV envelope protein. Both JEVpv and JEVrv exhibited high infectivity for the target cells, and JEVrv was able to propagate and form foci as did authentic JEV. Anti-JEV envelope antibodies neutralized infection of the viruses. Treatment of cells with inhibitors for vacuolar ATPase and clathrin-mediated endocytosis reduced the infectivity of JEVpv, suggesting that JEVpv enters cells via pH- and clathrin-dependent endocytic pathways. Although treatment of the particles of JEVpv, JEVrv, and JEV with cholesterol drastically reduced the infectivity as previously reported, depletion of cholesterol from the particles by treatment with methyl β-cyclodextrin enhanced infectivity. Furthermore, treatment of cells with sphingomyelinase (SMase), which hydrolyzes membrane-bound sphingomyelin to ceramide, drastically enhanced infection with JEVpv and propagation of JEVrv, and these enhancements were inhibited by treatment with an SMase inhibitor or C6-ceramide. These results suggest that ceramide plays crucial roles in not only entry but also egress processes of JEV, and they should assist in the clarification of JEV propagation and the development of novel therapeutics against diseases caused by infection with flaviviruses.
Japanese encephalitis virus (JEV) is a small, enveloped virus belonging to the family Flaviviridae and the genus Flavivirus, which also includes Dengue virus (DENV), West Nile virus (WNV), Yellow fever virus, and Tick-borne encephalitis virus (11). JEV is the most common agent of viral encephalitis, causing approximately 50,000 cases annually, of which 15,000 will die, and up to 50% of survivors are left with severe residual neurological complications. JEV has a single-stranded positive-sense RNA genome of approximately 11 kb, encoding a single large polyprotein, which is cleaved by the host- and virus-encoded proteases into three structural proteins, capsid (C), premembrane (PrM), and envelope (E), and seven nonstructural proteins. The structural proteins are components of viral particles, and the E protein is suggested to interact with a cell surface receptor molecule(s). Although a number of cellular components, including heat shock cognate protein 70 (33), glycosaminoglycans, such as heparin or heparan sulfate (21, 41), and laminin (3), have been shown to participate in JEV infection, the precise mechanisms by which these receptor candidates participate in JEV infection remain largely unclear.
In addition to the many studies identifying and characterizing receptor molecules in numerous viruses, data suggesting the involvement of membrane lipids, such as sphingolipids and cholesterol, in viral infection have also been accumulating. Lipid rafts consisting of sphingolipids and cholesterol and distributing to the outer leaflet of the cell membrane have been shown to be involved in the infection of not only many viruses but also several bacteria and parasites (24), in addition to playing roles in various functions such as lipid sorting, protein trafficking (26, 47), cell polarity, and signal transduction (38). With respect to cholesterol itself, various aspects of the life cycle of flaviviruses have been shown to involve this lipid, including the entry of DENV (34), hepatitis C virus (HCV) (16), and WNV (27), the membrane fusion of tick-borne encephalitis virus (40), and the replication of HCV (14, 17), WNV (23), and DENV (35). Recently Lee et al. (20) showed that treatment with cholesterol efficiently impairs both the entry and replication steps of JEV and DENV-2 but enhances infection with the Sindbis virus (22).
On the other hand, sphingolipids, including sphingomyelins and glycosphingolipids, are ubiquitous components of eukaryotic cell membrane structures, providing integrity to cellular membranes. Ceramide is one of the intermediates of sphingolipids and plays roles in cell differentiation, regulation of apoptosis and protein secretion, induction of cellular senescence, and other processes (2). Ceramide is generated from the hydrolysis of sphingomyelin by sphingomyelinase (SMase) or from catalysis by serine-palmitoyl-coenzyme A (CoA) transferase and ceramide synthase. Ceramide spontaneously self-associates to form ceramide-enriched microdomains and then to form larger ceramide-enriched membrane platforms which serve as the spatial and temporal organization for cellular signalosomes and for regulation of protein functions (2). The ceramide-enriched platforms have also been used by many pathogens to facilitate entry and infection (2). The acid SMase is activated not only by multiple stimuli, including receptor molecules, gamma irradiation, and some chemicals, but also by infection with some bacteria or viruses (36). Rhinovirus activates the SMase for generation of ceramide and forms ceramide-enriched membrane platforms that serve in the infection of target cells (10). Sindbis virus also activates the SMase and induces apoptosis through a continuous release of ceramide (15). In contrast to these viruses, ceramide inhibits infection with HIV (7) and HCV (48). Ceramide enrichment of the plasma membrane reduces expression of HCV receptor molecules through an ATP-independent internalization and impairs entry of HCV.
Pseudotype and recombinant viruses based on the vesicular stomatitis virus (VSV) bearing foreign viral envelope proteins have been shown to be powerful tools for the investigation of viral entry and the development of vaccines. These systems have been used to study infection with viruses that do not propagate readily (31, 43) or that are difficult to handle due to their high-level pathogenicity for humans (42). In addition, the systems allow us to focus on the investigation of entry mechanisms of particular viral envelope proteins by using control viruses harboring an appropriate protein on identical particles.
In the present study, we generated pseudotype (JEVpv) and recombinant (JEVrv) VSVs bearing the JEV envelope protein in human cell lines and determined the involvement of sphingolipids, especially ceramide, and cholesterol in infection of human cell lines with JEV. Both JEVpv and JEVrv exhibited infection of target cells via pH- and clathrin-dependent endocytosis. Treatment of cells with cholesterol impaired infection with JEVpv and JEVrv, as previously found in JEV infection (20). In contrast, treatment of cells with SMase drastically enhanced infection with both JEVpv and JEVrv and the production of infectious JEVrv particles. These results indicate that ceramide plays crucial roles in the entry and egress of JEV.
MATERIALS AND METHODS
Plasmids and cells.
A cDNA clone encoding the PrM and E proteins of the AT31 strain was generated by PCR amplification, cloned into pCAGGS/MCS-PM (43), and designated pCAGC105E (JEV). The plasmid used for construction of JEVrv was pVSVΔG-GFP2.6 (provided by M. A. Whitt, University of Tennessee), which has additional transcription units with multicloning sites (MCS) and green fluorescent protein (GFP) located between the M and L genes. The PrM-E gene, obtained from pCAGC105E (JEV) by digestion with BglII and EcoRI, was cloned into the SmaI site of pVSVΔG-GFP2.6 after blunting, and the construct was designated pΔG-JEV (PrM-E). Huh7, BHK, Vero, and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma, St. Louis, MO) containing 10% fetal bovine serum (FBS).
Viruses and chemicals.
Wild-type JEV was used as described previously (29). The virus was amplified on Huh7 cells and stored at −80°C. The infectious titer was determined by using a focus-forming assay as described below. Bafilomycin A1 from Streptomyces griseus was purchased from Fluka (Sigma). Chlorpromazine hydrochloride, sphingomyelinase (SMase), phospholipase C from Bacillus cereus, methyl-β-cyclodextrin (MβCD), a water-soluble cholesterol, and amitriptyline hydrochloride were obtained from Sigma. C6-ceramide and sphingomyelin were purchased from Biomol International (Plymouth Meeting, PA). Biotin-ceramide was purchased from Echelon Biosciences Inc. (Salt Lake City, UT).
Reverse genetics of VSV.
Recombinant VSVs were generated by a previously described method (43) with minor modifications. Briefly, BHK cells were grown to 90% confluence on 35-mm tissue culture plates and infected with a recombinant vaccinia virus encoding T7 RNA polymerase at a multiplicity of infection (MOI) of 5. After incubation at room temperature for 1 h, the cells were transfected with 4 μg of mixed plasmids encoding each component of VSV proteins (pBS-N/pBS-P/pBS-L/pBS-G, 3:5:1:8) and 2 μg of pΔG-Luci or pΔG-JEV (PrM-E) plasmid using the TransIT-LT1 transfection reagent (Mirus, Madison, WI). After 48 h of incubation, the supernatants were passed through a filter with a pore size of 0.22 μm (Milex-GS; Millipore, Tokyo, Japan) to remove vaccinia virus and inoculated into 293T cells that had been transfected with pCAGVSVG (25) 24 h previously. Recovery of progeny virus was assessed by the appearance of cytopathic effects at 24 to 36 h postinfection. VSV G-complemented (*G) recombinant viruses were stored at −80°C. The infectious titers of the recovered viruses were determined by a plaque assay.
Production and characterization of JEVpv, JEVrv, and JEV.
To generate JEVpv, Huh7 cells transiently expressing the PrM and E proteins by the transfection with pCAGC105E using TransIT-LT1 (Mirus) were infected with VSVΔG/Luc-*G, in which the G gene was replaced with the luciferase gene and was pseudotyped with the G protein, at an MOI of 0.1. The virus was adsorbed for 2 h at 37°C and then extensively washed four times with serum-free DMEM. After 24 h of incubation at 37°C with 10% FBS-DMEM, the culture supernatants were centrifuged to remove cell debris and stored at −80°C. To generate JEVrv, Huh7 cells were infected with VSVΔG/JEV-*G at an MOI of 5 for 2 h at 37°C and then extensively washed four times with serum-free DMEM. After 24 h of incubation at 37°C with 10% FBS-DMEM, the culture supernatants were collected and stored at −80°C. Schematic representations of the genome structures and the production of recombinant and pseudotype VSVs are shown in Fig. 1. The purification and concentration of the pseudotype or recombinant viruses were conducted as described previously (43). Purified viruses and infected cell lysates were analyzed by immunoblotting to detect the incorporation of the envelope protein with anti-JEV E mouse polyclonal antibody (E#2-1; unpublished). The infectivities of JEVpv, JEVrv, and JEV were assessed by both luciferase activity and a focus-forming assay, as described below. The relative light unit (RLU) value of luciferase was determined by using the Bright-Glo luciferase assay system (Promega Corporation, Madison, WI), following a protocol provided by the manufacturer. To examine the effects of oligosaccharide modification of the JEV E protein in cells or on the particles, the cell lysates and the purified particles were digested with endoglycosidase H (Endo H) or peptide-N-glycosidase F (PNGase F) (Boehringer Mannheim, Mannheim, Germany), following a protocol provided by the manufacturer, and analyzed by immunoblotting.
FIG. 1.

Schematic representation of the genome structures and production of recombinant and pseudotype VSVs. (A) The luciferase, PrME, and E1E2 genes were inserted into the full-length cDNA clone of VSV in place of the G gene and designated ΔG-Luc, ΔG-JEV, and ΔG-HCV, respectively. (B) Recombinant VSVs, JEVrv, HCVrv, and ΔG, bearing the JEV E protein, HCV E1/E2 proteins, and no envelope, respectively, were generated in 293T or Huh7 cells by infection with the respective recombinant VSV after complementation with VSV G protein (*G). (C) Pseudotype VSVs, JEVpv, VSVpv, HCVpv, and MLVpv, were generated by infection with VSVΔG/Luc-*G in 293T or Huh7 cells transiently expressing the respective foreign protein.
Pseudotype VSVs bearing HCVE1E2 (HCVpv), VSVG (VSVpv), and murine leukemia virus envelope (MLVpv) proteins were produced in 293T cells transfected with pCAGc60-p7 (H77), pCAGVSVG, and pFBASALF (provided by T. Miyazawa, Kyoto University), respectively, and used as controls. Recombinant HCV (HCVrv) was also used as a control as described previously (43). To neutralize infection with JEVpv, JEVrv, and JEV, viruses were preincubated with the indicated dilution of anti-JEV E monoclonal antibody (22A1; provided by E. Konishi, Kobe University) for 1 h at 37°C and then inoculated into Huh7 cells. After 1 h of adsorption, the cells were washed three times with DMEM containing 10% FBS, and infectivity was determined after 24 h of incubation at 37°C.
Focus-forming assays.
Cells infected with JEV, VSV, JEVrv, or HCVrv after treatment with the indicated reagents were cultured at 37°C with 0.8% methylcellulose in 10% FBS-DMEM for 24 or 48 h and fixed with 4% paraformaldehyde solution for 1 h. Cells were washed once with phosphate-buffered saline (PBS), treated with 0.5% Triton X-100 for 20 min for permeabilization, incubated with mouse monoclonal antibody to JEV (MsX Japanese encephalitis; Chemicon International Inc., Temecula, CA) for JEV or that to VSV N (10G4; provided by M. A. Whitt) for VSV, JEVrv, and HCVrv for 1 h, and stained by using a Vectastain Elite ABC anti-mouse IgG kit with a VIP substrate (Vector Laboratories, Burlingame, CA), following a protocol provided by the manufacturer.
Effects of chemicals on the infectivities of JEVpv, JEVrv, and JEV.
To examine the entry pathways of the viruses, cells treated with various concentrations of bafilomycin A1, chlorpromazine, MβCD, SMase, phospholipase C, or amitriptyline for 1 h at 37°C were inoculated with JEVpv, HCVpv, VSVpv, or MLVpv, and infectivity was determined by luciferase activity as described above. To examine the effects of cholesterol or SMase on the viral particles, purified virions incubated with various concentrations of water-soluble cholesterol or SMase for 1 h at 37°C were inoculated into the target cells. Viruses treated with SMase were ultracentrifuged (43) and resuspended in culture media to deplete any residual amount of SMase, and infectivity was determined by luciferase or focus-forming assay. To examine the effects of ceramide on the infection, 10 mM C6-ceramide or sphingomyelin dissolved in ethanol was diluted with medium at various concentrations and preincubated with JEVpv, HCVpv, VSVpv, or MLVpv for 1 h at 37°C. After treatment, the viruses were inoculated into Huh7 cells, washed with medium after 1 h of incubation at 37°C, and cultured for 24 h at 37°C, and the residual infectivity was determined by measuring luciferase activity. The effects of C6-ceramide on the infection of JEV were assessed by following the same protocol, and the residual infectivity was determined by focus-forming assay.
Ceramide binding assay.
To examine the interaction of JEV E protein and ceramide, purified viruses were incubated with 500 μl of lysis buffer (20 mM Tris-HCl, pH 7.4, containing 135 mM NaCl and 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN) and 10 μl of 1-mg/ml biotin-ceramide in dimethyl sulfoxide (DMSO) for 1 h at 37°C, and then 20 μl of streptavidin-Sepharose 4B (Zymed, Invitrogen, Carlsbad, CA) was added and the solution was kept at 4°C for 4 h. After washing with the lysis buffer three times, the pellets were analyzed by immunoblotting with anti-JEV E polyclonal antibody (E#2-1).
RESULTS
Construction and characterization of recombinant and pseudotype VSVs.
Recombinant VSVs were propagated in Huh7 cells by infection with VSVG-complemented (*G) recombinant VSVs possessing foreign genes of either JEV PrM/E, HCV E1/E2, or luciferase in place of the VSV G gene, as shown in Fig. 1A and B. The pseudotype VSVs, JEVpv, VSVpv, HCVpv, and MLVpv, were generated by infection with VSVΔG/Luc-*G in 293T or Huh7 cells transiently expressing the respective foreign protein (Fig. 1C).
To examine the properties of the JEV E proteins incorporated into JEV, JEVrv, and JEVpv particles, the E proteins expressed in Huh7 cells and incorporated into the viral particles were digested with Endo H or PNGase F and examined by immunoblotting (Fig. 2A). Although E proteins in the lysates of cells infected with JEV, JEVrv, or JEVpv were sensitive to both Endo H and PNGase F treatments, those incorporated into the viral particles were resistant to Endo H, suggesting that both JEVrv and JEVpv particles selectively incorporate the matured E proteins modified to the complex- or hybrid-type glycans as seen in the authentic JEV particles. Next, to examine the infectivity of JEVrv and JEVpv for the target cells, HCVpv, MLVpv, VSVpv, VSV, HCVrv, and ΔG were prepared as controls (Fig. 2B). Both JEVpv and JEVrv were infectious for Huh7, BHK, and Vero cells, whereas HCVpv and HCVrv were infectious for Huh7 cells but not for BHK and Vero cells, as previously reported (1). Although JEVpv and JEVrv generated in 293T cells were also infectious, these viruses were slightly more infective when generated in Huh7 cells, even though the efficiency of transfection of the expression plasmids into 293T cells was higher than that of transfection into Huh7 cells (data not shown). To determine the specificity of infection of JEVpv, JEVrv, and JEV, a neutralization assay was performed by using anti-E antibody (22A1). The infectivities of JEVpv and JEVrv but not of VSVpv and VSV for Huh7 cells were clearly inhibited by anti-E antibody in a dose-dependent manner (Fig. 2C). These results suggest that the JEVrv and JEVpv generated in this study had characteristics comparable to those of authentic JEV.
FIG. 2.

Characterization of JEVrv and JEVpv. (A) JEV E proteins expressed in cells incorporated into the viral particles were treated with endoglycosidase H (H) or peptide-N-glycosidase F (F) and examined by immunoblotting using anti-E polyclonal antibody. “-” indicates an untreated sample. (B) Infectivities of recombinant viruses (left panel) and pseudotype viruses (right panel) were determined in Huh7, BHK, and Vero cells by a focus-forming assay and measurement of luciferase activity (RLU), respectively. VSV without envelope (ΔG) was used as a negative control. ffu, focus-forming units. (C) Neutralization of JEVrv (left panel) or JEVpv (right panel) infection by anti-E polyclonal antibody. Viruses were incubated with the indicated dilution of antibody for 1 h at room temperature and inoculated into Huh7 cells. Residual infectivities are expressed as percentages. VSV and VSVpv were used as controls. The results shown are from three independent assays, with error bars representing standard deviations.
Entry pathways of JEVpv.
Previous studies showed that JEV infection was inhibited by treatment with inhibitors of vacuolar acidification, such as ammonium chloride, concanamycin A, and bafilomycin A1, suggesting that JEV enters target cells via pH-dependent endocytosis (30). Other flaviviruses, including WNV, DENV, and HCV, exhibit similar entry mechanisms (18, 45). To compare the entry pathway of JEV with those of other viruses, Huh7 cells were pretreated with various concentrations of bafilomycin A1 and then the cells were inoculated with JEVpv, HCVpv, VSVpv, and MLVpv (Fig. 3A). As expected, bafilomycin A1 treatment did not affect the infectivity of MLVpv-bearing envelope proteins of MLV, which enters cells through a pH-independent direct fusion of the viral membrane and plasma membrane. In contrast, infections with HCVpv and VSVpv, which enter cells through pH-dependent endocytosis, were inhibited by treatment with bafilomycin A1 in a dose-dependent manner. Similarly, infection with JEVpv was clearly inhibited by treatment with bafilomycin A1 in a dose-dependent manner, suggesting that JEVpv enters cells through pH-dependent endocytosis, as seen in JEV infection.
FIG. 3.

Entry pathways of the pseudotype VSVs. Huh7 cells were pretreated with various concentrations of bafilomycin A1 (A), chlorpromazine (B), or methyl-β-cyclodextrin (C) for 1 h and inoculated with the pseudotype viruses, JEVpv, HCVpv, VSVpv, and MLVpv. Luciferase activities were determined at 24 h postinfection. The results shown are from three independent assays, with error bars representing standard deviations.
To further examine the entry pathway of JEVpv, Huh7 cells were pretreated with various concentrations of chlorpromazine, an inhibitor of clathrin-mediated endocytosis, or MβCD, an inhibitor of caveolar/raft-mediated endocytosis, and infected with the pseudotype viruses. The infectivity of MLVpv was not affected by the treatment with either chlorpromazine or MβCD, as we expected. Treatment of cells with chlorpromazine slightly reduced the infectivity of JEVpv, HCVpv, and VSVpv in a dose-dependent manner (Fig. 3B), whereas treatment of cells with MβCD reduced the infectivity of HCVpv and VSVpv but increased the infectivity of JEVpv (Fig. 3C). These results suggest that JEVpv enters cells via clathrin-mediated endocytosis, as previously reported for infection with JEV (30), and that caveola/raft plays a different role in the entry of JEV than in the entry of HCV and VSV.
Effects of cholesterol on the entry and egress of JEV.
Recently it was shown that entry of flaviviruses, including JEV and DENV, was drastically inhibited by treatment of the particles with cholesterol (20). To examine the effect of cholesterol on entry of JEV, the pseudotype viruses were inoculated into Huh7 cells after treatment with various concentrations of cholesterol. The infectivity of JEVpv but not that of HCVpv, VSVpv, or MLVpv was severely impaired by treatment with cholesterol in a dose-dependent manner (Fig. 4A). Next, to examine the effect of cholesterol on the propagation of JEV, the recombinant viruses were inoculated into Huh7 cells after treatment with various concentrations of cholesterol. Infectivities of JEV and JEVrv but not those of VSV and HCVrv were inhibited by the treatment with cholesterol (Fig. 4B, upper panel). Suppression of the propagation of JEV and JEVrv was further confirmed by a focus-forming assay (Fig. 4B, lower panels). These results confirmed that JEV entry was suppressed by cholesterol, as previously reported (20), and raise the possibility that cholesterol participates not only in entry via the E protein but also in the assembly of the E protein. These data also support the notion that JEVpv and JEVrv are comparable to JEV in terms of the properties of the E protein involved in the entry and egress processes.
FIG. 4.
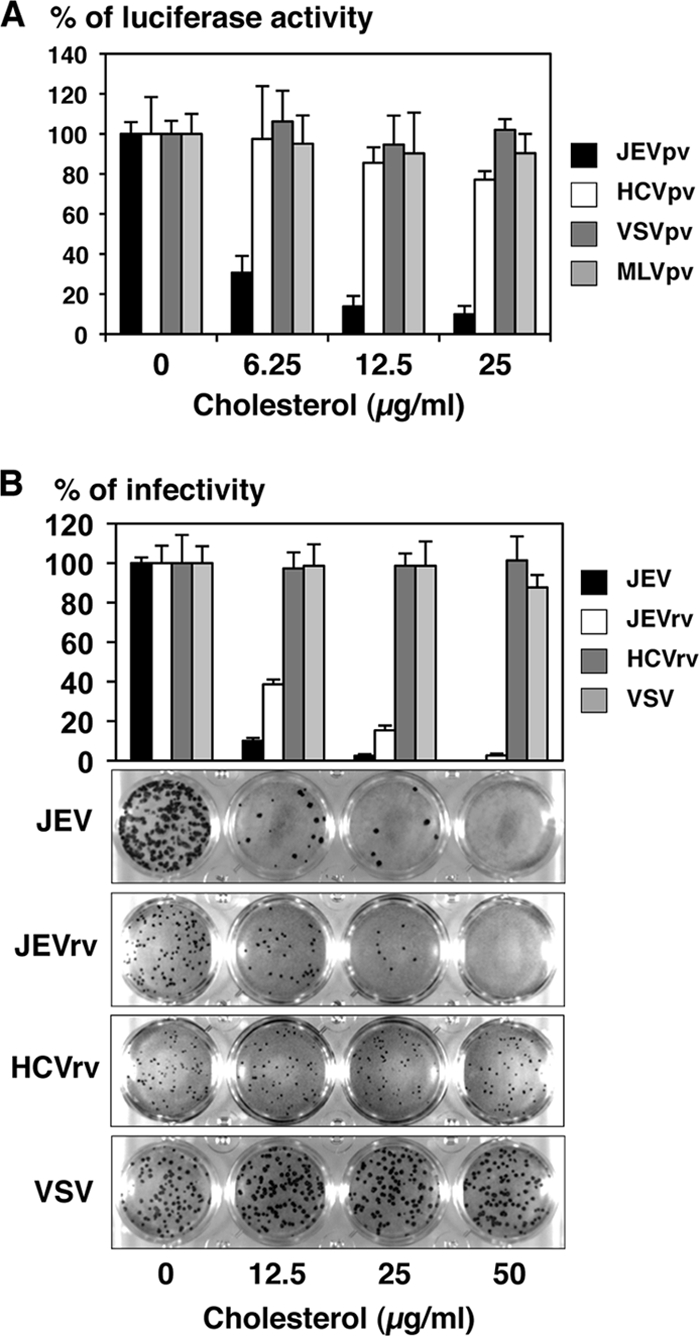
Effects of cholesterol on infection with recombinant and pseudotype VSVs. (A) The pseudotype viruses were incubated with various concentrations of cholesterol for 1 h at room temperature and inoculated into Huh7 cells, and luciferase activities were determined at 24 h postinfection. (B) JEV, JEVrv, HCVrv, and VSV were incubated with various concentrations of cholesterol for 1 h at room temperature and inoculated into Huh7 cells, and residual infectivities were determined by focus-forming assay in a culture medium containing 1% methylcellulose at 48 h postinfection for JEV, JEVrv, and HCVrv and at 24 h postinfection for VSV. Foci of infected cells were detected by immunohistochemical staining (lower panel). The rate of focus formation of the viruses was analyzed by counting foci. The results shown are from three independent assays, with error bars representing standard deviations.
Effects of SMase on infection with JEVpv, JEVrv, and JEV.
Because infection with enveloped viruses was initiated by the interaction of viral and host membrane lipids, we next examined the involvement of membrane lipids in the entry of JEV. Sphingolipid is a major component of eukaryotic lipid membranes, and sphingomyelin is one of the most abundant sphingolipids, with a wide presence across the cell membrane. SMase is known to cleave sphingomyelin, yielding phosphorylcholine and ceramide. To examine the effect of SMase on viral infection, cells were infected with viruses after treatment with various concentrations of SMase, and the infectivities of the viruses were assessed by the luciferase or focus-forming assay. Infection with JEVpv was drastically enhanced by SMase treatment of Huh7 cells, whereas such treatment exhibited no effect on infection with VSVpv and MLVpv and suppressed HCVpv infection (Fig. 5A). The enhancement of JEVpv infection by SMase treatment was also observed in other cell lines, including BHK and Vero cells (data not shown). Although the effect was not as evident as in JEVpv infection, SMase treatment exhibited a slight but substantial enhancement of the infectivity of JEV and JEVrv in Huh7 cells, in contrast to having no effect on VSV infection and a suppressive effect on HCVrv infection (Fig. 5B). The difference in the magnitude of enhancement of infectivity by treatment with SMase between infection with JEVpv and that with JEV or JEVrv might be attributable to the difference in the viral systems based on pseudotype (JEVpv) and replication-competent (JEV and JEVrv) viruses, which allow single and multiple rounds of infection, respectively. The effects of SMase may be more critical for the entry step than for other, later steps of infection. Suppression of HCVpv and HCVrv infection by treatment with SMase was consistent with previously reported data on infection of HCV-pseudotyped retroviral particles (HCVpp) and JFH1 virus (48).
FIG. 5.

Effects of SMase and amitriptyline treatment of cells on infection with pseudotype and recombinant VSVs. Huh7 cells were pretreated with various concentrations of SMase for 1 h, and then pseudotype viruses (A) or recombinant viruses (B) were inoculated. The infectivities were determined by luciferase activity measurement or focus-forming assay, and changes in infectivities are expressed as percentages. (C) The purified pseudotype particles were treated with various concentrations of SMase for 1 h and inoculated into Huh7 cells after removal of SMase by ultracentrifugation. Infectivities were determined at 24 h postinfection by measuring luciferase activity, and changes in infectivities are expressed as percentages. (D) Huh7 cells were pretreated with various concentrations of amitriptyline, an inhibitor for the acid SMase, for 1 h, and then pseudotype viruses were inoculated. Infectivities were determined at 24 h postinfection by measuring luciferase activity, and changes in infectivities are expressed as percentages. The results shown are from three independent assays, with error bars representing standard deviations.
Next, we examined the effect of SMase on the viral particles. Treatment of pseudotype particles of JEVpv, VSVpv, and MLVpv with various concentrations of SMase had no significant effect on their infectivity for Huh7 cells (Fig. 5C), whereas the infectivity of HCVpv particles was impaired by the treatment in a dose-dependent manner, as reported previously (1), suggesting that SMase treatment enhances the infectivity of JEVpv by modifying the molecules on target cells rather than the molecules on viral particles. To further determine the involvement of SMase in infection of JEVpv, cells were pretreated with various concentrations of amitriptyline, an inhibitor of acid SMase. The infectivity of JEVpv but not that of other viruses was decreased by the treatment with amitriptyline in a dose-dependent manner (Fig. 5D). A similar effect was observed with treatment with another SMase inhibitor, imipramine (data not shown). Collectively, these results suggest that entry of JEV into the target cells is enhanced by SMase treatment, which modifies the cell surface sphingolipids into a more competent state for interaction with the JEV envelope protein, thereby enabling its entry.
Effects of SMase on propagation of JEVrv and JEV.
We next examined the effects of SMase on the propagation of JEV. Recombinant VSV is capable of replicating by using the VSV genome and producing infectious particles bearing a foreign envelope protein encoded in place of the original G protein, and thus, it is feasible to assess the efficiency of not only entry but also egress of the recombinant viruses possessing foreign envelope genes of different origins, irrespective of their replication efficiency within the target cells. To examine the effects of SMase on viral propagation, cells were treated with various concentrations of the enzyme, inoculated with the recombinant viruses, and then cultured for up to 48 h in the presence of SMase. Production of JEVrv was dramatically enhanced by cultivation in the presence of SMase, in contrast to the suppression of HCVrv propagation (Fig. 6A). Although the effect of SMase treatment on the production of JEV was not as great as that seen in JEVrv propagation, treatment with SMase resulted in a substantial enhancement of JEV but not of VSV propagation (Fig. 6B). These results suggest that SMase treatment induces robust propagation of JEVrv mainly through enhancement of the entry step although also partly through enhancement of the egress step.
FIG. 6.
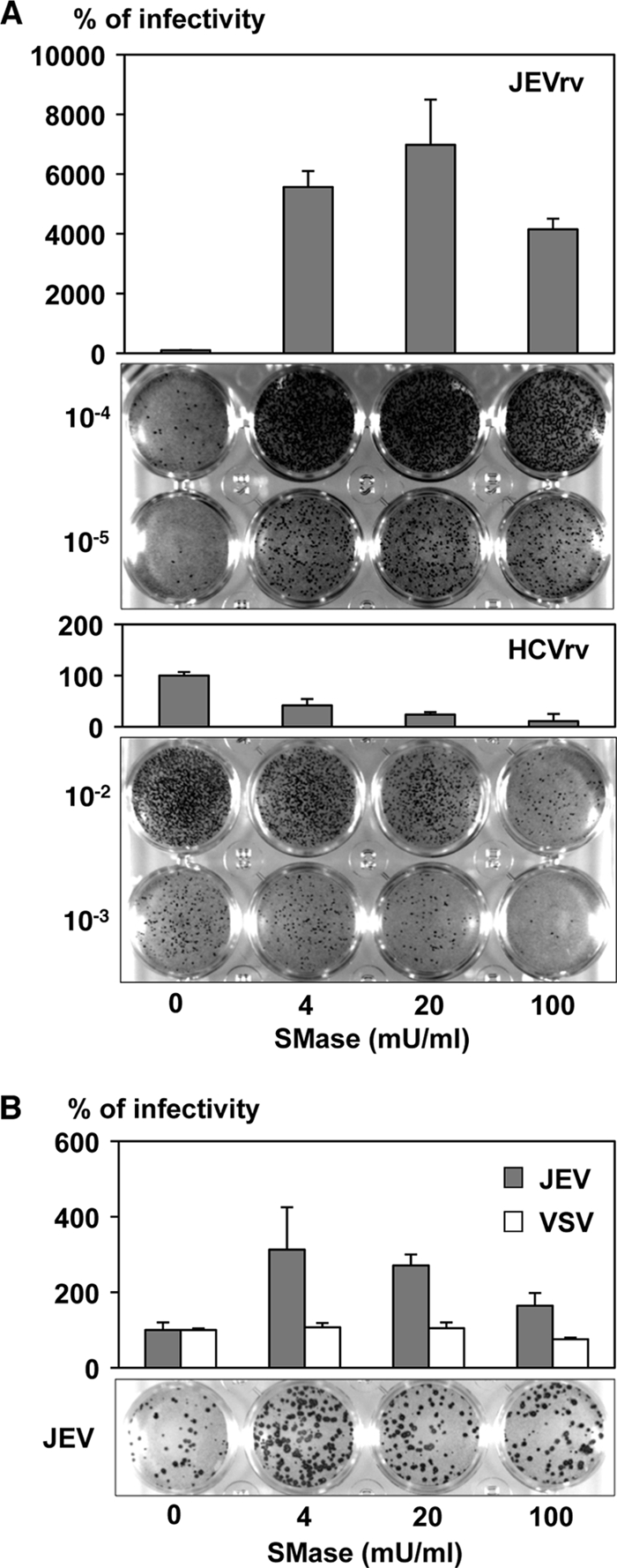
Effects of SMase on the propagation of JEVrv and JEV. Huh7 cells were pretreated with various concentrations of SMase for 1 h and inoculated with JEVrv or HCVrv (A) or JEV or VSV (B), and infectivities were determined by focus-forming assay in a culture medium containing 1% methylcellulose at 48 h after infection with JEVrv, HCVrv, and JEV and at 24 h after infection with VSV. Titers were determined by counts of foci detected by immunohistochemical staining (lower panels). The results shown are from three independent assays, with error bars representing standard deviations.
Involvement of ceramide in infection with JEV.
Because treatment of cells with SMase induces production of ceramide, we next examined the effect of ceramide on the infectivity of the viruses. Treatment of the pseudotype particles with C6-ceramide inhibited the infectivity of JEVpv for Huh7 cells in a dose-dependent manner, whereas no clear reduction of infectivity was observed with treatment of HCVpv, VSVpv, and MLVpv with ceramide (Fig. 7A). In contrast, treatment of the pseudotype particles with sphingomyelin, which is a substrate for SMase and is catalyzed into ceramide, did not affect the infectivity of the viruses, suggesting that the enhancement of infectivity of JEVpv by treatment with SMase was due to the generation of ceramide. Propagation of JEV but not of VSV was also suppressed by treatment of the viral particles with C6-ceramide in a dose-dependent manner (Fig. 7B). Finally, to confirm the interaction of the JEV E protein with ceramide, purified JEV and JEVrv particles were incubated with biotin-ceramide and streptavidin-Sepharose 4B and examined by pull-down assay (Fig. 7C). The E proteins of both JEV and JEVrv were precipitated with the ceramide beads. These results indicate that the interaction of the JEV E protein with ceramide plays a crucial role in the entry of JEV.
FIG. 7.

Involvement of ceramide in infection with JEV. (A) Effects of C6-ceramide or sphingomyelin in infection with JEVpv. Purified pseudotype viruses were pretreated with various concentrations of C6-ceramide (upper) or sphingomyelin (lower) for 1 h and then inoculated into Huh7 cells. The infectivities were determined at 24 h postinfection by luciferase activity, and changes in infectivities are expressed as percentages. (B) Effects of C6-ceramide in infection of JEVrv and JEV. JEV and VSV were pretreated with various concentrations of C6-ceramide for 1 h, and then the viruses were inoculated into Huh7 cells. At 24 h postinfection, the infectivities were determined by focus-forming assay. (C) Binding of JEV and JEVrv to ceramide beads. Purified viruses were preincubated with (+) or without (−) biotin-ceramide resolved in DMSO and streptavidin-Sepharose 4B. After washing, residual pellets were analyzed by immunoblotting. Inputs are purified viruses. The results shown are from three independent assays, with error bars representing standard deviations.
DISCUSSION
Ceramide has been shown to play a crucial role in various cell signaling pathways through the clustering and activation of the receptor molecules in lipid rafts. Although the generation of ceramide inhibits the infectivity of HIV and HCV by the rearrangement of the entry receptor molecules (7, 48), rhinovirus and Sindbis virus generate ceramide by activating SMase for their entry and cell survival, respectively (10, 15). In this study, we demonstrated for the first time that ceramide plays crucial roles not only in the entry pathway of JEV but also in the egress through a direct interaction with the E envelope proteins.
To examine the roles of the E protein in the infectivity of JEV, we employed pseudotype and recombinant VSVs bearing JEV envelope proteins as surrogate systems in addition to authentic JEV. VSV assembles and buds from the plasma membrane, and therefore the surrogate viruses bearing the foreign envelope proteins being expressed on the plasma membrane exhibited more-efficient incorporation of the envelope proteins. Although the E protein of JEV, as well as that of other flaviviruses, including HCV, is mainly retained on the endoplasmic reticulum (ER) membrane, the E protein was incorporated into JEVpv and JEVrv particles and exhibited infectivity comparable to that of authentic JEV. Further studies are needed to clarify the mechanisms of incorporation of the foreign envelope proteins on the ER membrane into VSV particles. In general, glycoproteins are modified into the complex type during the translocation from the ER to the Golgi apparatus. Although the JEV E glycoproteins were modified mainly into the high-mannose type in cells infected with JEVpv, JEVrv, or JEV, viruses possessing the E proteins were modified into the complex type within the particles. These results suggest that the E proteins of JEV and the surrogate viral particles are modified into the complex type after budding into the ER lumen during translocation into the Golgi apparatus. Recently assembly of DENV in the ER was revealed by three-dimensional architecture using electron tomography (49).
A number of viruses utilize cholesterol-rich membrane microdomains or lipid rafts for their entry, assembly, or egress processes (5). Cholesterol-rich membrane microdomains have been shown to be required for the entry but not for the replication of WNV through cholesterol depletion by treatment with MβCD (27). Entry of HCV was also shown to be partially required for cellular cholesterol (1, 16), which is consistent with the present data that infection with HCVpv was partially inhibited by treatment of cells with MβCD. Lee et al. recently reported that the infectivity of JEV, especially the replication step, was inhibited by treatment of cells with MβCD or the cholesterol chelation antibiotic filipin III (20). Furthermore, treatment of the viral particles with cholesterol inhibited the infectivity of JEV, in contrast to the enhancement of the infectivity of Sindbis virus by the same treatment (20, 22). Our data also indicated that the infectivity of JEVpv and JEVrv, as well as that of JEV, was completely inhibited by treatment of the particles with cholesterol in a dose-dependent manner, supporting the notion that the presence of an abundant amount of cholesterol increases the rigidity of the E protein of JEV particles and inhibits the membrane fusion event, as suggested by Lee et al. (20).
According to the current models, SMase alters the biophysical properties of the membrane bilayer by generating ceramide through the hydrolysis of sphingomyelin. Genetic disorders of SMase or ceramide metabolism are critically involved in human genetic diseases, such as Niemann-Pick disease (37) and Wilson's disease (19). In vivo studies of the function of SMase or ceramide in infections with pathogens are accumulating (9, 44, 46), and acid SMase-deficient mice have been shown to be unable to eliminate the pathogens because of failure to undergo apoptosis or phagolysosomal fusion, ultimately a massive release of cytokines and death by sepsis. It has recently been shown that acid SMase is a key regulator of cytotoxic granule secretion by primary T lymphocytes (13). The reduction of the cytolytic activity of CD8+ cytotoxic T lymphocytes in acid SMase-deficient mice resulted in a significantly delayed clearance of lymphocytic choriomeningitis virus infection. Recently it was shown that entry of HCV is inhibited by SMase treatment through the downregulation of CD81, a major receptor of HCV, because enrichment of ceramide on the plasma membrane induces internalization of CD81 (48). HIV infection is also inhibited by ceramide enrichment through a restriction of the lateral diffusion of CD4 (6). Sindbis virus and rhinovirus activate the SMase and induce generation of ceramide in the endosomal membrane. Inhibition of SMase by genetic manipulation or pharmacological agents prevents infection with rhinoviruses, suggesting that SMase and ceramide-enriched membrane platforms play an important role in viral infection (10).
In this study, we have shown that entry of JEVpv, JEVrv, and JEV was specifically enhanced by treatment of cells with SMase. Treatment of cells with amitriptyline, an inhibitor interfering with the binding of SMase to the lipid bilayer, impaired the uptake of rhinovirus (10) and Neisseria gonorrhoeae (8). The entry of JEVpv was also inhibited by treatment with the inhibitor. Furthermore, the infections of JEVpv and JEV were inhibited by treatment with C6-ceramide but not by treatment with sphingomyelin, and JEV and JEVrv were coprecipitated with the ceramide beads, suggesting that the interaction of ceramide with the JEV E protein plays a crucial role in the early steps of infection. Ceramide is known to bind to the ceramide transport protein (CERT), which transports ceramide from the ER to the Golgi apparatus (12), and thus, it might be feasible to speculate that CERT participates in the translocation or maturation of the JEV E protein. Further studies are needed to clarify the interaction among ceramide, CERT, and the JEV E protein. Recently Aizaki et al. reported that the infectivity of HCV particles was decreased by treatment with MβCD or SMase, suggesting that cholesterols or sphingolipids incorporated into the virions are important for the infectivity of HCV (1). In this study, SMase treatment of HCVpv particles but not of JEVpv particles reduced infectivity, suggesting that incorporation of cholesterols and sphingolipids into the viral particles was different among flaviviruses.
The discrepancy between the drastic increase in the production of infectious particles of JEVrv and the marginal increase in that for JEV induced by SMase treatment in ceramide-enriched cells may indicate that ceramide enrichment enhances the entry and egress steps but negatively regulates genomic replication of JEV. Previously it was reported that digestion of sphingomyelin by SMase induces cholesterol redistribution (32), an increase in intracellular cholesterol esterification (4), and a decrease in cholesterol biosynthesis (39). Furthermore, ceramide has been shown to selectively displace cholesterol from lipid rafts and decrease the association of the cholesterol binding protein caveolin-1 (28, 50). Although we have not determined the cholesterol composition of the membranes of cells treated with SMase, cholesterol depletion induced by SMase treatment may also participate in the enhancement of JEV entry.
JEV initiates infection by interacting with receptor and/or coreceptor molecule(s), probably in cooperation with ceramide located in the ceramide-enriched platforms. The ceramide-enriched membrane domains facilitate signal transduction through reorganization and clustering of cell surface receptor molecules. Although the entry receptor(s) of JEV has not been well characterized yet, modification of the distribution, organization, and steric conformation of the receptor molecule(s) by treatment with SMase may facilitate entry of JEV. Generation of ceramide by SMase treatment has been shown to promote vesicular fusion processes and fusion of phagosomes, thereby engulfing bacteria with late endosomes and resulting in efficient intracellular bacterial killing (46).
In conclusion, we have demonstrated that the entry and egress processes of JEV were enhanced by treatment with SMase by using pseudotype and recombinant VSVs. The interaction of cellular ceramide and the E glycoproteins facilitates infection and propagation of JEV. Modification of sphingolipids on the plasma membrane of the target cells might be a novel target for the development of antivirals against JEV infection.
Acknowledgments
We thank H. Murase for her secretarial work. We also thank M. A. Whitt and T. Miyazawa for providing plasmids and antibodies.
This research was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare; the Ministry of Education, Culture, Sports, Science, and Technology; the Global Center of Excellence Program; and the Foundation for Biomedical Research and Innovation.
Footnotes
Published ahead of print on 6 January 2010.
REFERENCES
- 1.Aizaki, H., K. Morikawa, M. Fukasawa, H. Hara, Y. Inoue, H. Tani, K. Saito, M. Nishijima, K. Hanada, Y. Matsuura, M. M. Lai, T. Miyamura, T. Wakita, and T. Suzuki. 2008. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 82:5715-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bollinger, C. R., V. Teichgraber, and E. Gulbins. 2005. Ceramide-enriched membrane domains. Biochim. Biophys. Acta 1746:284-294. [DOI] [PubMed] [Google Scholar]
- 3.Boonsanay, V., and D. R. Smith. 2007. Entry into and production of the Japanese encephalitis virus from C6/36 cells. Intervirology 50:85-92. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee, S. 1993. Neutral sphingomyelinase increases the binding, internalization, and degradation of low density lipoproteins and synthesis of cholesteryl ester in cultured human fibroblasts. J. Biol. Chem. 268:3401-3406. [PubMed] [Google Scholar]
- 5.Chazal, N., and D. Gerlier. 2003. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 67:226-237, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finnegan, C. M., S. S. Rawat, E. H. Cho, D. L. Guiffre, S. Lockett, A. H. Merrill, Jr., and R. Blumenthal. 2007. Sphingomyelinase restricts the lateral diffusion of CD4 and inhibits human immunodeficiency virus fusion. J. Virol. 81:5294-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finnegan, C. M., S. S. Rawat, A. Puri, J. M. Wang, F. W. Ruscetti, and R. Blumenthal. 2004. Ceramide, a target for antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 101:15452-15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grassme, H., E. Gulbins, B. Brenner, K. Ferlinz, K. Sandhoff, K. Harzer, F. Lang, and T. F. Meyer. 1997. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell 91:605-615. [DOI] [PubMed] [Google Scholar]
- 9.Grassme, H., V. Jendrossek, A. Riehle, G. von Kurthy, J. Berger, H. Schwarz, M. Weller, R. Kolesnick, and E. Gulbins. 2003. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9:322-330. [DOI] [PubMed] [Google Scholar]
- 10.Grassme, H., A. Riehle, B. Wilker, and E. Gulbins. 2005. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 280:26256-26262. [DOI] [PubMed] [Google Scholar]
- 11.Gubler, D., G. Kuno, and L. Markoff. 2007. Flaviviruses, p. 1153-1252. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 12.Hanada, K., K. Kumagai, S. Yasuda, Y. Miura, M. Kawano, M. Fukasawa, and M. Nishijima. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature 426:803-809. [DOI] [PubMed] [Google Scholar]
- 13.Herz, J., J. Pardo, H. Kashkar, M. Schramm, E. Kuzmenkina, E. Bos, K. Wiegmann, R. Wallich, P. J. Peters, S. Herzig, E. Schmelzer, M. Kronke, M. M. Simon, and O. Utermohlen. 2009. Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary T lymphocytes. Nat. Immunol. 10:761-768. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda, M., K. Abe, M. Yamada, H. Dansako, K. Naka, and N. Kato. 2006. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 44:117-125. [DOI] [PubMed] [Google Scholar]
- 15.Jan, J. T., S. Chatterjee, and D. E. Griffin. 2000. Sindbis virus entry into cells triggers apoptosis by activating sphingomyelinase, leading to the release of ceramide. J. Virol. 74:6425-6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapadia, S. B., H. Barth, T. Baumert, J. A. McKeating, and F. V. Chisari. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 81:374-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapadia, S. B., and F. V. Chisari. 2005. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U. S. A. 102:2561-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnan, M. N., B. Sukumaran, U. Pal, H. Agaisse, J. L. Murray, T. W. Hodge, and E. Fikrig. 2007. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J. Virol. 81:4881-4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang, P. A., M. Schenck, J. P. Nicolay, J. U. Becker, D. S. Kempe, A. Lupescu, S. Koka, K. Eisele, B. A. Klarl, H. Rubben, K. W. Schmid, K. Mann, S. Hildenbrand, H. Hefter, S. M. Huber, T. Wieder, A. Erhardt, D. Haussinger, E. Gulbins, and F. Lang. 2007. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 13:164-170. [DOI] [PubMed] [Google Scholar]
- 20.Lee, C. J., H. R. Lin, C. L. Liao, and Y. L. Lin. 2008. Cholesterol effectively blocks entry of flavivirus. J. Virol. 82:6470-6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, E., and M. Lobigs. 2002. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J. Virol. 76:4901-4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu, Y. E., T. Cassese, and M. Kielian. 1999. The cholesterol requirement for Sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 73:4272-4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackenzie, J. M., A. A. Khromykh, and R. G. Parton. 2007. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2:229-239. [DOI] [PubMed] [Google Scholar]
- 24.Manes, S., G. del Real, and A. C. Martinez. 2003. Pathogens: raft hijackers. Nat. Rev. Immunol. 3:557-568. [DOI] [PubMed] [Google Scholar]
- 25.Matsuura, Y., H. Tani, K. Suzuki, T. Kimura-Someya, R. Suzuki, H. Aizaki, K. Ishii, K. Moriishi, C. S. Robison, M. A. Whitt, and T. Miyamura. 2001. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology 286:263-275. [DOI] [PubMed] [Google Scholar]
- 26.Mayor, S., and H. Riezman. 2004. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol. 5:110-120. [DOI] [PubMed] [Google Scholar]
- 27.Medigeshi, G. R., A. J. Hirsch, D. N. Streblow, J. Nikolich-Zugich, and J. A. Nelson. 2008. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of alphavbeta3 integrin. J. Virol. 82:5212-5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Megha and E. London. 2004. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J. Biol. Chem. 279:9997-10004. [DOI] [PubMed] [Google Scholar]
- 29.Mori, Y., T. Yamashita, Y. Tanaka, Y. Tsuda, T. Abe, K. Moriishi, and Y. Matsuura. 2007. Processing of capsid protein by cathepsin L plays a crucial role in replication of Japanese encephalitis virus in neural and macrophage cells. J. Virol. 81:8477-8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nawa, M., T. Takasaki, K. Yamada, I. Kurane, and T. Akatsuka. 2003. Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J. Gen. Virol. 84:1737-1741. [DOI] [PubMed] [Google Scholar]
- 31.Perez, M., R. Clemente, C. S. Robison, E. Jeetendra, H. R. Jayakar, M. A. Whitt, and J. C. de la Torre. 2007. Generation and characterization of a recombinant vesicular stomatitis virus expressing the glycoprotein of Borna disease virus. J. Virol. 81:5527-5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porn, M. I., and J. P. Slotte. 1995. Localization of cholesterol in sphingomyelinase-treated fibroblasts. Biochem. J. 308(Part 1):269-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren, J., T. Ding, W. Zhang, J. Song, and W. Ma. 2007. Does Japanese encephalitis virus share the same cellular receptor with other mosquito-borne flaviviruses on the C6/36 mosquito cells? Virol. J. 4:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reyes-Del Valle, J., S. Chavez-Salinas, F. Medina, and R. M. Del Angel. 2005. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J. Virol. 79:4557-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothwell, C., A. Lebreton, C. Young Ng, J. Y. Lim, W. Liu, S. Vasudevan, M. Labow, F. Gu, and L. A. Gaither. 2009. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 389:8-19. [DOI] [PubMed] [Google Scholar]
- 36.Schenck, M., A. Carpinteiro, H. Grassme, F. Lang, and E. Gulbins. 2007. Ceramide: physiological and pathophysiological aspects. Arch. Biochem. Biophys. 462:171-175. [DOI] [PubMed] [Google Scholar]
- 37.Schuchman, E. H. 2007. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis. 30:654-663. [DOI] [PubMed] [Google Scholar]
- 38.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31-39. [DOI] [PubMed] [Google Scholar]
- 39.Slotte, J. P., and E. L. Bierman. 1988. Depletion of plasma-membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membranes and intracellular cholesterol pools in cultured fibroblasts. Biochem. J. 250:653-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stiasny, K., C. Koessl, and F. X. Heinz. 2003. Involvement of lipids in different steps of the flavivirus fusion mechanism. J. Virol. 77:7856-7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su, C. M., C. L. Liao, Y. L. Lee, and Y. L. Lin. 2001. Highly sulfated forms of heparin sulfate are involved in Japanese encephalitis virus infection. Virology 286:206-215. [DOI] [PubMed] [Google Scholar]
- 42.Takada, A., C. Robison, H. Goto, A. Sanchez, K. G. Murti, M. A. Whitt, and Y. Kawaoka. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 94:14764-14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tani, H., Y. Komoda, E. Matsuo, K. Suzuki, I. Hamamoto, T. Yamashita, K. Moriishi, K. Fujiyama, T. Kanto, N. Hayashi, A. Owsianka, A. H. Patel, M. A. Whitt, and Y. Matsuura. 2007. Replication-competent recombinant vesicular stomatitis virus encoding hepatitis C virus envelope proteins. J. Virol. 81:8601-8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teichgraber, V., M. Ulrich, N. Endlich, J. Riethmuller, B. Wilker, C. C. De Oliveira-Munding, A. M. van Heeckeren, M. L. Barr, G. von Kurthy, K. W. Schmid, M. Weller, B. Tummler, F. Lang, H. Grassme, G. Doring, and E. Gulbins. 2008. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 14:382-391. [DOI] [PubMed] [Google Scholar]
- 45.Tscherne, D. M., C. T. Jones, M. J. Evans, B. D. Lindenbach, J. A. McKeating, and C. M. Rice. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 80:1734-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Utermohlen, O., J. Herz, M. Schramm, and M. Kronke. 2008. Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology 213:307-314. [DOI] [PubMed] [Google Scholar]
- 47.Viola, A., and N. Gupta. 2007. Tether and trap: regulation of membrane-raft dynamics by actin-binding proteins. Nat. Rev. Immunol. 7:889-896. [DOI] [PubMed] [Google Scholar]
- 48.Voisset, C., M. Lavie, F. Helle, A. Op De Beeck, A. Bilheu, J. Bertrand-Michel, F. Terce, L. Cocquerel, C. Wychowski, N. Vu-Dac, and J. Dubuisson. 2008. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol. 10:606-617. [DOI] [PubMed] [Google Scholar]
- 49.Welsch, S., S. Miller, I. Romero-Brey, A. Merz, C. K. Bleck, P. Walther, S. D. Fuller, C. Antony, J. Krijnse-Locker, and R. Bartenschlager. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 5:365-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu, C., M. Alterman, and R. T. Dobrowsky. 2005. Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J. Lipid Res. 46:1678-1691. [DOI] [PubMed] [Google Scholar]