

Abstract
The UL16 protein of herpes simplex virus is capsid associated and was previously identified as a binding partner of the membrane-associated UL11 tegument protein (J. S. Loomis, R. J. Courtney, and J. W. Wills, J. Virol. 77:11417-11424, 2003). In those studies, a less-prominent, ∼65-kDa binding partner of unknown identity was also observed. Mass spectrometry studies have now revealed this species to be UL21, a tegument protein that has been implicated in the transport of capsids in the cytoplasm. The validity of the mass spectrometry results was tested in a variety of coimmunoprecipitation and glutathione S-transferase pull-down experiments. The data revealed that UL21 and UL16 can form a complex in the absence of other viral proteins, even when the assays used proteins purified from Escherichia coli. Moreover, UL11 was able to pull down UL21 only when UL16 was present, suggesting that all three proteins can form a complex. Deletion analyses revealed that the second half of UL21 (residues 268 to 535) is sufficient for the UL16 interaction and packaging into virions; however, attempts to map a subdomain of UL16 were largely unsuccessful, with only the first 40 (of 373) residues being found to be dispensable. Nevertheless, it is clear that UL16 must have two distinct binding sites, because covalent modification of its free cysteines with N-ethylmaleimide blocked binding to UL11 but not UL21. These findings should prove useful for elucidating the molecular machinery used to transmit a signal into a virion when it attaches to cells, a recently discovered mechanism in which UL16 is a central player.
Herpes simplex virus (HSV) contains more than 40 different virally encoded proteins that are found in three distinct layers: the capsid containing the viral DNA, the host-derived lipid envelope with embedded glycoproteins, and the tegument, an assortment of proteins located between the nucleocapsid and the envelope (22). While these regions are often discussed as separate structures, there is now clear evidence that the virion as a whole is a machine with interconnected parts that quickly rearrange on the inside in response to glycoprotein-binding events on the outside. Specifically, tegument protein UL16 is triggered to be released from the capsid when HSV attaches to host cells prior to membrane fusion, and the signal responsible for this can be sent in a cell-free manner by binding virions to immobilized heparin (21). It appears that glycoprotein C is involved in transmitting the signal (at least in a cell-free system), but all the other molecular “cogs” that drive this part of the HSV machine are unknown. To identify these components, we have been investigating UL16 and the network of molecular interactions in which it participates.
Our interest in UL16 began when we identified it as a binding partner of UL11 (17), a small tegument protein (only 96 amino acids) that is conserved among all herpesviruses. UL11 is peripherally bound to membranes via two fatty acids, myristate and palmitate (16), and trafficks through lipid raft domains (6, 12). It accumulates at the trans-Golgi network (TGN), where virus budding takes place (16, 30), and mutants that lack UL11 are defective for the production of virions, resulting in an increased number of unenveloped capsids in the cytoplasm (5, 9, 19). The UL11-UL16 interaction has since been confirmed by other groups (15, 37), and more recently, we have found that the interaction is direct and requires free cysteines present within UL16 (41). That is, chemical modification of free cysteines in UL16 with N-ethylmaleimide (NEM) blocks the interaction with UL11. On the UL11 side of the interaction, LI and acidic cluster motifs are needed for binding (17, 41).
UL16 is a 373-amino-acid protein that is also conserved among herpesviruses and exhibits dynamic capsid-binding properties. Although it is found in both the cytoplasm and the nucleus of the infected cell, it is only stably associated with capsids isolated from the cytoplasm (20, 24, 26). This finding, combined with the ability of UL11 to accumulate at the site of budding, led us to hypothesize that the UL11-UL16 interaction provides a bridging function to assist the capsid in acquiring its envelope (17). However, sometime after budding—as the virus egresses from the cell—the interaction of UL16 with the capsid is destabilized (20). And, as mentioned earlier, binding of the virion to its attachment receptors on the host cell surface (heparan sulfate) further disrupts the association of UL16 with the capsid (21). Free cysteines appear to play a critical role in this outside-in signaling event, because treatment of extracellular virions with NEM prior to cell binding prevents the release of UL16 from the capsid (21).
While UL16 was the most abundant protein pulled out of infected cell lysates in our search for UL11 binding partners, a much less prominent, but highly reproducible, ∼65-kDa species was also observed (17). Like UL16, this unknown protein was absent when either the LI or acidic cluster motifs were eliminated from the glutathione S-transferase (GST)-UL11 construct used in the experiment. This suggested that the unknown protein was obtained by either (i) competing with UL16 for binding to the same motifs within UL11 or (ii) binding to UL11 indirectly through an interaction with UL16. Because the LI and acidic cluster motifs of UL11 are recognized by host proteins for trafficking through lipid rafts (6, 16), the first hypothesis seemed likely; however, because UL16 participates in a complex signaling pathway within the virion, it was possible that the unknown protein would be a virus-encoded component. The purpose of the experiments described in this report was to identify this unknown protein and to determine how it fits into the UL16 network of interactions.
MATERIALS AND METHODS
Viruses, cells, and antibodies.
The KOS strain of HSV-1 was used as the wild type in this study (32). The recombinant KOS virus that expresses UL16-cyan fluorescent protein (CFP) has been described (41). HSV mutants that lacked the UL16 (4) or UL21 genes (3) were derived from the F strain. For experiments with pseudorabies virus (PRV), the Becker strain was used (17). All viruses were grown in Vero cells maintained in Dulbecco's modified Eagle's medium (DMEM; GIBCO) supplemented with 5% fetal bovine serum, penicillin (65 μg/ml), and streptomycin (131 μg/ml). Following infection, cells were grown in DMEM supplemented with 2% fetal bovine serum, 25 mM HEPES buffer, glutamine (0.3 μg/ml), penicillin, and streptomycin.
The UL16- and UL11-specific antibodies were produced in rabbits that were immunized with GST-UL16 or GST-UL11, respectively (6, 16, 17, 20). Antibodies specific for UL21 were prepared commercially (Cocalico Biologicals, Inc., Reamstown, PA) in rabbits by immunizing with GST-UL21 purified from bacteria. Antibodies specific for green fluorescent protein (GFP) and the very similar CFP were made against purified His6-GFP protein in rabbits (41). This antiserum also recognizes the N-terminal tag in His6-UL16 (41). Goat polyclonal antibodies raised against purified GST protein (Rockland) were used to detect GST-tagged proteins. All of the antibodies were used at a dilution of 1:3,000 (in 1% nonfat milk dissolved in 20 mM Tris, pH 7.6, 135 mM NaCl, and 0.1% Tween 20) for immunoblotting, unless otherwise noted.
UL21 expression vectors.
To express GST-UL21 in bacteria, the UL21 gene was amplified from viral DNA with forward (5′-CCGCGTGGATCCATGGAGCTTAGCTACGCC) and reverse (5′-CACGATGCGGCCGCTCTACACAGACTGTCC) primers that introduce BamHI and NotI sites, respectively (underlined). These sites were used to insert the resulting PCR product into the multiple cloning site of pGEX-4T-3 (Amersham). Similar methods were used to construct mutants that lack amino acids 2 to 139 or 268 to 535 of UL21.
To express GFP-UL21 in mammalian cells, a modified form of pEGFP-N2 (Clontech) was constructed to place suitable cloning sites after the GFP coding sequence. For this, the following two oligonucleotides were annealed and ligated between the BsrGI and NotI sites of the parental plasmid: 5′-GTACTCAACTAGTGCGCGCACCGGTCGATCGTGTACACCCTAATAGGC and 5′-GGCCGCCTATTAGGGTGTACACGATCGACCGGTGCGCGCACTAGTTGA. After annealing, the first oligonucleotide has a single-stranded end that is complementary to the BsrGI site but does not recreate that site upon ligation. The second oligonucleotide has an end that is complementary to NotI and does recreate that site upon ligation. The oligonucleotide pair also inserts new SpeI (italics) and BsrGI (bold) sites. The UL21 gene was inserted into this construct after amplifying with forward (5′-AGCTTCACTAGTATGGAGCTTAGCTACGCC) and reverse (5′-CCGGTGGATCCTGTACACTATTACACAGACTGTCCGTG) primers and then trimming the ends of the product with SpeI (italics) and BsrGI (bold). Various N- and C-terminal deletion mutants were created in a similar manner using oligonuleotides that amplify the desired portions of UL21.
GST-UL16 expression vectors.
A vector that expresses full-length GST-UL16 in bacteria has been described (41). To introduce UL16 deletions, DNA was amplified from a set of previously described UL16-GFP expression plasmids (41) with forward primers that contained a BamHI site and reverse primers that contained a NotI site. These sites were then used to insert the resulting PCR products into pGEX-4T-3.
Purification of GST-fusion proteins.
Bacterial strain BL21 containing plasmids for the GST-fusion proteins were incubated overnight at 37°C and then diluted 1:10 in yeast extract-tryptone medium. Protein expression was induced by the addition of 1.0 mM isopropyl-beta-d-thiogalactopyranoside (IPTG). Cells expressing GST-UL11 were grown 4 h at 37°C. We suspect that rapid production of GST-UL16 and GST-UL21 results in inefficient folding; therefore, those cultures were grown at room temperature for 24 h with IPTG treatments at time zero and at 8 h. Cultures were centrifuged at 10,000 × g for 10 min at 4°C. Cell pellets were suspended in phosphate-buffered saline (PBS), sonicated, and lysed for 30 min on ice with 1% Triton X-100 in the presence of Complete Mini protease inhibitors (Roche Applied Science). The lysate was centrifuged at 14,500 × g for 10 min at 4°C, and the supernatant was incubated with glutathione-Sepharose 4B beads (GE Healthcare) at room temperature for 30 min. The beads were pelleted at 1,000 × g for 2 min, washed three times with 0.1% Triton- containing PBS, and then suspended in 200 μl PBS. Yields were determined by SDS-PAGE followed by Coomassie blue staining.
Radiolabeling and analysis of infected cells.
Vero cells were infected with HSV or PRV at multiplicities of infection (MOIs) of 100 and 50, respectively. At 5 h postinfection, the cells were starved for 15 min in methionine-free medium and labeled for 3 h with l-[35S]methionine-cysteine (60 Ci/ml; 1,000 Ci/mmol). Cells were lysed in NP-40 lysis buffer with protease inhibitors on ice, precleared overnight with glutathione-Sepharose beads, and then incubated with purified GST derivatives on glutathione-Sepharose beads for 2 h at room temperature. The beads were washed three times in NP-40 lysis buffer, resuspended in sample buffer, separated in SDS-10% polyacrylamide gels, and transferred to nitrocellulose. The blots were then subjected to autoradiography for 8 h.
To test the necessity of UL16 for the UL11-UL21 interaction, experiments were performed in a similar manner to those above but with several changes. Cells were infected with wild-type (WT) or the HSV.ΔUL16 mutant at an MOI of 250. Nitrocellulose blots were subjected to autoradiography for 2 days to detect the radiolabeled proteins. To confirm that the radiolabeled proteins pulled down were in fact UL16 and UL21, the enhanced chemiluminescence method of immunoblot analysis was performed according to the manufacturer's instructions (Amersham) with UL16 or UL21 antiserum.
Mass spectrometry.
Cells infected for 7 h at an MOI of 100 were treated with NP-40 buffer, and the postnuclear supernatant was precleared with empty glutathione-Sepharose beads overnight. Beads with GST-UL11 bound were added to the cleared lysates and rocked for 2 h at room temperature. The beads were washed, resuspended in sample buffer, and subjected to SDS-PAGE followed by zinc staining per the manufacturer's instructions (Bio-Rad 161-0440). The 65-kDa band was excised, destained, and stored at −80°C.
Samples were reduced with 50 mM dithiothreitol at 56°C for 45 min and then alkylated with 55 mM iodoacetamide for 1 h at room temperature. The material was dried in a speed-vac, rehydrated in a 12.5 ng/μl modified sequencing-grade trypsin solution (Promega, Madison, WI), and incubated in an ice bath for 40 to 45 min. Excess trypsin solution was removed and replaced with 40 to 50 μl of 50 mM ammonium bicarbonate, 10% acetonitrile, pH 8.0, and the mixture was incubated overnight at 37°C. Peptides were extracted two times with 25 μl 50% acetonitrile, 5% formic acid and dried in a speed-vac. Digests were resuspended in 20 μl buffer A (5% acetonitrile, 0.1% formic acid, 0.005% heptafluorobutyric acid [HFBA]), and 3 to 6 μl was loaded onto a 12-cm by 0.075-mm fused silica capillary column packed with 5-μm-diameter C18 beads (The Nest Group, Southboro, MA) using an N2 pressure vessel at 1,100 lb/in2. Peptides were eluted over 55 min, by applying a 0 to 80% linear gradient of buffer B (95% acetonitrile, 0.1% formic acid, 0.005% HFBA) at a flow rate of 150 μl/min with a precolumn flow splitter, resulting in a final flow rate of ∼200 nl/min directly into the source. In some cases, the gradient was extended to 150 min to acquire more tandem mass spectrometry (MS/MS) spectra. An LCQ DecaXP linear ion trap (ThermoFinnigan, San Jose, CA) was run in an automated collection mode with an instrument method composed of a single segment and five data-dependent scan events with a full MS scan followed by four MS/MS scans of the highest-intensity ions. Normalized collision energy was set at 35, activation Q was 0.250, with minimum full scan signal intensity at 1 × 105 with no minimum MS2 intensity specified. Dynamic exclusion was turned on utilizing a 3-min repeat count of 2 with the mass width set at 1.0 m/z. Sequence analysis was performed with TurboSequest (ThermoFinnigan, San Jose, CA) using an indexed nonredundant protein database (nr) from the National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/).
Coimmunoprecipitation of UL11, UL16, and UL21 from infected cells.
Vero cells were infected at an MOI of 10 with wild-type HSV or HSV.UL16-CFP. At 20 h postinfection, the cells were scraped into PBS, pelleted by centrifugation at 1,000 × g for 5 min, and resuspended in NP-40 lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl [pH 8.0]) with protease inhibitors (Sigma product number P8340). After 15 min on ice, nuclei were removed by centrifugation for 4 min at 18,000 × g, and the supernatants were precleared overnight with protein A-agarose beads (Roche) at 4°C with rocking. Five microliters of antibody specific for UL11, UL16, or UL21 was added for 1 h at 4°C, and the immune complexes were collected with protein A-agarose beads (Roche) for 5 h with rocking. Beads and their bound proteins were washed three times with NP-40 lysis buffer, resuspended in sample buffer, separated in SDS-10% polyacrylamide gels, and analyzed by immunoblotting as described above, except that True-Blot horseradish peroxidase (HRP)-conjugated secondary antibody (eBioscience) was used at a dilution of 1:7,500. True-Blot HRP preferentially binds to native rabbit IgG and thereby reduces background from the heavy and light chains of the antibodies used for the immunoprecipitation.
Mapping of interaction domains using GST pull-down assays.
Lipofectamine 2000 (Invitrogen) was used to transfect Vero cells with plasmids that express wild-type or deletion mutants of GFP-UL21. At 20 h posttransfection, cells were lysed with NP-40 buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl [pH 8.0]), precleared with empty glutathione-Sepharose 4B beads overnight at 4°C, and then incubated for 2 h at room temperature with wild-type or mutant GST-UL16 proteins isolated on glutathione-Sepharose beads. The beads were washed three times with NP-40 buffer, and bound proteins were separated by SDS-10% PAGE, transferred to nitrocellulose, and immunoblotted with anti-GFP serum.
In vitro binding of purified GST-UL21 to His6-UL16.
To determine whether UL16 and UL21 have the abilities to directly interact, a protocol similar to that used to detect interactions between UL11 and UL16 was used (41). GST-UL21 and His6-UL16 derivatives were expressed in the BL21 strain of Escherichia coli. The GST-tagged proteins were purified as described above. His6-UL16 was induced with IPTG for 24 h at room temperature and purified by means of nickel beads according to the recommendations of the manufacturer (Novagen). Beads with GST-fusion proteins attached were mixed with various amounts of soluble His6-UL16 in 0.5% NP-40 lysis buffer and rocked at room temperature for 2 h. The GST-beads were then recovered by centrifugation, washed three times, and suspended in sample buffer for SDS-PAGE. Proteins were transferred to nitrocellulose, Ponceau stained, and analyzed by immunoblotting with the anti-His6-GFP antibody.
Confocal microscopy of GFP constructs.
To determine the steady-state location of wild-type and mutant GFP-UL21 in transfected cells, confocal microscopy was used. Vero cells were seeded on coverslips in 35-mm plates and transfected using Lipofectamine 2000 (Invitrogen). After 20 h, 0.5 mg/ml of Hoechst stain (Invitrogen) was added to the medium of each plate and incubated at 37°C for 10 min. The medium was then removed and the cells were washed twice with PBS containing 3% bovine serum albumin. The cells were fixed in 3% paraformaldehyde in PBS for 20 min at 4°C. Three more washes were performed followed by affixation of the coverslips to slides. Cells were imaged using the GFP and Hoechst channels and a 60× oil immersion objective of a Leica TCS SP2 AOBS confocal microscope.
Analysis of UL21 packaging.
To map the region of UL21 required for virion packaging, a transfection-infection assay was used. Vero cells in 100-mm dishes were transfected with 10 μg of DNA for each of the indicated UL21 constructs or with 1 μg of DNA for the GFP-only control. At 24 h posttransfection, the cells were infected with wild-type HSV or HSV.ΔUL21 at an MOI of 2. After an additional 20 to 24 h, the growth media were collected and cell debris was removed by centrifugation for 10 min at 1,000 × g. Extracellular virions (in 6 ml) were pelleted through a 30% (wt/vol) sucrose cushion for 1 h at 83,500 × g in an SW41 rotor and dissolved in sample buffer. To examine expression levels, the corresponding cell monolayers were scraped into PBS, and a small sample (1/170) from each plate was dissolved in sample buffer. The cell and virion samples were separated by SDS-PAGE and analyzed by immunoblotting using antibodies specific for GFP or the major capsid protein, VP5 (a loading control).
To analyze UL21 packaging in the UL16-null virus, Vero cells were infected with wild-type HSV or HSV.ΔUL16 at an MOI of 10. At 24 h postinfection, media were collected, cleared at 1,000 × g for 5 min, and then centrifuged for 1 h at 83,500 × g in an SW32 rotor. The pellets were dissolved in sample buffer, and equal quantities of virus were loaded into an SDS-PAGE gel for Western blotting analyses.
NEM treatment of UL16, UL11, and UL21.
In a variation of the pull-down experiment described above, insect cells were infected with a baculovirus that expresses UL16-GFP (41) and lysed in NP-40 buffer. Following a 4-min centrifugation at 18,000 × g, the clarified supernatant was mixed with bead-bound GST-UL11 or GST-UL21 and incubated for 2 h at room temperature. NEM at a final concentration of 10 mM was added to the lysates for 30 min either prior to or after the incubation with GST proteins. In some samples, NEM was added to GST-fusion proteins and then washed away with PBS prior to mixing with the UL16-GFP-containing lysates. The beads with GST-fusion proteins and bound UL16-GFP were washed three times with NP-40 lysis buffer, dissolved in sample buffer, and separated in 10% polyacrylamide gels. The proteins were visualized by immunoblotting with anti-GST and anti-GFP antibodies.
RESULTS
Identification of the unknown binding partner.
UL16 was previously identified as a binding partner of UL11 in GST pull-down and coimmunoprecipitation experiments (17). In those studies, GST-UL11 consistently pulled down a higher-molecular-mass, ∼65-kDa species from infected cell lysates (Fig. 1, left panel, middle lane). To identify the protein(s) within this band, larger amounts were obtained, digested with trypsin, and analyzed by mass spectrometry. Evidence for peptides from two proteins was found. The majority of the resolved data corresponded to peptides from UL21 (48% coverage of 535 amino acids), a capsid-bound protein that has been reported to interact with microtubules and may play a role in intracellular transport (7, 33). The results also indicated the presence of peptides from UL48 (17% coverage of 490 amino acids), which is also capsid bound and similar in molecular weight to UL21 (20, 40). Because the UL16 and UL21 homologs from PRV have been shown to interact in some manner (10), we suspected that UL21 was the relevant binding partner and UL48 was a comigrating contaminant. However, further comparisons suggested that there are some dramatic differences between HSV and PRV with regard to these proteins. For example, whereas the UL11 proteins from HSV and PRV gave similar protein profiles when used in pull-down experiments with HSV-infected cell lysates (Fig. 1, left panel), they exhibited strikingly different patterns when used with PRV-infected cell lysates (Fig. 1, right panel). UL11PRV pulled down proteins with sizes consistent for UL21PRV and UL16PRV (60 and 36 kDa, respectively), but the protein that was pulled down by UL11HSV most strongly and reproducibly was an ∼28-kDa species, which mass spectrometry revealed to be the US2 protein from PRV (54% coverage of 256 amino acids). While no significance can be placed on this heterologous virus protein interaction, it is clear from these results that distinct differences exist in the UL16 interaction networks of these two alphaherpesviruses. Thus, it was necessary to ascertain for HSV whether UL21 is indeed a binding partner of either UL16 or UL11.
FIG. 1.
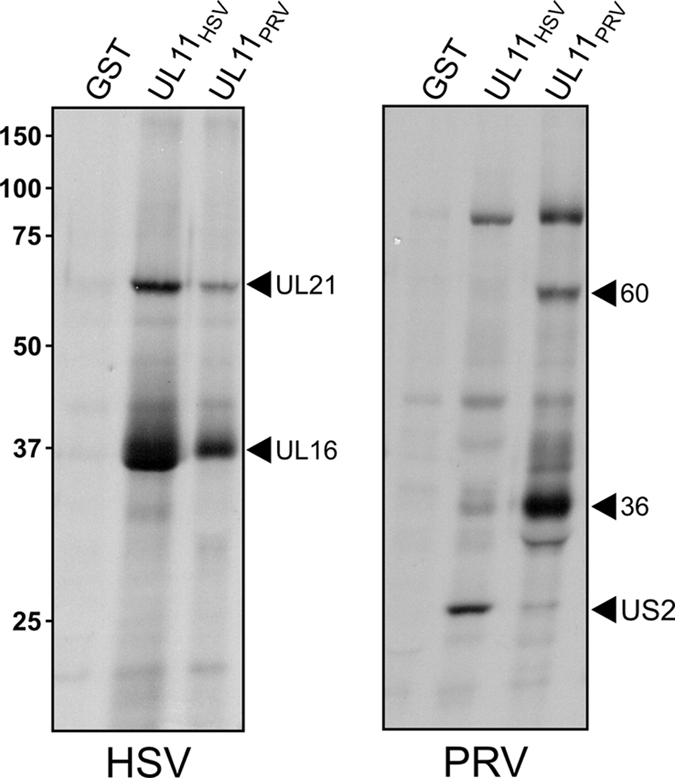
Distinct interaction profiles observed with GST-UL11 homologs of HSV and PRV. Vero cells were infected with HSV or PRV, and 5 h later, newly synthesized proteins were radiolabeled with [35S]methionine-cysteine for 3 h. The cells were lysed with NP-40-containing buffer, and after discarding the nuclei, glutathione beads that displayed the indicated GST-fusion proteins were added. Bound proteins were separated by SDS-PAGE, transferred to nitrocellulose, and detected by autoradiography.
Coimmunoprecipitation of UL16 and UL21.
To look for UL16 within UL21 complexes that are produced during an infection, coimmunoprecipitations from HSV-infected cell lysates were performed. For this, a rabbit antiserum was produced against GST-UL21 purified from bacterial cells, and it proved to be specific in test immunoprecipitation and immunoblotting assays (data not shown). When this antibody was used to collect UL21 from infected cell lysates, UL16 was found to be present (Fig. 2A), again suggesting that these two proteins interact in some manner. UL16 was also immunoprecipitated with an antibody against UL11, which was expected since those two proteins are known to directly interact (41).
FIG. 2.

Coimmunoprecipitation of UL16 and UL21 from infected cells. Vero cells infected with HSV (A) or HSV.UL16-CFP (B) were disrupted in NP-40 lysis buffer 20 h postinfection. Proteins were immunoprecipitated (IP) with antibodies specific for UL11, UL16, UL21, or GFP or no herpesvirus proteins (preimmune [pre]). The proteins were separated by SDS-PAGE and analyzed by immunoblotting with anti-UL16 or anti-UL21 serum. A portion (1/15) of the lysate was analyzed for protein expression.
To confirm that UL21 is in a complex with UL16, the converse immunoprecipitation was done. Discouragingly, we did not detect UL21 when proteins were collected with an antibody specific for UL16 (Fig. 2B, lane 2). Further experimentation revealed that this failure was the result of an antibody problem rather than the lack of a UL16-UL21 complex. That is, UL21 was indeed found when cells were infected with a recombinant virus that expresses UL16-CFP (41), and complexes were immunoprecipitated with antibodies against GFP (Fig. 2B, lane 1). Thus, the UL16-specific antibody either cannot access the epitopes within the complex or inadvertently disrupts the UL16-UL21 complex.
UL11 pulls down UL21 only in the presence of UL16.
Because UL21 was detected when complexes are collected with GST-UL11 (Fig. 1), it was possible that these two proteins directly interact. Alternatively, UL21 might interact indirectly to UL11, with UL16 serving to connect them (e.g., within a heterotrimer). To distinguish between these two possibilities, the GST-UL11 pull-down experiments were repeated with a virus lacking the UL16 open reading frame (4). For this, cells were infected with wild-type HSV or mutant ΔUL16 and then metabolically labeled at 5 h postinfection with [35S]methionine-cysteine. Cell lysates were mixed with equal amounts of purified GST constructs (Fig. 3, right panel), and bound proteins were separated by SDS-PAGE, transferred to nitrocellulose, and analyzed by autoradiography. A GST construct was used that contained only the first half of UL11 (mutant Δ51-96) but retained the ability to interact with UL16 (17), and it yielded the same pattern of binding partners as full-length UL11 when used with the WT virus (Fig. 3, left panel, lane 1). Immunoblot experiments confirmed that the two radiolabeled proteins were indeed UL21 and UL16 (data not shown). In the absence of UL16 expression, UL21 was not pulled down by GST-Δ51-96 (Fig. 3A, left panel), and its absence was confirmed by immunoblotting (data not shown). Consistent with this finding (but in a separate experiment), UL11-GFP from transfected cells did not bind to GST-UL21 (data not shown). Based on these results, it seemed likely that UL21 interacts with UL16 and not with UL11.
FIG. 3.

GST-UL11 pulls down UL21 only when UL16 is present. Vero cells infected for 5 h with wild-type HSV or HSV.ΔUL16 were radiolabeled with [35S]methionine-cysteine for 3 h, washed, and resuspended in NP-40 lysis buffer. The indicated GST constructs were added, and bound proteins were separated by SDS-PAGE, transferred to nitrocellulose, and subjected to autoradiography (left panel). The blot was stained with Ponceau S to visualize the amounts of purified GST proteins present (right panel).
UL21 and UL16 can interact even in the absence of other viral proteins.
To test the hypothesis that these two proteins interact in a manner that does not require any other viral protein, purified GST-UL16 was used in an attempt to pull down UL21 that had been expressed by itself via transient transfection. At the time these experiments were initiated, an antibody specific for UL21 was not available, and therefore, a GFP tag was added to the N terminus of the protein with the hope that it would not interfere with the assay (Fig. 4A). The expected ∼90-kDa chimera was detected in cell lysates by immunoblotting with GFP antiserum, but large amounts of N-terminally derived, smaller species were also found (Fig. 4B, lane 2). GST-UL16 readily pulled down the full-length GFP-UL21 but not the shorter products (Fig. 4C, lane 2) or GFP when it was expressed by itself (Fig. 4B and C, right-most lanes). The ∼52-kDa species running below GFP-UL21 (Fig. 4C, lane 2) is likely a degradation product that arose subsequent to the pull down because it did not preexist in the cell lysates (Fig. 4B). These results demonstrate that no other viral proteins are required for the interaction of UL21 with UL16. Additionally, GST-UL11 was not able to pull down UL21-GFP (data not shown), consistent with the idea that UL16 is required to link these two proteins.
FIG. 4.

The UL16-binding site is located in the second half of UL21. (A) Diagrams of UL21 and the N- and C-terminal truncation mutants, all of which were constructed as GFP-fusion proteins. A summary of all the binding results is provided on the right. (B) Expression levels of GFP-UL21 derivatives. Vero cells were transfected with the indicated constructs, and 20 h later, they were lysed in sample buffer, separated by SDS-PAGE, and transferred to nitrocellulose for immunoblotting with antibodies specific for GFP. (C) GST-UL16 pull-down assays. Vero cells transfected with the indicated constructs were harvested in NP-40-containing lysis buffer, and after discarding the nuclei, glutathione beads that displayed the GST-UL16 fusion protein were added. Bound proteins were separated by SDS-PAGE, transferred to nitrocellulose, and detected with GFP-specific antibodies.
The second half of UL21 is sufficient for binding to UL16.
In an effort to map a domain of UL21 that is important for the interaction with UL16, numerous N-terminal and C-terminal deletion mutants were generated, each fused to GFP (Fig. 4A). For all the mutants, proteins of the expected sizes were expressed at levels similar to or greater than the wild type, but abundant amounts of shorter products were again observed (Fig. 4B and data not shown). The transfected cell lysates were mixed with equal amounts of purified GST-UL16 (Ponceau S staining) (data not shown), and proteins that bound were detected by immunoblotting. Removal of any N-terminal segment up to amino acid 268 did not disrupt the interaction of UL21 with UL16 (Fig. 4C). In contrast, larger N-terminal deletions and all C-terminal deletions—even removal of just the last 44 amino acids—resulted in a complete loss of UL16 binding (Fig. 4C and data not shown). Thus, the UL16-binding site is located in the second half of UL21.
Each half of UL21 is sufficient for accumulation in the nucleus.
Having GFP on UL21 and all of the deletion mutants made it possible to find out where they accumulate in the cell without the need for an antibody. This was of interest because previous immunofluorescence experiments had provided rather different results, with studies of infected cells showing UL21 to be mostly cytoplasmic (3) and studies of transfected cells suggesting that the protein is located in both the cytoplasm and nucleus (33). In an attempt to shed light on this discrepancy, the GFP-tagged constructs (Fig. 4A) were examined by confocal microscopy. Full-length GFP-UL21 was found to be strongly (but not exclusively) localized to the nuclei of transfected cells (Fig. 5, top), even though its size (∼90 kDa) is much larger than the ∼50-kDa limit allowed for passive entry through the nuclear pore complex (8, 18). Thus, these experiments confirm that UL21 can enter the nucleus in the absence of other viral proteins (33) even though its sequence lacks an obvious nuclear localization signal (data not shown). To ascertain whether the localization pattern of UL21 changes in the presence of other viral proteins, cells that had been transfected for 18 h were infected with HSV for 16 h and then examined. At the start of the infection, GFP-UL21 was again found to be present mostly in the nucleus, but the amount in the cytoplasm seemed to increase later (Fig. 5, images below the dotted line).
FIG. 5.

Nuclear localization of GFP-UL21 in the absence of other viral proteins. Confocal microscopy results for GFP-tagged UL21 constructs are shown. Vero cells were seeded on coverslips and subsequently transfected with the indicated GFP-UL21 constructs. After 20 h, the cells were stained with Hoechst dye to label the nuclei, washed, paraformaldehyde fixed, affixed to slides, and examined with a confocal microscope to visualize the GFP signals alone (left panels) or in combination with the nuclear signals (right panels). For the bottom panels (below the dotted line), cells that had been transfected for 18 h were infected with HSV and examined at the indicated times postinfection (PI).
Examination of the mutants revealed that each half of UL21 behaves like the full-length molecule and accumulates strongly in the nucleus when expressed in the absence of other viral proteins (Fig. 5, NΔ267 and CΔ268). Both of these constructs have predicted sizes of 56 kDa, which approaches the limit for passive nuclear entry; hence, it is unclear whether they each have a nuclear localization signal or instead have the ability to be retained in the nucleus once they enter.
While mutants with shorter deletions in the N-terminal half of UL21 were also localized to nuclei (data not shown), those with shorter deletions in the C-terminal half gave a variety of patterns. For example, mutant CΔ492 failed to enter the nucleus (Fig. 5), even when the transfected cells were infected with HSV (data not shown). In contrast, mutant CΔ424 was localized to nuclei when expressed by itself but became mostly cytoplasmic when the transfected cells were infected with HSV (Fig. 5, images below the dotted line). Further studies are needed to elucidate the mechanistic reasons for these intracellular localization patterns.
The second half of UL21 is sufficient for virion packaging.
The GFP-UL21 mutants were also examined for their abilities to be packaged into virions in a transfection-infection assay. Cells transfected with the expression vectors were subsequently infected with either wild-type HSV or a mutant virus that lacks the UL21 gene but is still viable (3). In this case, the results were clear-cut. Deletion of any portion of the first half of UL21 (i.e., through residue 268 in mutant NΔ267 [Fig. 4A]) did not abolish packaging into the UL21-null mutant, but all deletions in the second half did (Fig. 6 and data not shown). It is unknown why mutants NΔ45 and NΔ78 showed reduced packaging. Since larger deletions recovered packaging, it seems likely that these mutants may have folding defects that reduce packaging efficiency. Similar results were obtained with the wild-type virus (data not shown). Thus, the domain that is needed for packaging overlaps precisely with the domain that is needed for the formation of UL21-UL16 complexes.
FIG. 6.

The second half of UL21 is sufficient for packaging into virions. Vero cells were transfected to express the indicated GFP-UL21 constructs or GFP alone and then infected with HSV at 24 h posttransfection. After another 20 h, extracellular virions were purified from the medium by centrifugation through a sucrose cushion. Virions and infected cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for GFP and VP5.
The interaction of UL21 with UL16 is direct but not essential for UL21 packaging.
Although the experiments described above showed that no other viral proteins are needed for UL21 and UL16 to form complexes, they did not rule out the involvement of host proteins. To address this, GST-UL21 and two derivatives that lack portions of the N-terminal sequence (NΔ140) or C-terminal sequence (CΔ268) were purified from bacteria and mixed with increasing concentrations of a His6-tagged derivative of UL16, which was also purified from bacteria and is known to be competent for binding directly to UL11 (41). The amounts of purified GST derivatives present in the assay were revealed by Ponceau S staining (Fig. 7A), and bound His6-UL16 was detected with antiserum that recognizes its tag (Fig. 7B). As expected, the GST-only control protein and the mutant lacking the putative UL16-binding domain interacted poorly or not at all with His6-UL16. However, the full-length GST-UL21 construct was also very limited in this regard, even though it was the most abundant fusion protein in the experiment (Fig. 7A). On the other hand, the construct with a deletion in the N-terminal half of UL21 was able to interact with His6-UL16 in an efficient, dose-dependent manner, in spite of being the least concentrated of the fusion proteins. Thus, it is clear that UL21 and UL16 can interact in a direct manner.
FIG. 7.

UL21 and UL16 directly interact, but this is not essential for UL21 packaging. The indicated GST-fusion proteins were expressed in bacteria and purified on glutathione-Sepharose beads. These were incubated with increasing amounts of soluble His6-UL16 (100, 200, or 400 ng), which was also purified from bacteria. The beads were washed, and the associated proteins were separated by SDS-PAGE and then transferred to nitrocellulose. (A) The amounts of input His6-UL16 and each of the GST-fusion proteins were revealed by Ponceau S staining. (B) The amount of His6-UL16 bound to the beads was measured by immunoblotting with a rabbit antibody that recognizes the His6 tag. (C) Media from Vero cells infected with either wild-type or ΔUL16 HSV were collected 24 h postinfection and centrifuged to pellet the virions. Samples of the cell lysates and virions were separated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies.
It is not known why full-length GST-UL21 lacked binding activity in this assay. The purified protein seemed to be properly folded because it was able to interact with baculovirus-expressed UL16-GFP produced in insect cells (see below) or mammalian cells (data not shown). Perhaps the UL16-binding domain of the purified protein is negatively regulated by the N-terminal half of UL21 and is activated upon exposure to lysates from eukaryotic cells. Further experimentation will be needed to find the explanation for this unexpected result.
Because UL16 is known to be capsid associated (20, 21), we hypothesized that UL21 might be packaged as a result of the direct interaction of these two proteins. To test this, virions were isolated from the media of Vero cells infected with either wild-type or ΔUL16 HSV and probed for UL21. We found that in the absence of UL16, the packaging of UL21 was reduced but not eliminated (Fig. 7C) and, hence, the UL16-UL21 interaction is not essential for packaging.
Deletion analysis of UL16.
In an attempt to identify the UL21-binding site within UL16, N- and C-terminal deletions were made in the context of a previously described GST-fusion protein. Specifically, UL16 deletion mutants NΔ40, NΔ80, NΔ120, CΔ46, CΔ70, and CΔ125 were used (41). The proteins were purified from bacteria, and equal amounts were mixed with lysates of radiolabeled Vero cells that had been transfected with a UL21-GFP expression vector. Only the wild-type GST-UL16 and NΔ40 deletion mutant were found to be able to pull down UL21-GFP (data not shown). These results were not surprising, considering the first 40 amino acids of UL16 are the least conserved, and all the other deletion mutants were previously found to disrupt binding to UL11 in similar experiments (41). Thus, UL16 appears to be sensitive to deletion and may not have separable domains for binding to UL11 and UL21.
NEM-modified UL16 is competent for binding to UL21.
Recent studies have shown that free cysteines in UL16 (i.e., those not in disulfide bonds) are important for the interaction with UL11. Thus, when UL16 is treated with NEM (a membrane-permeable compound that covalently modifies free cysteines), the interaction with UL11 is blocked. In contrast, NEM treatment of UL11 (or substitution of all four of its cysteines with serines) has no effect on UL16 binding (41).
To test whether free cysteines are important for the UL16-UL21 interaction, a recombinant baculovirus was used that expresses UL16-GFP (41). NEM was added to infected cell lysates, either before or after incubation with purified GST-UL11, GST-UL21, or GST-only constructs. Alternatively, NEM was added to the GST-fusion proteins and washed away prior to being mixed with the lysates containing UL16-GFP. Despite being able to completely block the UL11-UL16 interaction (Fig. 8, left panels), NEM had no effect on the binding of GST-UL21 to UL16-GFP (middle panels). Moreover, NEM did not disrupt any complexes once they had formed. These results offer clear evidence that UL16 contains distinct binding sites for UL11 and UL21.
FIG. 8.

NEM modification of UL16 blocks the interaction with UL11 but not UL21. Insect cells infected with a baculovirus that expresses UL16-GFP were lysed with NP-40 buffer and either untreated (lane 1 of each panel) or treated for 30 min with NEM, either before or after 2 h of incubation with the indicated bead-bound GST-fusion protein (lanes 2 and 4 of each panel). Alternatively, NEM was added to the GST-fusion protein and washed away prior to mixing with UL16-GFP (lane 3 in each panel). The bead-bound GST fusion and associated proteins were washed, dissolved in sample buffer, separated in SDS-10% polyacrylamide gels, and visualized by immunoblotting with anti-GST (bottom) and anti-GFP (top) antibodies.
DISCUSSION
The experiments described here have revealed new and fundamental information regarding the interaction network in which the conserved UL16 tegument protein operates. In particular, the previously unidentified 65-kDa protein that we found first in pull-down experiments using GST-UL11 (17) has proven to be the UL21 tegument protein. The results show that UL21 does not interact with UL11 but instead binds to it indirectly through UL16. This finding is consistent with the observation that the UL16 and UL21 homologs of PRV form some sort of a physical complex in infected cells, as determined by coimmunoprecipitation assays (10). However, the experiments presented here go further in showing that no other viral proteins are needed for the interaction; indeed, UL16 and UL21 were found to directly bind to one another. Further still, these studies have shown that the second half of UL21 contains the binding site for UL16, and UL16 contains distinct binding sites for UL21 and UL11. Whether these findings will also apply to the homologs of PRV remains to be seen. Based on the findings presented here, there appear to be striking differences in the UL11-UL16-UL21 interaction networks of HSV compared to PRV, and further comparisons are warranted.
Is a stable UL11-UL16-UL21 complex produced?
Although complexes containing UL11, UL16, and UL21 have been found in GST pull-down experiments such as the ones described here, it is unknown whether a tripartite complex actually exists in the infected cell or within the virion. That is, the loading of proteins in a cell lysate onto an exogenously added GST-fusion construct can reveal much about molecular interactions but little about their timing. In contrast, coimmunoprecipitation assays can reveal the composition of native complexes, and in such experiments with PRV, UL11 was not detected in complex with UL16 and UL21 (10), which argues against a stable complex containing all three proteins. In the case of HSV, background bands obscured the presence of proteins the size of UL21 when UL11-UL16 complexes were collected (17). It is possible that stable tripartite complexes exist for both HSV and PRV but are disrupted when cells are lysed with buffers that contain NP-40, as is the case for the interaction of UL16 with the capsid (20, 21). On the other hand, changes have been found to occur within the tegument of HSV during virion egress (20, 25) and cell binding (21), and thus, it is easy to imagine that the interactions among UL11, UL16, and UL21 are dynamic (e.g., binding of UL16 to UL11 might trigger the release of UL16 from UL21). If this is the case, then the tripartite complex might only exist transiently.
Potential roles for the UL11-UL16-UL21 network of interactions.
It is difficult to create molecular models with these three proteins because so little is known about them, but a few clues are available. Beginning in the nucleus, UL16 and UL21 have been proposed to participate in DNA packaging/capsid maturation events. Specifically, UL16 of HSV colocalizes with sites of capsid assembly (20, 24, 26), and UL21 deletion mutants of PRV accumulate capsids lacking DNA (7, 38). Moreover, the UL16 and UL21 homologs of varicell-zoster virus have been reported to interact with components of the DNA packaging machinery in yeast two-hybrid experiments (36). Thus, it is conceivable that these proteins are part of the DNA-packaging complex that associates with the unstable procapsid (1, 2, 13, 14, 27-29, 31, 34, 35, 39, 42). In the case of HSV, however, deletion of the UL21 gene did not result in defects in the cleavage and packaging of viral DNA (3), even though it appears to be important for virulence in the host animal (11, 23), and it is unknown whether UL16 and UL21 actually interact within the nucleus.
Although both proteins have been found to be associated with capsids, UL16 has been found only on capsids purified from the cytoplasm (20), whereas UL21 has also been reported to copurify with capsids from the nucleus (7, 33). Obviously, if these two proteins were to stably interact within the nucleus, then both would be expected to be found on nuclear capsids. It may be the case that their interaction does not take place in the nucleus, is transient, or is lost when the nucleus is disrupted. Whatever the explanation, both proteins are found on cytoplasmic capsids. It is tempting to hypothesize that the C-terminal domain of UL21 contains the capsid binding domain since it is also is the domain required for packaging (this study); however, further experiments will be required to address this question.
Potential roles for the UL16 and UL21 proteins in the cytoplasm have been suggested from studies of the HSV homologs. UL21 has been shown to physically interact in some manner with microtubules in vitro and to cause the outgrowth of long cellular processes when overexpressed in nonneuronal cells (33). These observations raise the possibility that UL21 could be involved with capsid transport to the TGN, perhaps via its N-terminal domain. In contrast, UL16 has been shown to interact with the UL11 protein, which accumulates at the TGN, and it is possible that this interaction facilitates the budding process by linking capsids to the membrane (17, 41). Based on the recent observation that the interaction of UL16 with capsids is destabilized during egress (20), we speculated that the UL11-UL16 interaction might destabilize UL16 on the capsid during virus budding (41). In support of this hypothesis, it is interesting that NEM blocks the interaction of UL16 with UL11 (41) and stabilizes the UL16-capsid interaction (20), but this modification has no effect on the UL16-UL21 interaction (this study). If UL21 and UL16 are important for transport of capsids to the TGN, then it is easy to imagine that the virus would need to release this machinery to enable proper transport during subsequent virus entry events (i.e., toward the nucleus). Lastly, it will be of interest to ascertain whether UL21 is needed for the internal rearrangements of the tegument that occur following virion release (25) and binding to cell attachment receptors (21).
Acknowledgments
We thank Joel Baines at Cornell University for kindly providing the HSV.ΔUL16 and HSV.Δ21 deletion viruses.
This work was funded by a grant from the NIH to J.W.W. (AI071286) and a grant from the NIH to O.J.S. (CA0176595). A.L.H. was supported by a fellowship from the American Heart Association (0525542U). D.G.M. was supported by a training grant from the NIH (CA60395).
Footnotes
Published ahead of print on 30 December 2009.
REFERENCES
- 1.Addison, C., F. J. Rixon, and V. G. Preston. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J. Gen. Virol. 71:2377-2384. [DOI] [PubMed] [Google Scholar]
- 2.Baines, J. D., C. Cunningham, D. Nalwanga, and A. Davison. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J. Virol. 71:2666-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines, J. D., A. H. Koyama, T. Huang, and B. Roizman. 1994. The UL21 gene products of herpes simplex virus 1 are dispensable for growth in cultured cells. J. Virol. 68:2929-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines, J. D., and B. Roizman. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 65:938-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baines, J. D., and B. Roizman. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baird, N. L., P. C. Yeh, R. J. Courtney, and J. W. Wills. 2008. Sequences in the UL11 tegument protein of herpes simplex virus that control association with detergent-resistant membranes. Virology 374:315-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Wind, N., F. Wagenaar, J. Pol, T. Kimman, and A. Berns. 1992. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J. Virol. 66:7096-7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dingwall, C., and R. A. Laskey. 1986. Protein import into the cell nucleus. Annu. Rev. Cell Biol. 2:367-390. [DOI] [PubMed] [Google Scholar]
- 9.Fulmer, P. A., J. M. Melancon, J. D. Baines, and K. G. Kousoulas. 2007. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J. Virol. 81:3097-3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klupp, B. G., S. Bottcher, H. Granzow, M. Kopp, and T. C. Mettenleiter. 2005. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J. Virol. 79:1510-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klupp, B. G., B. Lomniczi, N. Visser, W. Fuchs, and T. C. Mettenleiter. 1995. Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212:466-473. [DOI] [PubMed] [Google Scholar]
- 12.Koshizuka, T., Y. Kawaguchi, N. Nozawa, I. Mori, and Y. Nishiyama. 2007. Herpes simplex virus protein UL11 but not UL51 is associated with lipid rafts. Virus Genes 35:571-575. [DOI] [PubMed] [Google Scholar]
- 13.Lamberti, C., and S. K. Weller. 1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 226:403-407. [DOI] [PubMed] [Google Scholar]
- 14.Lamberti, C., and S. K. Weller. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, J. H., V. Vittone, E. Diefenbach, A. L. Cunningham, and R. J. Diefenbach. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347-354. [DOI] [PubMed] [Google Scholar]
- 16.Loomis, J. S., J. B. Bowzard, R. J. Courtney, and J. W. Wills. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209-12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loomis, J. S., R. J. Courtney, and J. W. Wills. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417-11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macara, I. G. 2001. Transport into and out of the nucleus. Microbiol. Mol. Biol. Rev. 65:570-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacLean, C. A., A. Dolan, F. E. Jamieson, and D. J. McGeoch. 1992. The myristylated virion proteins of herpes simplex virus type 1: investigation of their role in the virus life cycle. J. Gen. Virol. 73:539-547. [DOI] [PubMed] [Google Scholar]
- 20.Meckes, D. G., Jr., and J. W. Wills. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 81:13028-13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meckes, D. G., Jr., and J. W. Wills. 2008. Structural rearrangement within an enveloped virus upon binding to the host cell. J. Virol. 82:10429-10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2009. Herpesvirus assembly: an update. Virus Res. 143:222-234. [DOI] [PubMed] [Google Scholar]
- 23.Michael, K., B. G. Klupp, A. Karger, and T. C. Mettenleiter. 2007. Efficient incorporation of tegument proteins pUL46, pUL49, and pUS3 into pseudorabies virus particles depends on the presence of pUL21. J. Virol. 81:1048-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nalwanga, D., S. Rempel, B. Roizman, and J. D. Baines. 1996. The UL16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226:236-242. [DOI] [PubMed] [Google Scholar]
- 25.Newcomb, W. W., and J. C. Brown. 2009. Time-dependent transformation of the herpesvirus tegument. J. Virol. 83:8082-8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oshima, S., T. Daikoku, S. Shibata, H. Yamada, F. Goshima, and Y. Nishiyama. 1998. Characterization of the UL16 gene product of herpes simplex virus type 2. Arch. Virol. 143:863-880. [DOI] [PubMed] [Google Scholar]
- 27.Patel, A. H., F. J. Rixon, C. Cunningham, and A. J. Davison. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217:111-123. [DOI] [PubMed] [Google Scholar]
- 28.Poon, A. P., and B. Roizman. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67:4497-4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salmon, B., and J. D. Baines. 1998. Herpes simplex virus DNA cleavage and packaging: association of multiple forms of UL15-encoded proteins with B capsids requires at least the UL6, UL17, and UL28 genes. J. Virol. 72:3045-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schimmer, C., and A. Neubauer. 2003. The equine herpesvirus 1 UL11 gene product localizes to the trans-Golgi network and is involved in cell-to-cell spread. Virology 308:23-36. [DOI] [PubMed] [Google Scholar]
- 31.Sherman, G., and S. L. Bachenheimer. 1987. DNA processing in temperature-sensitive morphogenic mutants of HSV-1. Virology 158:427-430. [DOI] [PubMed] [Google Scholar]
- 32.Smith, K. O. 1964. Relationship between the envelope and the infectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814-816. [DOI] [PubMed] [Google Scholar]
- 33.Takakuwa, H., F. Goshima, T. Koshizuka, T. Murata, T. Daikoku, and Y. Nishiyama. 2001. Herpes simplex virus encodes a virion-associated protein which promotes long cellular processes in over-expressing cells. Genes Cells 6:955-966. [DOI] [PubMed] [Google Scholar]
- 34.Taus, N. S., and J. D. Baines. 1998. Herpes simplex virus 1 DNA cleavage/packaging: the UL28 gene encodes a minor component of B capsids. Virology 252:443-449. [DOI] [PubMed] [Google Scholar]
- 35.Taus, N. S., B. Salmon, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115-125. [DOI] [PubMed] [Google Scholar]
- 36.Uetz, P., Y. A. Dong, C. Zeretzke, C. Atzler, A. Baiker, B. Berger, S. V. Rajagopala, M. Roupelieva, D. Rose, E. Fossum, and J. Haas. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239-242. [DOI] [PubMed] [Google Scholar]
- 37.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagenaar, F., J. M. Pol, N. de Wind, and T. G. Kimman. 2001. Deletion of the UL21 gene in pseudorabies virus results in the formation of DNA-deprived capsids: an electron microscopy study. Vet. Res. 32:47-54. [DOI] [PubMed] [Google Scholar]
- 39.Weller, S. K., E. P. Carmichael, D. P. Aschman, D. J. Goldstein, and P. A. Schaffer. 1987. Genetic and phenotypic characterization of mutants in four essential genes that map to the left half of HSV-1 UL DNA. Virology 161:198-210. [DOI] [PubMed] [Google Scholar]
- 40.Wolfstein, A., C. H. Nagel, K. Radtke, K. Dohner, V. J. Allan, and B. Sodeik. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227-237. [DOI] [PubMed] [Google Scholar]
- 41.Yeh, P. C., D. G. Meckes, Jr., and J. W. Wills. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 82:10693-10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu, D., and S. K. Weller. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J. Virol. 72:7428-7439. [DOI] [PMC free article] [PubMed] [Google Scholar]