

Abstract
Neurotropic coronavirus induces acute encephalomyelitis and demyelination in mice. Infection of BALB/c (H-2d) mice expressing a dominant negative gamma interferon (IFN-γ) receptor specifically in oligodendrocytes was examined to determine the influence of IFN-γ signaling on pathogenesis. Inhibition of IFN-γ signaling in oligodendrocytes increased viral load, infection of oligodendrocytes, oligodendrocyte loss, demyelination, and axonal damage resulting in increased mortality. IFN-γ levels and the inflammatory response were not altered, although the level of tumor necrosis factor (TNF) mRNA was increased. These data indicate that IFN-γ signaling by oligodendroglia reduces viral replication but affects both demyelination and tissue destruction in a host-specific manner.
Infection of mice with a sublethal neurotropic JHM strain of mouse hepatitis virus (JHMV) results in an acute encephalomyelitis accompanied by demyelination that progresses to a chronic infection (3). Oligodendrocytes are a major target of infection during acute replication and a reservoir during viral persistence, which is characterized by viral RNA, viral antigen, and ongoing demyelination (3). Infection of gamma interferon (IFN-γ)-deficient mice and immunodeficient recipients of memory T-cell subsets suggests a critical requirement for IFN-γ in controlling virus replication in oligodendrocytes but not in astrocytes or microglia/macrophages (5, 19). Infection of C57BL/6 transgenic (TG) mice expressing a dominant negative (dn) IFN-γ receptor 1 (IFN-γR1) specifically in oligodendrocytes under the control of the proteolipid protein promoter (10) demonstrated increased numbers of infected oligodendrocytes without altering the extent of demyelination or tissue destruction (11). These data revealed that IFN-γ signaling to oligodendrocytes is critical for control of virus replication but that the extent of tissue destruction and morbidity is independent of the viral load. This notion is supported by recent studies suggesting that immune modulatory effects of IFN-γ are more critical to pathogenesis than virus replication per se (5, 12, 20, 24). As BALB/c mice mount increased Th1 responses to mouse hepatitis virus infection (13) and H-2d-restricted virus-specific CD8 T cells are induced more rapidly than are H-2b-restricted CD8 T cells (2), we determined whether a distinct genetic background would alter pathogenesis imposed by the oligodendrocyte-driven dnIFN-γR1 transgene. The TG C57BL/6 line expressing high levels of the dnIFN-γR1 (10) was backcrossed nine times onto BALB/cByJ mice (Jackson Laboratory, Bar Harbor, ME) and intercrossed to produce homozygous mice. TG mRNA was detected only in brain and spinal cord (Fig. 1A), demonstrating specific central nervous system (CNS) expression. Oligodendrocytes isolated from the CNS of TG mice and wild-type (WT) BALB/c mice (National Cancer Institute, Frederick, MD) were analyzed for IFN-γR1 expression by using anti-CD119 monoclonal antibody (MAb) (10). CD119 expression was increased >2-fold on oligodendrocytes derived from the TG mice (Fig. 1B). In contrast, IFN-γR1 expression was not altered on microglia (data not shown), confirming promoter-specific transgene expression.
FIG. 1.

IFN-γ signaling in oligodendrocytes protects BALB/c mice. (A) Expression of the dnIFN-γR1 transgene mRNA in the CNS. RNA obtained from brain (BR), spinal cord (SC), spleen (SPL), liver (LIV), and lungs (LNG) of naïve homozygous TG and WT BALB/c mice, amplified by PCR by using transgene (top)- and hypoxanthine phosphoribosyltransferase (HPRT) (bottom)-specific primers as previously described (10). (B) Surface expression of IFN-γR1 (CD119; PharMingen, San Diego, CA) on oligodendrocytes obtained from spinal cords of naïve TG and WT animals by trypsin digestion and enrichment on 30%/70% Percoll (Pharmacia, Uppsala, Sweden) gradients as described previously (11, 17). Flow cytometry histogram of CD119 expression on CD45− O4+ oligodendrocytes from TG mice (dark gray) and WT mice (solid line). The dotted line represents isotype control staining. (C) Survival rates of TG and WT mice following JHMV infection. (D) Virus replication in brains of TG and WT mice determined by plaque assay of tissue homogenates on DBT cell monolayers. Each time point represents the average of ≥3 individuals ± the standard deviation. At all time points p.i., differences between TG and WT mice were statistically significant (P < 0.05).
Homozygous dnIFN-γR1 TG and WT mice were infected by intracranial injection of 500 PFU of the 2.2v-1 variant of JHMV, and clinical symptoms were scored daily in a blinded fashion as previously described (9). Both TG and WT mice exhibited symptoms of encephalitis by 7 days postinfection (p.i.) and partial hind-limb paralysis by 9 days p.i. However, clinical symptoms were increased in TG mice compared to those in WT mice at all time points p.i., with a significant increase by day 9 p.i. (clinical scores were 2.8 ± 0.9 for TG mice and 1.7 ± 1.0 for WT mice; P < 0.05), correlating with ∼80% mortality in TG mice by day 14 p.i. (Fig. 1C). CNS virus replication in TG mice exceeded replication in WT mice as early as day 5 p.i., and the increase was sustained until day 14 p.i. (Fig. 1D).
The number of virus-infected cells, identified by antinucleocapsid MAb J.3.3 (11, 19), with a morphology consistent with oligodendrocytes, was increased in the white matter of TG mice compared to that in WT mice (TG, 102.2 cells/mm2; WT, 43.5 cells/mm2; P < 0.05) (Fig. 2A and B). Demyelination was also increased in infected TG mice (Fig. 2C and D). Although quantitative analysis of demyelination, carried out as previously described (11), showed that myelin loss in the white matter was ∼2-fold greater in TG than in WT mice at day 10 p.i. (TG, 60% ± 23% of spinal cord white matter; WT, 31% ± 1%), the difference did not reach statistical significance (P = 0.08). Axonal damage was measured by dual staining with MAb SMI-31 and MAb SMI-32. SMI-31 identifies undamaged axons, while SMI-32 identifies remaining damaged axons within areas of demyelination. Therefore, dual stains define both the area of myelin loss and damaged axons within the lesion. Axonal loss was confined to demyelinated lesions in both groups (Fig. 2E and F). Increased tissue damage further coincided with an increased number of nuclei expressing activated caspase 3 in white-matter tracks of TG mice, suggesting an association between increased oligodendrocyte infection and apoptosis (TG, 96.2 cells/mm2; WT, 58.3 cells/mm2; P < 0.05) (Fig. 2G and H). Finally, TG mice exhibited a loss of white-matter oligodendrocytes in areas adjacent to myelin loss (Fig. 2I and J), consistent with increased oligodendrocyte infection and caspase 3-positive cells adjacent to the demyelinated lesions. Quantification of oligodendrocytes, via reactivity with an anti-adenomatous polyposis coli (APC) MAb as previously described (11), showed an ∼10-fold decrease in oligodendrocytes in the areas adjacent to the lesions in TG mice compared to in WT mice (TG, 5 cells/mm2; WT, 55 cells/mm2) at day 10 p.i. These results suggest that demyelinated lesions expand due to increased infection and apoptosis of oligodendrocytes.
FIG. 2.

IFN-γ regulates CNS immunopathology. Longitudinal sections of paraffin-embedded spinal cords from TG and WT mice at day 10 p.i. examined for viral antigen detected with MAb J.3.3 (A and B); for demyelination via luxol fast blue stain (C and D); for axonal integrity via anti-SMI-31/32 (Covance, Princeton, NJ) (E and F); for apoptotic cells, visualized with anti-activated caspase 3 (Cell Signaling, Beverly, MA) (G and H). Oligodendrocytes were visualized in white-matter tracks with an antibody specific for APC (Abcam Inc., Cambridge, MA). Oligodendrocytes adjacent to demyelinated lesions were determined by counting the number of APC-positive cells within 500 μm from the edge of each area of demyelination (I and J). All stains were carried out as previously described (11, 12) on sections from three to six mice scored in a blinded fashion. Panels represent areas reflecting the average blinded score value of each group. Size bar for A, B, C, D, G, and H = 200 μm; size bar for E, F, I, and J = 50 μm.
To ensure that transgene expression did not affect inflammatory cell recruitment into the CNS, TG and WT mice were compared using flow cytometry as previously described (11, 29). The total numbers of CD45hi inflammatory cells were similar in the two groups at all time points p.i. (Fig. 3A), with the exception of a slight increase in TG mice at day 5 p.i. The levels of neutrophil (Ly6G+) and monocyte (CDllb+) infiltration were also similar (data not shown). The levels of recruitment of CD4 T cells (Fig. 3B), CD8 T cells (Fig. 3C), and virus-specific tetramer+ CD8 T cells within the CD45hi infiltrates were also very similar in the two groups (Fig. 3D). In addition, no differences in T-cell or B-cell frequencies or expression of activation makers were detected in cervical lymph nodes (data not shown), excluding a peripheral impairment of T-cell activation and expansion. Similar IFN-γ protein levels in the CNS of infected TG and WT mice at 7 and 10 days p.i. (Fig. 4A) further supported T-cell effector activity and eliminated the possibility that IFN-γ was sequestered by binding to the dn receptor. Expression of inducible nitric oxide synthase (iNOS) and interleukin-1β (IL-1β) mRNA were also similar in TG and WT mice, confirming no gross differences in inflammatory responses despite increased viral load (Fig. 4B and C). In contrast, increased expression of tumor necrosis factor (TNF) mRNA in the TG mice (Fig. 4D) is consistent with enhanced microglia/monocyte activation, potentially attributed to clearance of apoptotic cells. Thus, TNF may contribute to enhanced neuronal damage and morbidity. Similar inflammation and IFN-γ levels, irrespective of increased viral replication in TG mice versus WT mice, suggested that dysregulated IFN-γ signaling by oligodendrocytes and/or altered oligodendrocyte/CD8 T-cell interaction(s) enhanced pathogenesis. This notion is supported by IFN-γ-dependent upregulation of major histocompatibility complex (MHC) class I antigen presentation components (17) and a concomitant strong induction of the B7-H1 inhibitory molecule, specifically on oligodendrocytes (20). Indeed, mRNA expression analysis of CD45− O4+ oligodendrocytes purified by a fluorescence-activated cell sorter (FACS) revealed that MHC class I heavy-chain mRNA was reduced ∼2.4-fold and B7-H1 mRNA was reduced ∼6-fold in TG mice versus WT mice at both days 7 and 10 p.i. (Fig. 4E and F). The reduction in B7-H1 mRNA being more pronounced than that in MHC class I mRNA is consistent with enhanced susceptibility of TG oligodendrocytes to CD8 T-cell-mediated cytolysis and subsequent apoptosis.
FIG. 3.

CNS inflammation during acute JHMV encephalomyelitis in TG and WT mice. Inflammatory cells from homogenized brain were enriched using Percoll gradients as previously described (4, 5, 11). Percentages of CD45hi inflammatory cells within total recovered cells (A); percentages of CD4 T cells (B) and CD8 T cells (C) within the CD45hi population; and virus-specific CD8 T cells within the CD8 T-cell population (D) determined via an Ld tetramer containing the immunodominant virus nucleocapsid protein-derived epitope (1, 2). Data represent mean ± standard error of the mean (SEM) of the results for three experiments per time point, with pooled samples from three individuals per experiment.
FIG. 4.

Altered gene expression by impaired IFN-γ signaling in oligodendrocytes. (A) Homogenates of individual brains from infected TG and WT mice were used to determine IFN-γ levels by enzyme-linked immunosorbent assay as described previously (29). RNA was extracted from the brains of TG and WT mice at 10 days p.i. using the Trizol reagent. Expression of iNOS (B), IL-1β (C), and TNF mRNA (D) relative to that of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was determined by quantitative reverse transcription-PCR (qRT-PCR), using previously described primers (12, 17). Each time point represents the average for ≥3 individuals ± the standard deviation. (E) Expression of MHC class I and B7-H1 (F) mRNA determined by qRT-PCR as described previously (17) in CD45− O4+ oligodendrocytes purified from groups of six to seven mice at days 7 and 10 p.i. Expression levels were normalized to GAPDH by using the following formula: 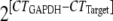 ×1,000, where CT is the threshold cycle. Expression levels in naïve mice were subtracted. Statistically significant differences for TNF, MHC class I, and B7-H1 mRNA are denoted by a single asterisk (P < 0.05) or a double asterisk (P < 0.005).
×1,000, where CT is the threshold cycle. Expression levels in naïve mice were subtracted. Statistically significant differences for TNF, MHC class I, and B7-H1 mRNA are denoted by a single asterisk (P < 0.05) or a double asterisk (P < 0.005).
The present data support the concept that IFN-γ signaling is critical for oligodendrocyte control and/or resistance to JHMV infection (11, 19). However, while increased oligodendrocyte infection in dnIFN-γR1 TG mice on the C57BL/6 background did not exacerbate tissue destruction and morbidity (11), BALB/c mice expressing the identical transgene displayed significantly enhanced pathology. Severity of infectious disease is linked to numerous factors, including differential innate and adaptive immune responses determined by the host genetic disposition (6, 18, 22, 23, 27). For example, innate immunity controls resistance of C57BL/6, but not BALB/c, mice to infection with the mouse-adapted human severe acute respiratory syndrome (SARS) coronavirus (21, 25). In addition, the balance of Th1- and Th2-associated cytokines influences control of infectious diseases (7, 15, 16, 26), while host-dependent cytotoxic T-lymphocyte responses regulate CNS viral persistence (8, 20). Although some outcomes correlate with MHC alleles (14, 22), the mechanisms and genes controlling differences in pathogenesis remain poorly defined. The absence of demyelination in JHMV-infected immunodeficient mice on both the H-2b and H-2d genetic backgrounds, but an induction of demyelination following T-cell transfer (4, 5, 28), suggests that demyelination is not directly mediated by viral infection of oligodendrocytes but, rather, requires an adaptive immune component. Increased pathology in dnIFN-γR1 BALB/c mice may thus reflect host-dependent oligodendrocyte-T-cell interactions, host-dependent phagocytic functions, or a combination of these factors. IFN-γR1 expression by oligodendrocytes derived from dnIFN-γR1 BALB/c mice is reduced by ∼50% compared to that derived from C57BL/6 dnIFN-γR1 mice (data not shown). Thus, subtle differences in the ability of oligodendrocytes to respond to IFN-γ, which may influence the expression of inhibitory molecules, i.e., B7-H1 (20), coupled with overexpression of TNF-α might facilitate a more vigorous, yet destructive, immune response. These data suggest that IFN-γ signaling in oligodendrocytes is associated with a more neuroprotective phenotype in BALB/c mice than that in C57BL/6 mice; however, it remains to be determined how distinct genetic regulation of factors controlling antigen presentation components, costimulatory or inhibitory ligands, or their respective receptors on T cells may alter the strength of CD8 T-cell effector functions and their consequences on the local environment.
Acknowledgments
This work was supported by National Institutes of Health grants NS 18146 and AI 47249.
We thank Wen Wei for technical assistance.
Footnotes
Published ahead of print on 30 December 2009.
REFERENCES
- 1.Bergmann, C., M. McMillan, and S. Stohlman. 1993. Characterization of the Ld-restricted cytotoxic T-lymphocyte epitope in the mouse hepatitis virus nucleocapsid protein. J. Virol. 67:7041-7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergmann, C. C., J. D. Altman, D. Hinton, and S. A. Stohlman. 1999. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J. Immunol. 163:3379-3387. [PubMed] [Google Scholar]
- 3.Bergmann, C. C., T. E. Lane, and S. A. Stohlman. 2006. Coronavirus infection of the central nervous system: host-virus stand-off. Nat. Rev. Microbiol. 4:121-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann, C. C., B. Parra, D. R. Hinton, R. Chandran, M. Morrison, and S. A. Stohlman. 2003. Perforin-mediated effector function within the central nervous system requires IFN-gamma-mediated MHC up-regulation. J. Immunol. 170:3204-3213. [DOI] [PubMed] [Google Scholar]
- 5.Bergmann, C. C., B. Parra, D. R. Hinton, C. Ramakrishna, K. C. Dowdell, and S. A. Stohlman. 2004. Perforin and gamma interferon-mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J. Virol. 78:1739-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brinton, M. A. 2001. Host factors involved in West Nile virus replication. Ann. N. Y. Acad. Sci. 951:207-219. [DOI] [PubMed] [Google Scholar]
- 7.Castilow, E. M., M. R. Olson, and S. M. Varga. 2007. Understanding respiratory syncytial virus (RSV) vaccine-enhanced disease. Immunol. Res. 39:225-239. [DOI] [PubMed] [Google Scholar]
- 8.Dethlefs, S., M. Brahic, and E. L. Larsson-Sciard. 1997. An early, abundant cytotoxic T-lymphocyte response against Theiler's virus is critical for preventing viral persistence. J. Virol. 71:8875-8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleming, J. O., M. D. Trousdale, F. A. el-Zaatari, S. A. Stohlman, and L. P. Weiner. 1986. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J. Virol. 58:869-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.González, J. M., C. C. Bergmann, B. Fuss, D. R. Hinton, C. Kangas, W. B. Macklin, and S. A. Stohlman. 2005. Expression of a dominant negative IFN-gamma receptor on mouse oligodendrocytes. Glia 51:22-34. [DOI] [PubMed] [Google Scholar]
- 11.González, J. M., C. C. Bergmann, C. Ramakrishna, D. R. Hinton, R. Atkinson, J. Hoskin, W. B. Macklin, and S. A. Stohlman. 2006. Inhibition of interferon-gamma signaling in oligodendroglia delays coronavirus clearance without altering demyelination. Am. J. Pathol. 168:796-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapil, P., R. Atkinson, C. Ramakrishna, D. J. Cua, C. C. Bergmann, and S. A. Stohlman. 2009. Interleukin-12 (IL-12), but not IL-23, deficiency ameliorates viral encephalitis without affecting viral control. J. Virol. 83:5978-5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyuwa, S., K. Fujiwara, and K. Yamanouchi. 1984. Genetic control of delayed-type hypersensitivity to mouse hepatitis virus in mice. Jpn. J. Exp. Med. 54:217-219. [PubMed] [Google Scholar]
- 14.Lévy-Leblond, E., D. Oth, and J. M. Dupuy. 1979. Genetic study of mouse sensitivity to MHV3 infection: influence of the H-2 complex. J. Immunol. 122:1359-1362. [PubMed] [Google Scholar]
- 15.Lipoldová, M., and P. Demant. 2006. Genetic susceptibility to infectious disease: lessons from mouse models of leishmaniasis. Nat. Rev. Genet. 7:294-305. [DOI] [PubMed] [Google Scholar]
- 16.Lundberg, P., C. Ramakrishna, J. Brown, J. M. Tyszka, M. Hamamura, D. R. Hinton, S. Kovats, O. Nalcioglu, K. Weinberg, H. Openshaw, and E. M. Cantin. 2008. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J. Virol. 82:7078-7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malone, K. E., S. A. Stohlman, C. Ramakrishna, W. Macklin, and C. C. Bergmann. 2008. Induction of class I antigen processing components in oligodendroglia and microglia during viral encephalomyelitis. Glia 56:426-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monteyne, P., F. Bihl, F. Levillayer, M. Brahic, and J. F. Bureau. 1999. The Th1/Th2 balance does not account for the difference of susceptibility of mouse strains to Theiler's virus persistent infection. J. Immunol. 162:7330-7334. [PubMed] [Google Scholar]
- 19.Parra, B., D. R. Hinton, N. W. Marten, C. C. Bergmann, M. T. Lin, C. S. Yang, and S. A. Stohlman. 1999. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 162:1641-1647. [PubMed] [Google Scholar]
- 20.Phares, T. W., C. Ramakrishna, G. I. Parra, A. Epstein, L. Chen, R. Atkinson, S. A. Stohlman, and C. C. Bergmann. 2009. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J. Immunol. 182:5430-5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts, A., D. Deming, C. D. Paddock, A. Cheng, B. Yount, L. Vogel, B. D. Herman, T. Sheahan, M. Heise, G. L. Genrich, S. R. Zaki, R. Baric, and K. Subbarao. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez, M., J. Leibowitz, and C. S. David. 1986. Susceptibility to Theiler's virus-induced demyelination. Mapping of the gene within the H-2D region. J. Exp. Med. 163:620-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadler, A. J., and B. R. Williams. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savarin, C., C. C. Bergmann, D. R. Hinton, R. M. Ransohoff, and S. A. Stohlman. 2008. Memory CD4+ T-cell-mediated protection from lethal coronavirus encephalomyelitis. J. Virol. 82:12432-12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheahan, T., T. E. Morrison, W. Funkhouser, S. Uematsu, S. Akira, R. S. Baric, and M. T. Heise. 2008. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 4:e1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibuya, K., D. Robinson, F. Zonin, S. B. Hartley, S. E. Macatonia, C. Somoza, C. A. Hunter, K. M. Murphy, and A. O'Garra. 1998. IL-1 alpha and TNF-alpha are required for IL-12-induced development of Th1 cells producing high levels of IFN-gamma in BALB/c but not C57BL/6 mice. J. Immunol. 160:1708-1716. [PubMed] [Google Scholar]
- 27.Thach, D. C., T. Kimura, and D. E. Griffin. 2000. Differences between C57BL/6 and BALB/cBy mice in mortality and virus replication after intranasal infection with neuroadapted Sindbis virus. J. Virol. 74:6156-6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu, G. F., A. A. Dandekar, L. Pewe, and S. Perlman. 2000. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 165:2278-2286. [DOI] [PubMed] [Google Scholar]
- 29.Zuo, J., S. A. Stohlman, J. B. Hoskin, D. R. Hinton, R. Atkinson, and C. C. Bergmann. 2006. Mouse hepatitis virus pathogenesis in the central nervous system is independent of IL-15 and natural killer cells. Virology 350:206-215. [DOI] [PMC free article] [PubMed] [Google Scholar]