

Abstract
Long-term potentiation (LTP) requires postsynaptic depolarization that can result from EPSPs paired with action potentials or larger EPSPs that trigger dendritic spikes. We explored the relative contribution of these sources of depolarization to LTP induction during synaptically driven action potential firing in hippocampal CA1 pyramidal neurons. Pairing of a weak test input with a strong input resulted in large LTP (∼75% increase) when the weak and strong inputs were both located in the apical dendrites. This form of LTP did not require somatic action potentials. When the strong input was located in the basal dendrites, the resulting LTP was smaller (≤25% increase). Pairing the test input with somatically evoked action potentials mimicked this form of LTP. Thus, back-propagating action potentials may contribute to modest LTP, but local synaptic depolarization and/or dendritic spikes mediate a stronger form of LTP that requires spatial proximity of the associated synaptic inputs.
Introduction
Since its discovery (Bell et al., 1997; Markram et al., 1997; Bi and Poo, 1998), spike-timing dependent plasticity (STDP) has attracted considerable attention (Abbott and Nelson, 2000; Bi and Wang, 2002; Kepecs et al., 2002; Roberts and Bell, 2002; Tsodyks, 2002; Dan and Poo, 2004, 2006), in part because of its seemingly simple dependence on a single, easily quantifiable parameter. In the neocortex and hippocampus, however, other factors, such as spike frequency and synaptic strength, influence the induction of STDP, not only quantitatively, but also in a qualitative manner (Sjöström et al., 2001; Wittenberg and Wang, 2006). These factors have been shown to exert their influence by providing postsynaptic depolarization, which is known to promote long-term potentiation (LTP) of synaptic strength (Kelso et al., 1986; Gustafsson et al., 1987).
The action potential is an important source of depolarization for Hebbian LTP (Magee and Johnston, 1997; Sjöström and Häusser, 2006), but it is not required under all conditions (Lisman and Spruston, 2005). Simple and direct evidence of this is the fact that LTP can be induced under somatic voltage clamp, provided that the holding potential is sufficiently depolarized (Malinow and Tsien, 1990). LTP can also be induced by strong synaptic stimulation, even if somatic action potentials are prevented. In this case, dendritically initiated spikes provide an important source of the necessary postsynaptic depolarization (Golding et al., 2002; Remy and Spruston, 2007). Even when action potentials do occur, an important aspect of their function may be to trigger dendritic calcium spikes (Larkum et al., 1999), which facilitate the induction of LTP (Kampa et al., 2007). Dendritic spikes are likely to be effective sources of depolarization for LTP, because they are broader and larger than back-propagating action potentials (bAPs), especially in distal dendrites (Golding et al., 1999; Larkum et al., 2007).
An important question for understanding the induction requirements for LTP is which of these sources of depolarization, synaptic potentials, back-propagating action potentials, or dendritic spikes, is most important for the induction of LTP during synaptically driven activity? We addressed this question in CA1 pyramidal neurons in rat hippocampal slices. Physiologically relevant stimuli capable of inducing LTP (Larson and Lynch, 1986) were generated by stimulating presynaptic inputs just strongly enough to drive action potential firing and repeating the stimulus at theta frequency (5 Hz). By comparing LTP induced in this manner to that induced with action potentials evoked by somatic current injection or in the absence of action potentials, we evaluated the relative contribution of action potentials and other sources of depolarization during synaptically induced LTP. We find that the ability of action potentials to provide the depolarization necessary for LTP is less effective than synaptic potentials and dendritic spikes, which induce strong LTP, but only at synapses located on a common dendritic domain.
Materials and Methods
All experiments were performed under the approval of the Northwestern University Animal Care and Use Committee.
Hippocampal slice preparation.
Transverse hippocampal slices (300 μm) were prepared from 4 to 8 week-old Wistar rats anesthetized with halothane or isoflurane. Animals were perfused through the heart with ice-cold artificial CSF (ACSF) (see composition below) and the brain was removed rapidly into ice-cold ACSF, and then blocked for preparation of near-horizontal slices using a vibratome.
Hippocampal slice electrophysiology.
ACSF contained (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 25 dextrose. Recording temperature was 33 ± 2°C. SR95531 (4 μm) (Sigma-Aldrich) and 1 μm CGP52432 (Tocris Bioscience) were added to the ACSF for pharmacological blockade of GABAA and GABAB synaptic transmission. Somatic and dendritic whole-cell current-clamp recordings were made using patch-clamp electrodes pulled from borosilicate glass and filled with intracellular solution containing (in mm): 135 K-gluconate, 7.5 KCl, 10 Na2-phosphocreatine, 10 HEPES, 2 MgATP, 0.3 NaGTP, 0.1% biocytin. Recordings were made using one or two bridge amplifiers (BVC-700; Dagan). Electrode resistance in the bath for somatic recordings ranged from 2 to 4 MΩ, and series resistance ranged from 8 to 20 MΩ. The dendritic electrode resistance was 6–9 MΩ, and series resistance ranged from 15 to 40 MΩ. Electrophysiological traces were digitized by an ITC-16 board (Instrutech) under control of macros custom programmed in IGOR Pro (WaveMetrics). EPSP amplitude and initial slope were monitored by using 0.05 Hz synaptic stimulation. Bipolar electrodes fashioned from theta glass (Sutter Instruments) were used in conjunction with Dagan BSI-950 biphasic stimulators. Stimulating electrodes were positioned at least 75 μm away from the recorded neuron. For all LTP experiments, EPSPs were monitored for a baseline that was at least 5 min, and for 20–40 min after LTP induction. When noted, 10 μm TTX was pressure-applied through a patch pipette positioned near the soma and axon initial segment under visual guidance. The pressure injection preceded the stimulation by 2 s and lasted ∼5 s. The flow of solution was monitored by using 0.1% Fast Green in the TTX pipette and optimized to avoid TTX diffusion into proximal stratum radiatum (SR). TTX application prevented axonal action potential generation in response to large (1.5–3 nA, 5 ms) somatic current injections. Synaptic waveforms for current injections were generated using a function of the form: I(t) = C × (e(−t/τ2) − e(−t/τ1)), where C is a scaled factor adjusted to give the desired maximum amplitude, and τ1 and τ2, the rise and decay time constants, respectively, were 0.5 ms and 40–60 ms, such that the resulting EPSP was similar in shape and duration to synaptically evoked EPSPs.
Data analysis and statistics.
Analysis of electrophysiology data were performed using custom programs in IGOR Pro (Wavemetrics). Synaptic strength was quantified by the initial slope of the EPSP. For each response, a fit of the EPSP from 20 to 50% of its peak amplitude was verified by visual inspection to provide an appropriate measure of synaptic strength; improper fits and data contaminated by spontaneous EPSPs or other sources of noise were excluded from the final analysis. All statistical analyses were performed using Prism 4 software (GraphPad Software). The magnitude of LTP for each cell was determined by the change in average initial slope from the baseline period (5 min before pairing) to a comparable period 15–20 min after pairing. Pooled data from multiple cells were tested for significant potentiation using a one-way ANOVA with repeated measures. Statistical comparison of potentiation between two experimental groups was performed using two-way ANOVA with repeated measures. Comparisons between multiple groups was performed using one-way ANOVA with post hoc Bonferroni tests. Other comparisons were made using unpaired- and paired-sample Student's t tests, as indicated. For all statistical tests, significance was p < 0.05. All measurements are presented as mean ± SEM.
Results
Synaptic inputs onto different dendritic domains
To investigate the interaction between segregated excitatory inputs onto CA1 pyramidal neurons during plasticity, two inputs were activated via extracellular stimulating electrodes (Fig. 1A). One stimulating electrode was placed in distal stratum radiatum (dSR) and stimulation intensity was adjusted to give a subthreshold EPSP (2–4 mV amplitude measured at the soma). This relatively weak input was used as the test pathway. The second stimulating electrode was placed proximal to pyramidal cell somata, either in proximal stratum radiatum (pSR) or in stratum oriens (SO). The stimulus intensity of the proximal input was adjusted to give a relatively strong input, such that a single pulse alone gave 1–3 action potentials. In another set of experiments proximal synaptic stimulation was replaced by a synaptic current waveform injected through the somatic recording electrode.
Figure 1.

Induction paradigm and independence of input pathways. A, Schematic of induction paradigm. A weakly activated (2–4 mV somatic EPSP amplitude) dSR input (just below stratum lacunosum moleculare, SLM) was paired with a strong input [SR, SO, somatic current injection, somata in stratum pyramidale (SP)] that elicited an average of two action potentials, and repeated 40 times at 5 Hz. B, Protocol used to test for independence between pathways. First, the in-pathway facilitation ratio (initial EPSP slope ratio of response 2/response 1) was established for the weak pathway, and then cross-pathway facilitation (initial EPSP slope ratio of response 4/response 1) was determined by activating the strong pathway followed by the weak pathway (3, 4). Interpulse intervals were 50–150 ms. C, Results of cross-facilitation experiments. For all three conditions, in-pathway facilitation ratios were significantly greater than 1.0 (Student's t test, p < 0.01), whereas cross-facilitation ratios were not (p > 0.05). In-pathway facilitation ratios were greater than cross-facilitation ratios (paired t test, p < 0.01). Thus, there were few or no shared fibers between the different input pathways. D, Twenty to eighty percent rise times of EPSPs elicited by dSR stimulation were significantly longer than rise times of pSR or SO EPSPs (paired t test, p < 0.01). Cells were held at approximately −80 mV to prevent contamination of responses with action potentials.
In some experiments, pathway independence was determined by using a cross-facilitation protocol (Fig. 1B). Within-pathway facilitation was measured using intervals of 50–150 ms. To verify that the second, strong pathway did not directly activate fibers in the pathway of interest, a single stimulus was delivered to the strong pathway, followed by a second stimulus delivered to the test pathway 50–150 ms later (Fig. 1B). For dSR and SO inputs, in-path facilitation was significantly greater than 1.0 (p < 0.01, Student's t test), was significantly different from the cross-facilitation ratio (p < 0.01, paired t test), and the cross-facilitation ratios were not significantly different from 1.0 (Student's t test) (Fig. 1C, n = 7 for pSR-dSR; n = 6 for SO-dSR; n = 6 for pSR-SO). Thus, the activated pathways were indeed separate, with few or no shared fibers between the different pathways. Consistent with their expected dendritic locations, the 20–80% rise times of the dSR EPSPs, were greater than the rise times of pSR and SO EPSPs (2–4 mV EPSPs, dSR: 5.4 ± 0.3 ms; pSR: 4.0 ± 0.2 ms; SO: 3.6 ± 0.3 ms, p < 0.01, paired t tests).
Plasticity of remote synaptic inputs paired with suprathreshold somatic current injections
Previous studies in CA1 neurons have shown LTP when small EPSPs are paired at a relatively low frequency (≤5 Hz) with action potentials evoked by current injection (Meredith et al., 2003; Wittenberg and Wang, 2006; Campanac and Debanne, 2008). To compare the magnitude of LTP induced in this way to that induced with synaptically evoked action potentials, we paired a weak dSR input with action potentials elicited by somatic current injection, with the amplitude of the current injection set to trigger 1–3 action potentials when paired with the dSR input. Pairing was repeated 40 times at 5 Hz (Fig. 2A). Consistent with previous findings, we found that this protocol resulted in a modest amount of potentiation (Fig. 2B,C; 24.6 ± 0.1% increase at 15–20 min after induction, p < 0.01). An unpaired control input in pSR was unchanged (Fig. 2B,C).
Figure 2.

Plasticity was modest when weak inputs were paired with somatic current injections. A, Schematic of the induction paradigm: weak dSR was paired with current injection. Examples of membrane potential traces during pairing: first, fifth, and 40th responses in the series of 40 pairings at 5 Hz. B, An example experiment for an individual cell, including examples of membrane potential traces before and after pairing. C, Pooled data for all experiments with pairing as shown in A. Significant potentiation of the dSR input was observed (24.6 ± 0.1% increase at 15–20 min post induction; repeated measures one-way ANOVA, **p < 0.01). The unpaired pSR pathway was not significantly changed (−1.7 ± 0.1% change, repeated measures one-way ANOVA, p = 0.09). Input resistance (RN) was stable throughout the experiment. D, Schematic of the induction paradigm: weak pSR paired with somatic current injections. Examples of membrane potential traces during pairing (scale is the same as in A). E, An example experiment for an individual cell, including examples of membrane potential traces before and after pairing (scale is the same as in B). F, Pooled data for all experiments with pairing as shown in D. A trend toward potentiation of the pSR input was observed (30.7 ± 0.2% increase at 15–20 min post induction; repeated measures one-way ANOVA, p = 0.09) whereas the change of the unpaired pathway was not significantly different from baseline (26.1 ± 0.2%, repeated measures one-way ANOVA, p = 0.2). RN was stable throughout the experiment.
To compare LTP of proximal and distal inputs paired with action potentials elicited by current injection, we paired a weak pSR input with action potentials evoked by somatic current injection (Fig. 2D). Similar to the amount of LTP of dSR inputs, this protocol resulted in a trend toward potentiation (Fig. 2E,F; 30.7 ± 0.2% increase, p = 0.09). The unpaired dSR control input increased by 26.1 ± 0.2%, but was not significantly different from the control period (p = 0.2). Thus, when paired with weakly activated SR inputs at low frequency, action potentials produce little or no synaptic plasticity.
Associative plasticity of adjacent and remote synaptic inputs
We paired a weak test input in dSR (2–4 mV EPSPs) with synaptically driven action potentials produced by stimulation of synaptic inputs onto the basal dendrites in SO (Fig. 3A). As in the case with current injection, the SO stimulus intensity was set to trigger 1–3 action potentials when paired with the dSR input. After this pairing protocol we observed a significant but modest potentiation of the test EPSP evoked by stimulation of dSR (Fig. 3B,C; 33.4 ± 0.1% increase, p < 0.01). To monitor SO synapses before and after the pairing, stimulus intensity of the SO stimulating electrode was reduced during the baseline and monitoring period to result in a small EPSP (2–4 mV). The pairing resulted in potentiation of this input as well (Fig. 3B,C; 39.7 ± 0.2% increase, p < 0.01).
Figure 3.

Plasticity was strongest when weak inputs were paired with a strongly activated synaptic input colocalized on the apical dendrite. A, Schematic of the induction paradigm: weak dSR was paired with strong SO stimulation. Examples of membrane potential responses during pairing. B, An example experiment for an individual cell. Examples of membrane potential responses before and after pairing. C, Pooled data for all experiments with pairing as shown in A. Significant potentiation of both pathways was observed (dSR = 33.4 ± 0.1%, SO = 39.7 ± 0.2% increase at time 15–20 min post induction; repeated measures one-way ANOVA, **p < 0.01). Input resistance (RN) was stable throughout the experiment. D, Schematic of the induction paradigm: weak dSR paired with strong pSR. Examples of membrane potential responses during pairing (scale same as in A). E, An example experiment for an individual cell. F, Pooled data for all experiments with pairing as shown in D. Both pathways potentiated with this protocol (dSR = 82.5 ± 0.1%, pSR = 36.2 ± 0.1%, increase at 15–20 min post induction; repeated measures one-way ANOVA, **p < 0.01). pSR potentiation was significantly less than dSR potentiation (repeated measures two way ANOVA, p < 0.05). RN was stable throughout the experiment.
We compared the LTP above, induced in dSR when action potentials were evoked by a remote input (SO), to that induced by pairing of an adjacent (pSR) input (Fig. 3D). In this case, larger potentiation of the dSR input resulted (Fig. 3E,F; 82.5 ± 0.1% increase, p < 0.01), as well as potentiation of the strongly activated pSR pathway (36.2 ± 0.1% increase, p < 0.01). Surprisingly, the EPSPs from the strongly activated pSR input were potentiated less than the weakly activated dSR input (Fig. 3E,F; p < 0.05). Weak dSR paired with strong pSR in the presence of AP-5 (50 μm) did not result in plasticity (p > 0.05, repeated measures one-way ANOVA, weak dSR 106 ± 15% of baseline; strong pSR 86 ± 18%)
The potentiation of dSR paired with strong pSR activation was greater than the potentiation of dSR paired with either strong SO activation or somatic current injection (Fig. 4; p < 0.01), as well as the unpaired dSR pathway in experiments in which weak pSR was paired with current injections (Fig. 4; p < 0.05).
Figure 4.

Greater potentiation occurs in distal inputs when paired with strong activation of a colocalized synaptic input, than when paired with remote synaptic activation or somatic current injections. A, Schematic of the protocols used to induce plasticity. B, Potentiation ratios of dSR inputs. Significant potentiation of the distal input was observed for all conditions (one-way ANOVA with repeated measures, p < 0.01, not indicated), except for the case of unpaired dSR during weak pSR paired with current injection. Comparisons of potentiation across groups were made using one-way ANOVA with post hoc Bonferroni tests compared with the reference case of weak dSR paired with strong pSR (*p < 0.05, **p < 0.01).
The reduced potentiation of strongly activated pSR compared with weakly activated dSR (Fig. 3F) may be a result of a site dependence or strength dependence of plasticity. Thus, we investigated whether strongly activated dSR synapses are potentiated. Strongly activated dSR paired with weakly activated pSR resulted in a relatively strong potentiation of dSR, as well as potentiation of weakly activated pSR (Fig. 5A–C; pSR = 60.5 ± 0.2%, dSR = 76.6 ± 0.2%, increase, p < 0.01 for each). Thus, the test pathway in dSR was potentiated after cooperative activation of a large number of synapses in the distal pathway. This level of potentiation was greater than the case of weakly activated dSR paired with current injection (p < 0.05), and the case of strongly activated SO paired with weakly activated dSR (p < 0.01). Thus, strong synaptic depolarization, when located at the test input or near it on the same dendrite, resulted in LTP of greater magnitude than the case of current injection at the soma (Fig. 2) or synaptic inputs located remotely from the site of the test input (Fig. 3C).
Figure 5.
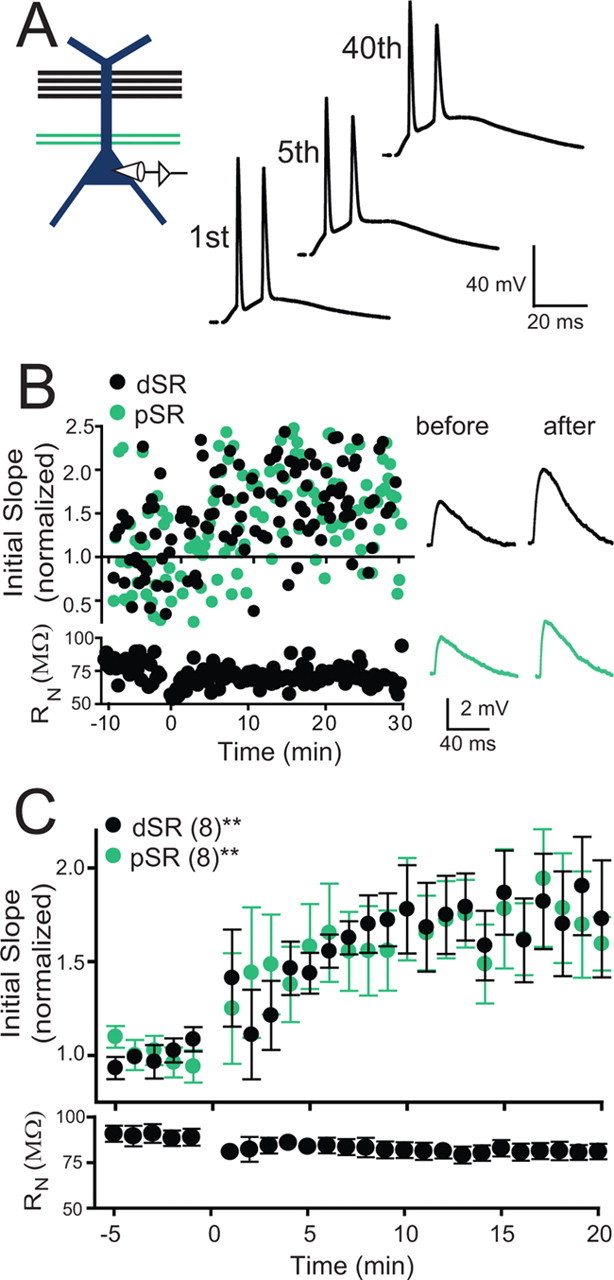
Reversing the strength of colocalized inputs on the apical dendrite results in large LTP of dSR inputs. A, Schematic of the induction paradigm: Strong dSR was paired with weak pSR stimulation. Examples of membrane potential responses during pairing. B, An example experiment for an individual cell. C, Pooled data for all experiments with pairing as shown in A. A significant potentiation of both pathways was observed (dSR = 76.6 ± 0.2%, pSR = 60.5 ± 0.2% increase at time 15–20 min post induction; repeated measures one-way ANOVA, **p < 0.01).
Dendritic events mediating short-range associative plasticity
The enhanced plasticity of strongly activated dSR or weakly activated dSR paired with strongly activated pSR, compared with pairing with strong SO or current injection, may be because the two inputs were located on the same dendrite, thus resulting in greater depolarization at the site of the weak dSR input. Alternatively, strong pSR may activate additional pathway-specific biochemical cascades that facilitate potentiation of the dSR input. Thus, we mimicked the synaptic depolarization at the site of the weak dSR input by recording in the distal region (200–300 μm), and pairing a synaptic waveform current injection through the dendritic recording electrode with weakly activated dSR synapses (Fig. 6A). Because EPSPs were recorded via the dendritic recording pipette, the stimulus intensity was adjusted to give larger EPSP amplitudes (4–8 mV), such that dendritic attenuation would result in somatic EPSP amplitudes similar to experiments with somatic recordings. In this condition, significant potentiation was observed (Fig. 6B,C; 76.6 ± 0.2% increase, p < 0.05). This potentiation was not different from that observed after pairing with strong pSR (Fig. 3F, p = 0.92), indicating that the greater depolarization at the site of the dSR synapses is responsible for the enhanced plasticity observed with strong pSR-weak dSR pairings, and not additional biochemical mechanisms recruited by strong pSR stimulation.
Figure 6.
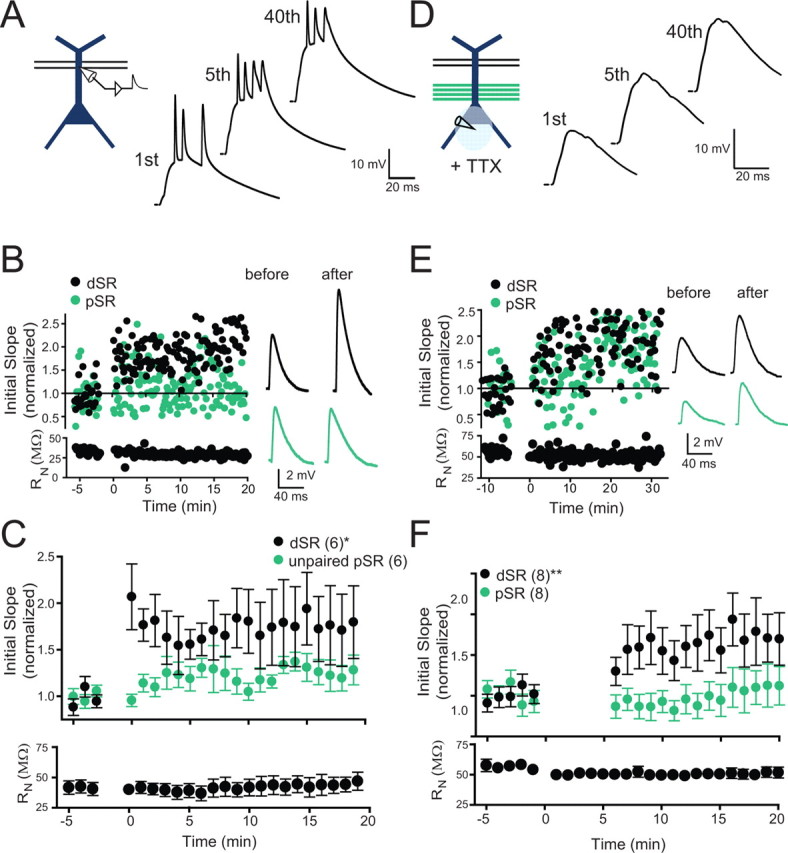
Local depolarization is sufficient for potentiation of weak dSR. A, Schematic of the induction paradigm: weak dSR paired with dendritic current injection. Examples of membrane potential responses during pairing. B An example experiment for an individual cell with membrane potential responses before and after pairing. C, Pooled data for all experiments with pairing as shown in D. Potentiation of dSR was observed with this protocol (76.6 ± 0.2%, increase at 15–20 min post induction; repeated measures one-way ANOVA, *p < 0.05). Input resistance (RN) was stable throughout the experiment. D, Schematic of the induction paradigm: weak dSR paired with strong pSR while locally applying TTX to block action potential initiation. Examples of membrane potential responses during pairing. E, An example experiment for an individual cell with membrane potential responses before and after pairing (scale same as in B). F, Pooled data for all experiments with pairing as shown in A. Local puff application of TTX near the soma did not prevent potentiation of the weak dSR input (75.5% increase at 15–20 min post induction; repeated measures one-way ANOVA, **p < 0.01). In some cases TTX application affected synaptic responses for up to 5 min after application, so these points are deleted from the summary plot. No potentiation of the strong pSR input was observed (but see main text for important caveat). RN was stable throughout the experiment.
To insure that potentiation during dendritic current injections was not a result of greater depolarization than the case of synaptic activation, the membrane potential in this condition was compared with the membrane potential measured in dendritic recordings during strong pSR paired with weak dSR inputs (compare Figs. 6A and 8A; p > 0.05, Student's t test, average membrane potential during 200 ms of induction pulse: pSR paired with dSR, −57.7 ± 2.6 mV; dendritic current injection paired with dSR, −49.5 ± 3.8 mV). Furthermore, there was no correlation in the amount of plasticity observed for the case of dendritic current injections paired with dSR with the level of depolarization measured at the recording electrode (slope not significantly different from zero, p = 0.87), indicating that there is a threshold level of depolarization required for plasticity of the weak dSR input, and that the depolarization levels tested here were above that level.
Figure 8.

Pairing of a strong pSR input with a weak dSR input resulted in significantly greater dendritic depolarization than pairing dSR with strong SO. A, Dendritic recordings were made on the apical dendrite (175–275 μm from the soma) and a weak dSR input was paired 5 or 40 times at 5 Hz with either strong pSR or SO activation, and these pairings were alternated between strong pSR and SO at least twice in any given cell. Examples of dendritic membrane potential responses during the first, fifth, and 40th pairings of dSR with pSR (black) or SO (gray). B, For the first action potential peak and maximum membrane voltage during pairings, strong activation of pSR resulted in greater depolarization compared with strong activation of SO (paired-sample Student's t tests, **p < 0.01). The integral of membrane voltage for each pairing was greater for the case of strong pSR activation when compared with strong SO activation (pSR: 2260 ± 320 mV · ms; SO: 1160 ± 135 mV · ms, paired-sample Student's t test, **p < 0.01)
We tested whether the somatic action potential is required for plasticity during pairing of weak and a strong input colocalized on the apical dendrites. To investigate this, we blocked the generation of somatic action potentials by localized pressure application of TTX near the soma while pairing weak dSR and strong pSR inputs (Fig. 6D). To insure that somatic sodium channels were blocked during TTX pressure application, a synaptic current waveform was injected through the somatic recording electrode less than one second before and after synaptic activation. The amplitude of the synaptic waveform resulted in a depolarization at least as great as that caused by the synaptic stimulation. Only cells in which the synaptic waveform current injections resulted in no regenerative events were used. A robust potentiation of the weak dSR input was observed (Fig. 6E). In the pooled data from all experiments, the weak dSR input potentiated significantly (Fig. 6F; 75.5 ± 0.3% increase, p < 0.01). Furthermore, this potentiation was not different from the control condition (83% vs 76%, p = 0.36), suggesting that under these conditions the back-propagating action potential is not a significant factor contributing to synaptic plasticity. In 3 of 8 experiments, the pSR input was robustly potentiated, whereas in 5 of 8 experiments pSR was strongly depressed, resulting in less potentiation of the strong pSR inputs than in the control condition. This result should be interpreted with caution, because of the possibility that pSR inputs are reduced as a result of diffusion of TTX from the cell body layer to the proximal synapses. The large potentiation of the dSR input, however, indicates that the back-propagating action potential is not required for potentiation of the distal input when paired with the strong proximal input.
Potentiation of the dSR pathway was not different when LTP was induced by either pairing with strong pSR activation, strong activation of dSR, during the blockade of somatic action potentials with somatically applied TTX when weak dSR was paired with strong pSR, or when synaptic inputs were mimicked by current injection through a dendritic recording electrode (Fig. 7A). Thus, potentiation of distal inputs requires a sufficient level of depolarization, which does not have to be synaptic, and does not require back-propagating action potentials.
Figure 7.

Potentiation of dSR inputs was similar when significant depolarization occurs on the same dendrite, and did not require back-propagating action potentials. A, Schematic of the protocols used to induce plasticity. B, Potentiation ratios of dSR inputs. Significant potentiation of the distal input was observed for all conditions (one-way ANOVA with repeated measures, p < 0.05, not indicated). Comparisons of potentiation across groups were made using one-way ANOVA with post hoc Bonferroni tests compared with the reference case of weak dSR paired with strong pSR (NS, not significant).
Depolarization during different induction conditions
The enhanced plasticity of the weak dSR input when it was colocalized with a strong input to pSR may be attributable to electrical cooperativity, with greater depolarization at the site of the dSR synapses when paired with strong pSR stimulation than with strong SO stimulation. To test this, we performed a series of dendritic recordings in which the weak dSR was paired with either a strong pSR or strong SO input in the same cell (n = 5, Fig. 8A). Stimulus intensities of both pSR and SO were adjusted such that 1–2 action potentials occurred with a single independent strong stimulus. Weak dSR was paired with either SO or pSR (either 2 × 40 or 8 × 5 times) at 5 Hz. pSR and SO pairings were alternated at least twice. The depolarization at the site of the dendritic recording during the induction protocol for the two conditions was compared. As expected, both the peak of the first spike and the maximum voltage reached during the pairings were larger during strong pSR stimulation than during strong SO stimulation (Fig. 8B). The total integral of depolarization was also greater for pSR than for SO (pSR: 2260 ± 320 mV · ms; SO: 1160 ± 135 mV · ms, paired-sample Student's t test, **p < 0.01). Thus, a likely mechanism for enhanced potentiation of dSR paired with pSR when compared with dSR paired with SO is the greater depolarization achieved during the dSR-pSR pairing.
Previous studies have shown that distal inputs can be potentiated in a manner independent of the back-propagating action potential, but requiring the generation of local dendritic spikes (Golding et al., 2002; Gordon et al., 2006; Remy and Spruston, 2007). In those studies, dendritic spikes were generated in response to high-frequency bursts (100 Hz) of one strongly activated pathway. In contrast, the form of plasticity described here involves a cooperative interaction between two input pathways, and is induced by synaptic activation at a frequency of 5 Hz. We therefore performed simultaneous somatic and dendritic recordings to measure the dendritic depolarization that occurs in this paradigm. The dendritic recordings were made at distances ranging from 45 to 175 μm from the soma (n = 7). Local regenerative dendritic events were observed in some recordings (4 of 7 cells). Some of these dendritically originating events occurred after a back-propagating action potential had invaded the dendrite, and the dendritic spike did not subsequently trigger additional action potentials (Fig. 9A, inset, 3 of 7 cells, 13 events total in all cells during 80 pairings in each cell). In two cells we observed a dendritic spike that preceded the somatic action potential (Fig. 9B; in one cell, 1 event on the first pairing, observed twice in two separate pairings of 40; in the second cell, 13 of the first 15 pairings). These events occurred in recordings made close to the soma (45 and 90 μm). Thus, a majority of the depolarization observed during these recordings was due to synaptic inputs or back-propagating action potentials, with local regenerative dendritic events observed occasionally. Because of the difficulty of observing dendritic spikes with a recording electrode at a single location, it cannot be concluded that they are absent in many cases. Rather, dendritic spikes clearly occur in some cases, and may occur in other cases where they are not observed.
Figure 9.

Regenerative events originating in the dendrites were detected in some cases. A, Simultaneous somatic and dendritic recording (interelectrode distance of 175 μm) during pairing of weak dSR with strong pSR. Regenerative dendritic events were observed once during 80 pairings in this cell. Similar events were observed in 3 of 7 cells (1, 5, and 10 events of 80 pairings in each cell, respectively). B, Example recording with an interelectrode distance of 45 μm. A dendritic spike that preceded the somatic action potential was observed in two trials in this cell (first two examples shown) and 15 trials in another cell (90 μm). Dendritic spikes that clearly preceded the somatic action potential were not observed in the other 5 cells.
Discussion
In general, the largest and most robust potentiation observed in this study resulted from cooperative synaptic inputs onto the same apical dendritic domain. This resulted in a greater local depolarization in the apical dendrites and a robust form of LTP that did not depend on back-propagating action potentials.
LTP exhibits the property of cooperativity, which means that multiple synapses must be synchronously activated in order for potentiation to occur (McNaughton et al., 1978; Levy and Steward, 1979). Direct current injection via a recording electrode can eliminate the requirement for activation of many synapses (Gustafsson et al., 1987), suggesting that the basis of cooperativity is the requirement for sufficient postsynaptic depolarization. This feature of LTP is also responsible for the property of associativity, by which a strong pathway can lead to the potentiation of weaker, but coincidentally activated pathway (Levy and Steward, 1979; Barrionuevo and Brown, 1983; Kelso and Brown, 1986).
The depolarization required for associative LTP can be provided by synaptic potentials, back-propagating action potentials, or dendritic spikes. Although it is well known that action potentials are not required for the induction of LTP (Kelso et al., 1986), under appropriate conditions they can induce LTP when paired with small EPSPs, even unitary events from single presynaptic axons (Markram et al., 1997; Bi and Poo, 1998; Sjöström et al., 2001). Under natural conditions, however, action potentials result from synaptic excitation. We therefore compared the relative efficacy of action potentials and other sources of depolarization during the induction of associative LTP in the hippocampus. We found that action potentials evoked by current injection can result in modest potentiation of pSR or dSR (∼25% potentiation). This potentiation, however, was not significantly greater than the unpaired controls for each pathway (30.7 ± 0.2% vs 9.9 ± 0.1% for pSR and 24.6 ± 0.1% vs 26.1 ± 0.2% for dSR; repeated measures ANOVA, p = 0.11 for pSR and p = 0.59 for dSR). Thus, modest potentiation may occur during pairing with somatically elicited action potentials, but it is difficult to detect when compared with control, nonpaired pathways. By comparison, potentiation was significantly greater (∼75% potentiation) when the small EPSP was paired with colocalized synaptic inputs that were sufficient to induce spiking (Fig. 4).
Differences in protocols and experimental preparations make it difficult to directly compare the results reported here with previous studies, but our results are generally consistent with the modest degree of potentiation when small EPSPs are paired with action potentials at low frequency (Sjöström et al., 2001; Meredith et al., 2003; Wittenberg and Wang, 2006; Campanac and Debanne, 2008). Also consistent with previous studies (Watanabe et al., 2002; Frick et al., 2004; Buchanan and Mellor, 2007), we also find potentiation that occurs under these conditions and is not due to pairing of EPSPs and action potentials. Rather, action potential firing may cause some nonspecific potentiation, even in unpaired synaptic inputs (Buchanan and Mellor, 2007). Despite this observation, there is considerable evidence that action potentials can influence synaptic plasticity. In hippocampal neurons, back-propagating action potentials are required for LTP under some conditions (Magee and Johnston, 1997) and action potential firing can prolong the duration of LTP as well (Raymond, 2008). In neocortical pyramidal neurons, amplifying back-propagating action potentials via dendritic depolarization can convert LTD to LTP (Sjöström and Häusser, 2006) and calcium spikes triggered by back-propagating action potentials can lead to the induction of LTP (Kampa et al., 2006). Furthermore, action potentials occurring just before EPSPs can lead to LTD (Markram et al., 1997; Bi and Poo, 1998; Wittenberg and Wang, 2006). Thus, action potentials can influence plasticity in a variety of ways.
Our study is the first direct comparison of LTP induction after synaptic vs nonsynaptic postsynaptic depolarization during low frequency pairings. In contrast to the modest amount of associative LTP induced with current-evoked action potentials or spatially distributed strong and weak inputs, spatially restricted inputs produced approximately three times as much LTP with the same number of pairings (∼75% increase). This form of short-range associative LTP did not require action potential firing at all, suggesting that other sources of depolarization are responsible for this form of LTP. Furthermore, substituting dendritic current injection for the strong proximal input could mimic the large LTP of distal inputs produced by pairing with a strong proximal input, suggesting that the strong input facilitates LTP simply by providing depolarization, and not by providing some other local biochemical effect. Other forms of LTP that do not require action potentials firing have been reported previously (Golding et al., 2002; Dudman et al., 2007; Remy and Spruston, 2007). Our results extend these observations by quantifying the relative amount of LTP induced under conditions when action potentials are elicited with current injection versus synaptic stimulation.
Dendritic recordings revealed that the apical dendritic response, near the potentiated synapses in dSR, was consistently larger during the short-range associative pairing than during the long-range pairing. One simple explanation for this result is that the synaptically mediated depolarization is larger at the weak input during the short-range pairing. In addition, however, dendritic recordings revealed the presence of apparently regenerative dendritic responses during the short-range pairing. Although these events were not abundant, dendritic spikes are likely to occur more often than can be easily observed in recordings from the main apical dendrite. Direct evidence supports the notion that activation of a few as 10 spines can trigger dendritic spikes in apical oblique branches, but that these spikes attenuate to just a few millivolts over distances of ∼100 μm (Losonczy and Magee, 2006). Furthermore, dendritic spikes are very powerful inducers of LTP, as even a single dendritic spike can lead to >50% LTP (Remy and Spruston, 2007). Thus, our data indicate that the strong synaptic input, although just above threshold for action potential firing, is strong enough to evoke local dendritic spikes, which together with the synaptic depolarization, are sufficient to contribute to the induction of powerful LTP that is not affected by the back-propagating action potentials. The larger depolarization occurring near the strong input likely results in the activation of more calcium entry through NMDA receptors and calcium channels, which may explain the enhanced LTP observed during short-range associations (Sjöström and Nelson, 2002).
In addition to the associative potentiation of the weak input, we also observed LTP of the strong input itself. Interestingly, however, this potentiation was greater for distal apical inputs (dSR, ∼75% increase) than for proximal apical or basal inputs (∼35% increase). This finding suggests that there are inherent differences in the proximal versus distal synapses, or in modulatory receptors (metabotropic) that are activated in these regions. Intriguingly, this could lead to greater potentiation of distal inputs, thus explaining observations that some distal synapses are physically larger and have larger conductances compared with proximal synapses (Magee and Cook, 2000; Nicholson et al., 2006), and leading to some compensation for the effect of dendritic distance on the somatic EPSP.
Although our study focused entirely on LTP, the results also have implications for LTD induction rules. Previous studies demonstrating that spikes preceding EPSPs lead to LTD have all been performed using current-evoked action potentials. Our finding that synaptic depolarization has a stronger potentiating influence than back-propagating action potentials suggests that the location of the strong synaptic input driving the action potentials will influence the induction of LTD at synapses activated after the spiking. It is likely that this potentiating influence would reduce LTD or overcome it, suggesting that whether synapses depress or not will depend on their location relative to the strong input. This effect is also likely to influence the presynaptic-postsynaptic timing rules that govern the induction STDP.
In summary, we directly compared the LTP of a weak test input paired with action potentials evoked by somatic current injection, a strong synaptic input onto a separate dendritic compartment, or a strong input onto the same dendritic compartment. We find that action potentials evoked by current injection or strong inputs onto a separate dendritic compartment have a modest potentiating effect, whereas synaptic potentials and/or dendritic spikes induced by a strong input on the same dendrite as the test input provide a more effective signal for the induction of LTP. Although considerable evidence from previous studies suggests an important role for back-propagating action potentials in regulating the induction of synaptic plasticity, a key point is that when action potentials are evoked by synaptic input, as expected under natural conditions in vivo, the potentiating effect of the synaptic potential and dendritic spikes exceeds that of the action potential, resulting in larger potentiation of colocalized synapses that comprise the strong input.
Footnotes
We thank Tyler Best, Yujin Kim, Shannon Moore, Jin-yong Park, Stefan Remy, and Mark Sheffield for helpful discussions and comments on this manuscript.
The authors declare no competing interests.
References
- Abbott and Nelson, 2000.Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Barrionuevo and Brown, 1983.Barrionuevo G, Brown TH. Associative long-term potentiation in hippocampal slices. Proc Natl Acad Sci U S A. 1983;80:7347–7351. doi: 10.1073/pnas.80.23.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell et al., 1997.Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- Bi and Poo, 1998.Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi and Wang, 2002.Bi GQ, Wang HX. Temporal asymmetry in spike timing-dependent synaptic plasticity. Physiol Behav. 2002;77:551–555. doi: 10.1016/s0031-9384(02)00933-2. [DOI] [PubMed] [Google Scholar]
- Buchanan and Mellor, 2007.Buchanan KA, Mellor JR. The development of synaptic plasticity induction rules and the requirement for postsynaptic spikes in rat hippocampal CA1 pyramidal neurones. J Physiol. 2007;585:429–445. doi: 10.1113/jphysiol.2007.142984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac and Debanne, 2008.Campanac E, Debanne D. Spike timing-dependent plasticity: a learning rule for dendritic integration in rat CA1 pyramidal neurons. J Physiol. 2008;586:779–793. doi: 10.1113/jphysiol.2007.147017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan and Poo, 2004.Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Dan and Poo, 2006.Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Dudman et al., 2007.Dudman JT, Tsay D, Siegelbaum SA. A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron. 2007;56:866–879. doi: 10.1016/j.neuron.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick et al., 2004.Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Golding et al., 1999.Golding NL, Jung HY, Mickus T, Spruston N. Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci. 1999;19:8789–8798. doi: 10.1523/JNEUROSCI.19-20-08789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding et al., 2002.Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Gordon et al., 2006.Gordon U, Polsky A, Schiller J. Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J Neurosci. 2006;26:12717–12726. doi: 10.1523/JNEUROSCI.3502-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson et al., 1987.Gustafsson B, Wigström H, Abraham WC, Huang YY. Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J Neurosci. 1987;7:774–780. doi: 10.1523/JNEUROSCI.07-03-00774.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa et al., 2006.Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574:283–290. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa et al., 2007.Kampa BM, Letzkus JJ, Stuart GJ. Dendritic mechanisms controlling spike-timing-dependent synaptic plasticity. Trends Neurosci. 2007;30:456–463. doi: 10.1016/j.tins.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Kelso and Brown, 1986.Kelso SR, Brown TH. Differential conditioning of associative synaptic enhancement in hippocampal brain slices. Science. 1986;232:85–87. doi: 10.1126/science.3952501. [DOI] [PubMed] [Google Scholar]
- Kelso et al., 1986.Kelso SR, Ganong AH, Brown TH. Hebbian synapses in hippocampus. Proc Natl Acad Sci U S A. 1986;83:5326–5330. doi: 10.1073/pnas.83.14.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs et al., 2002.Kepecs A, van Rossum MC, Song S, Tegner J. Spike-timing-dependent plasticity: common themes and divergent vistas. Biol Cybern. 2002;87:446–458. doi: 10.1007/s00422-002-0358-6. [DOI] [PubMed] [Google Scholar]
- Larkum et al., 1999.Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Larkum et al., 2007.Larkum ME, Waters J, Sakmann B, Helmchen F. Dendritic spikes in apical dendrites of neocortical layer 2/3 pyramidal neurons. J Neurosci. 2007;27:8999–9008. doi: 10.1523/JNEUROSCI.1717-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson and Lynch, 1986.Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- Levy and Steward, 1979.Levy WB, Steward O. Synapses as associative memory elements in the hippocampal formation. Brain Res. 1979;175:233–245. doi: 10.1016/0006-8993(79)91003-5. [DOI] [PubMed] [Google Scholar]
- Lisman and Spruston, 2005.Lisman J, Spruston N. Postsynaptic depolarization requirements for LTP and LTD: a critique of spike timing-dependent plasticity. Nat Neurosci. 2005;8:839–841. doi: 10.1038/nn0705-839. [DOI] [PubMed] [Google Scholar]
- Losonczy and Magee, 2006.Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Magee and Cook, 2000.Magee JC, Cook EP. Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat Neurosci. 2000;3:895–903. doi: 10.1038/78800. [DOI] [PubMed] [Google Scholar]
- Magee and Johnston, 1997.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Malinow and Tsien, 1990.Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Markram et al., 1997.Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- McNaughton et al., 1978.McNaughton BL, Douglas RM, Goddard GV. Synaptic enhancement in fascia dentata: cooperativity among coactive afferents. Brain Res. 1978;157:277–293. doi: 10.1016/0006-8993(78)90030-6. [DOI] [PubMed] [Google Scholar]
- Meredith et al., 2003.Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson et al., 2006.Nicholson DA, Trana R, Katz Y, Kath WL, Spruston N, Geinisman Y. Distance-dependent differences in synapse number and AMPA receptor expression in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:431–442. doi: 10.1016/j.neuron.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Raymond, 2008.Raymond CR. Different requirements for action potentials in the induction of different forms of long-term potentiation. J Physiol. 2008;586:1859–1865. doi: 10.1113/jphysiol.2008.151035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy and Spruston, 2007.Remy S, Spruston N. Dendritic spikes induce single-burst long-term potentiation. Proc Natl Acad Sci U S A. 2007;104:17192–17197. doi: 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts and Bell, 2002.Roberts PD, Bell CC. Spike timing dependent synaptic plasticity in biological systems. Biol Cybern. 2002;87:392–403. doi: 10.1007/s00422-002-0361-y. [DOI] [PubMed] [Google Scholar]
- Sjöström and Häusser, 2006.Sjöström PJ, Häusser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron. 2006;51:227–238. doi: 10.1016/j.neuron.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Sjöström and Nelson, 2002.Sjöström PJ, Nelson SB. Spike timing, calcium signals and synaptic plasticity. Curr Opin Neurobiol. 2002;12:305–314. doi: 10.1016/s0959-4388(02)00325-2. [DOI] [PubMed] [Google Scholar]
- Sjöström et al., 2001.Sjöström PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Tsodyks, 2002.Tsodyks M. Spike-timing-dependent synaptic plasticity - the long road towards understanding neuronal mechanisms of learning and memory. Trends Neurosci. 2002;25:599–600. doi: 10.1016/s0166-2236(02)02294-4. [DOI] [PubMed] [Google Scholar]
- Watanabe et al., 2002.Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg and Wang, 2006.Wittenberg GM, Wang SS. Malleability of spike-timing-dependent plasticity at the CA3-CA1 synapse. J Neurosci. 2006;26:6610–6617. doi: 10.1523/JNEUROSCI.5388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]