

Abstract
Purpose
CCL2 plays an important role in vascular inflammation by inducing leukocyte recruitment and activation. The authors had previously found that the blockade of NAD(P)H oxidase in turn blocks leukocyte adhesion to retinal vessels during diabetes and uveitis. In this study, the role of NAD(P)H oxidase in CCL2 production was assessed.
Methods
Studies were performed in three mouse models with lipopolysaccharide (LPS)-induced uveitis, ischemic retinopathy, and streptozotocin diabetes and in cytokine- and LPS-treated cells. CCL2 mRNA and protein expression were measured by quantitative PCR and ELISA. NF-κB activity was detected by reporter gene assay. Kinase phosphorylation was determined by immunoblotting.
Results
Expression of CCL2 was increased in the retinas of all three mouse models. The effect was strongest in the LPS-treated mice, with a peak mRNA increase at 3 hours. This increase was abrogated by administration of the NAD(P)H oxidase inhibitor apocynin. Apocynin also blocked CCL2 production in endothelial cells (ECs), retinal microglia, and Müller cells stimulated with TNF-α, VEGF, or LPS. Studies using human ECs demonstrated that TNF-α–induced CCL2 production was also inhibited by the NAD(P)H oxidase inhibitor DPI, the antioxidant N-acetyl-l-cysteine, or the superoxide scavenger Tiron, further indicating that inhibition occurs through the NAD(P)H/ROS pathway. Analysis of downstream signals showed that inhibition of NAD(P)H oxidase partially inhibited NF-κB activation but did not reduce CCL2 mRNA stability or prevent TNF-α–induced phosphorylation of p38MAPK. However, TNF-α–induced Akt phosphorylation was blocked, and inhibiting Akt dramatically decreased CCL2 production.
Conclusions
NAD(P)H oxidase activity is required for CCL2 production during retinal vascular inflammation. Akt and NF-κB are involved in this signaling pathway.
Vascular inflammation is believed to play a critical role in the pathogenesis of a variety of retinal diseases, such as diabetic retinopathy, uveitis, and ischemic retinopathy. A common feature associated with these diseases is increased leukocyte adhesion to the vessel wall, which can cause retinal capillary plugging and nonperfusion, leading to vascular injury, hyperpermeability, and neovascularization.1-5 The healthy vascular wall is smooth and resistant to the adhesion of leukocytes. However, in disease conditions, stimulators such as proinflammatory cytokines, oxidative stress, and endotoxin can cause endothelial cell expression of chemokines and adhesion molecules that will induce leukocyte activation and adhesion to the vessel wall.6,7
CCL2, formerly called monocyte chemoattractant protein-1, is a chemokine that is produced in response to different inflammatory stimuli, such as TNF-α, IL-1β, and endotoxin. It plays a critical role in inflammation by recruiting and activating monocytes, macrophages, T cells, and natural killer cells.6 CCL2 released by endothelial cells and leukocytes forms a gradient that directs a subset of leukocytes moving toward the site of inflammation and triggers firm adhesion of rolling leukocytes to the endothelium.8,9 In addition, CCL2 regulates the respiratory burst and the release of cytokines by leukocytes.10,11 As a consequence of its important function during vascular inflammation, CCL2 expression has been linked to a variety of acute and chronic inflammatory diseases. Abrogating CCL2 pathways by gene deletion or antibody neutralization has been shown to prevent the pathogenesis of many inflammatory conditions, including endotoxin-induced uveitis (EIU), retinal neovascularization, and atherosclerosis.12-14
Various reports have shown that reactive oxygen species (ROS) play essential roles in the vascular inflammation induced by growth factors and cytokines.15,16 Oxidative stress is known as a critical player in the pathogenesis of various diseases, including hypertension, atherosclerosis, and diabetes.17 NAD(P)H oxidase is the major source of ROS in the vasculature. In phagocytic cells, NAD(P)H oxidase is a multiprotein complex consisting of membrane-bound NOX2 (formerly known as gp91phox), p22phox, the cytoplasmic subunits p47phox and p67phox, and the small Rho GTPase Rac.17 Vascular endothelial cells express the same subunits and the homologues of NOX2, such as NOX1 and NOX4.17 We and others18,19 have shown that the diabetes-induced increase in VEGF expression and the breakdown of the blood–retinal barrier correlate with increases in oxidative stress. Recently, we also demonstrated that NAD(P)H oxidase activity has a primary role in retinal vascular inflammation and that the abrogation of NAD(P)H oxidase activity reduces leukocyte adhesion to the vessel and vascular hyperpermeability.20
Here we hypothesized that NAD(P)H oxidase is critical for CCL2 expression during vascular inflammation in the retina and determined potential downstream targets of NAD(P)H oxidase in the regulation of CCL2 production. Our in vivo and in vitro results show that the NAD(P)H oxidase/ROS pathway is required for CCL2 production in vivo and in different types of cells in response to inflammatory stimuli. NAD(P)H oxidase may regulate CCL2 expression through Akt and NF-κB but not by enhancing CCL2 mRNA stability.
Methods
Treatment of Animals
All procedures with animals were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the institutional animal care and use committee (Animal Welfare Assurance no. A3307-01).
Experiments were performed with C57Bl/6J mice. EIU was induced by injection of lipopolysaccharide (LPS) from Salmonella typhimurium (4 mg/kg in PBS, intraperitoneally; Sigma-Aldrich, St. Louis, MO). Age-matched control mice received vehicle alone. Diabetes was induced by repeated injection (normally three times) of streptozotocin (STZ; 50 mg/kg, dissolved in 0.1 M sodium citrate buffer [pH 4.5], intraperitoneally, once every other day) until diabetes was established. Mice with glucose levels greater than 300 mg/dL (measured by blood glucose meter) for 1 month were used for experiments. Ischemic retinopathy was induced as described.21 Mice maintained in normoxia (postnatal day 14) were used as controls. For apocynin treatment, mice were injected with apocynin (10 mg/kg in 0.9% saline, intravenously) 1 hour before the injection of LPS.
Tissue Culture
Primary rat microglia were isolated and grown in culture medium (Dulbecco modified Eagle medium [DMEM]/F12; Mediatech, Manassas, VA) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin solution (Invitrogen, Carlsbad, CA), as described.22 Briefly, retinas were collected and washed twice with ice-cold PBS and were digested with 0.125% trypsin at 37°C for 3 minutes. Digestion was stopped by the addition of culture medium, and tissues were triturated with a plastic pipette and washed twice. Cells were filtered through a nylon mesh (mesh opening, 100 μm; Nitex [Wildlife Supply Co., Buffalo, NY]), collected by centrifugation, resuspended in culture medium, and plated into 100-cm2 cell culture flasks at a density of 2×105 cells/cm2. After 2 weeks, microglial cells were harvested in culture medium by shaking the flasks at 100 rpm for 1.5 hours. Cell suspension was centrifuged, and the detached cells were replated in culture medium for experiments. The purity of the microglial cultures was approximately 98% as determined by immunocytochemical staining analysis for OX42, a macrophage/microglial marker. For treatments, cells were seeded at a density of 1 × 105 cells/well in 24-well plates in culture medium. One day after seeding, culture cells were washed in media (Cellgro Complete; Mediatech) and incubated in the same media with various treatments.
Rat Müller cells (rMC-1)23 were cultured in DMEM (Invitrogen) supplemented with 10% FBS. Primary bovine retinal endothelial cells (ECs) were isolated as described24 and were maintained in M199 supplied with bovine brain extract and 10% FBS. Human umbilical vein ECs (HUVECs) were purchased from Lonza (Basel, Switzerland) and were grown in Endothelial Cell Growth Medium (Lonza). These cells were used from passages 2 to 6 and were incubated in serum-free medium overnight before treatment.
Reverse Transcriptase–Polymerase Chain Reaction
Total RNA was isolated with a PCR kit (RNAqueous 4PCR; Applied Biosystems, Austin, TX) for retinal tissue or with reagent (TRIzol; Invitrogen) for cells, according to the manufacturer’s instructions. Total RNA was reverse transcribed with M-MLV reverse transcriptase (Invitrogen) to generate cDNA. Gene-specific primers were then used for semiquantitative PCR amplification to detect relative amounts of transcript. The PCR reaction was set up in 25 μL with cDNA reverse transcribed from 20 ng total RNA. The following PCR program was used: step 1, 2 minutes at 94°C; step 2, 30 seconds at 94°C; step 3, 30 seconds at 62°C; step 4, 15 seconds at 72°C; step 5, 24 repetitions of steps 2 to 4; step 6, 7 minutes at 72°C; step 7, hold at 4°C. This program was chosen based on a pioneer experiment performed by adding series-diluted cDNA from TNF-α–treated ECs to ensure that it achieved a linear amplification of CCL2. Alternatively, gene expression was determined by real-time quantitative PCR (Power SYBR Green PCR Master Mix; Applied Biosystems), which was performed on a thermocycler (StepOne Plus; Applied Biosystems). Cycle threshold, determined as the initial increase in fluorescence above background, was ascertained for each sample. Melt curves was performed on completion of the cycles to ensure that nonspecific products were absent. 18S or GAPDH was used as an internal control in the PCR reaction for normalization. Of note, GAPDH was not used as a control in diabetic animal studies. Primer sequences were as follows: human CCL2, forward, 5′-CTC GCT CAG CCA GAT GCA AT-3′; human CCL2, reverse, 5′-GGA CAC TTG CTG CTG GTG AT-3′; mouse CCL2, forward, 5′-GGC TCA GCC AGA TGC AGT TAA-3′; mouse CCL2, reverse, 5′-CCT ACT CAT TGG GAT CAT CTT GCT- 3′; rat CCL2, forward, 5′-CTC AGC CAG ATG CAG TTA ATG C-3′; rat CCL2, reverse, 5′-AGC CGA CTC ATT GGG ATC AT-3′; human intercellular adhesion molecule (ICAM)-1, forward, 5′-GCC AGG AGA CAC TGC AGA CA-3′; human ICAM-1, reverse, 5′-TGG CTT CGT CAG AAT CAC GTT-3′; 18S, forward, 5′- GTT GGT TTT CGG AAC TGA GGC-3′; 18S, reverse, 5′-GTC GGC ATC GTT TAT GGT CG-3′; GAPDH, forward, 5′-ACC ACA GTC CAT GCC ATC AC-3′; GAPDH, reverse, 5′-TCC ACC ACC CTG TTG CTG TA-3′.
ELISA
CCL2 levels in the conditioned medium were estimated with ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. To determine CCL2 expression in the mouse retina, retinas were dissected and lysed by sonication with lysis buffer (20 mM imidazole, 100 mM KCl, 1 mM MgCl2, 1% Triton X-100, 10 mM NaF, 1 mM Na3VO4, 1 mM EGTA, 1 mM EDTA, pH 6.8) supplemented with protease inhibitors. The lysate was cleared of debris by centrifugation, and the supernatant was used for ELISA with a mouse CCL2 ELISA kit (R&D Systems). Protein concentration in the lysates was determined by a BCA assay (Pierce Biotechnology, Rockford, IL). CCL2 concentration was normalized to total protein in the lysate.
Luciferase Reporter Gene
HUVECs were plated in a 24-well plate and transfected with reagent (Lipofectin; Invitrogen) according to the manufacturer’s instructions. Cells were cotransfected with 0.725 μg pNF-κB-luc, a multimerized NF-κB-luciferase (firefly luciferase) reporter gene plasmid, and 0.2 μg of pRL-CMV (Renilla luciferase) internal control plasmid to normalize for transfection efficiency. After 24 hours, the cells were incubated in serum-free medium and treated with vehicle or apocynin and then were stimulated with TNF-α for 6 hours. Renilla luciferase and firefly luciferase were measured (Dual Luciferase Assay System; Promega Corporation, Madison, WI), and Renilla values were used for normalization.
Western Blot Analysis
After incubation in serum-free media overnight, cells were stimulated with TNF-α at 37°C for the indicated times in the presence of vehicle (dimethyl sulfoxide) or apocynin. Then cells were lysed in sodium dodecyl sulfate (SDS) sample buffer and subjected to 10% SDS polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred onto a nitrocellulose membrane, and the membrane was incubated with primary antibodies from Cell Signaling Technology (Danvers, MA) against total p38 mitogen-activated protein kinase (p38MAPK; no. 9212), phosphorylated p38MAPK (no. 9211), total Akt (no. 9272), and phosphorylated Akt (no. 4058) followed by horseradish peroxidaseconjugated secondary antibody. Immunoreactive proteins were detected with the enhanced chemiluminescence (ECL) system (GE Healthcare Bio-Sciences Corp., Piscataway, NJ).
Statistical Analysis
Results are expressed as mean ± SEM. Group differences were evaluated with the use of one-way ANOVA followed by post hoc Student’s t-test (SigmaStat software; Aspire Software International, Ashburn, VA). Results were considered significant at P < 0.05.
Results
LPS-Induced CCL2 Expression Abrogation by Inhibition of NAD(P)H Oxidase
CCL2 expression in the retina was determined by ELISA. CCL2 protein levels were significantly elevated in the retinas of mice with diabetes, ischemic retinopathy, and endotoxin-induced uveitis (EIU). The effect was most prominent in the EIU model, where the expression of CCL2 was increased by more than 100-fold (Fig. 1). Thus, the EIU model was selected to test whether NAD(P)H oxidase is involved in CCL2 production in inflammation.
Figure 1.
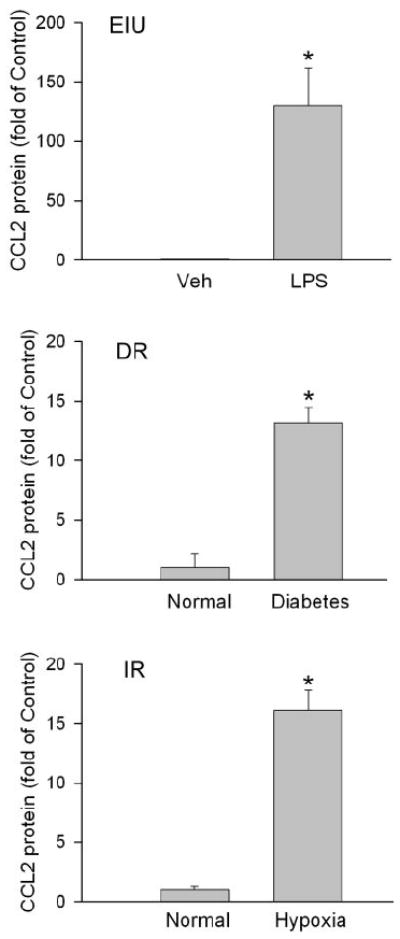
CCL2 expression increases in eye disease models. Endotoxin-induced uveitis (EIU), diabetic retinopathy (DR), and ischemic retinopathy (IR) mouse models were generated. CCL2 expression in the retina was determined by ELISA (n = 3–8). *P < 0.05 compared with vehicle or normal control.
LPS was administered to determine the optimal time for the study of CCL2 mRNA expression in EIU. As shown in Figure 2A, CCL2 mRNA was robustly increased after LPS injection and reached a peak at 3 hours. However, this increase was significantly reduced 47% by the inhibition of NAD(P)H oxidase with apocynin (Fig. 2B). Apocynin is a specific inhibitor for NAD(P)H oxidase that blocks assembly of the enzyme complex.25
Figure 2.
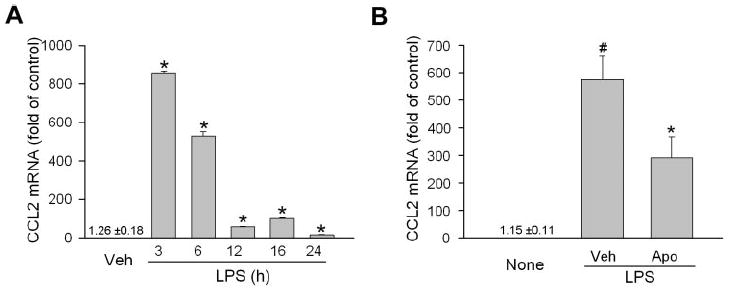
Inhibition of NAD(P)H oxidase blocks CCL2 production in the retina in EIU. (A) Male C57/BL6 mice were injected with LPS (4 mg/kg intraperitoneally) and were killed at the time indicated. CCL2 mRNA in the retina was then determined by quantitative PCR and normalized to vehicle control (n = 3). *P < 0.05 compared with vehicle control (saline). (B) Mice were pretreated with apocynin (Apo, 10 mg/kg intravenously) 1 hour before LPS injection. Three hours after LPS treatment, mice were killed, and CCL2 mRNA in the retina was determined by quantitative PCR and normalized to control mice without LPS treatment (None; n = 5–7). #P < 0.05 compared with None. *P < 0.05 compared with saline vehicle control.
Blockade of CCL2 Expression in Retinal Cells by Inhibition of NAD(P)H Oxidase
The role of NAD(P)H oxidase in CCL2 production was further studied in cultured cells including microglia, Müller cells, and endothelial cells (ECs), which are the major retinal cell types that mediate the immune response. Given that TNF-α induces ROS signal in ECs,26,27 is recognized as a potent initiator of inflammation, and is critically involved in the pathogenesis of EIU and ischemic retinopathy,4,28,29 experiments were conducted to determine whether NAD(P)H oxidase is required for CCL2 production induced by LPS and TNF-α. As shown in Figure 3A, the incubation of microglia with LPS resulted in a twofold increase of CCL2 mRNA. However, in the presence of apocynin, LPS-induced CCL2 expression was significantly attenuated. Similarly, TNF-α–induced CCL2 expression in Müller cells and ECs, whereas apocynin treatment completely abolished the effect of TNF-α (Figs. 3B–D). These results indicated that NAD(P)H oxidase plays a general role in controlling CCL2 production during inflammation.
Figure 3.
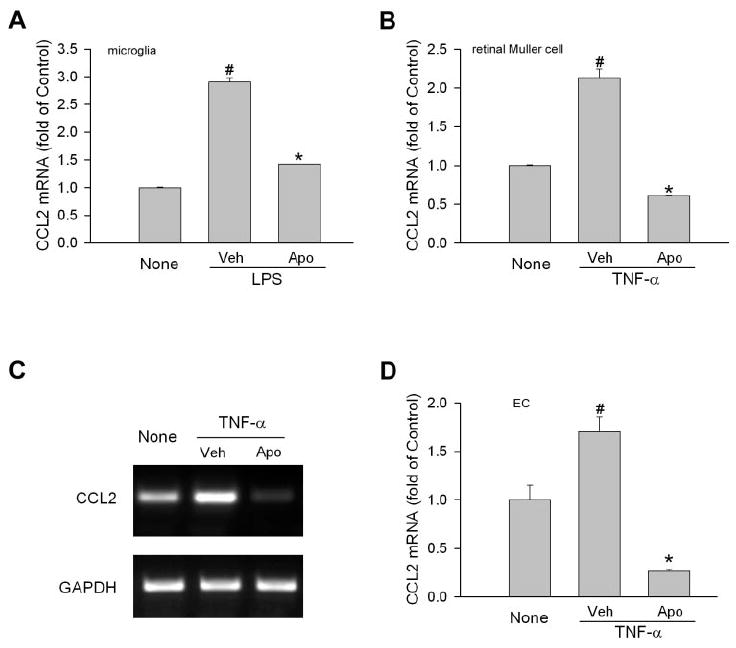
Involvement of NAD(P)H oxidase in CCL2 production in retinal cells. (A) Retinal microglial cells were pretreated with apocynin (Apo, 0.5 mM) or vehicle (Veh) for 0.5 hour Cells were then treated with LPS (30 ng/mL) for 3 hours. Total RNA was extracted, and CCL2 mRNA was measured by quantitative PCR. Cells without treatment (None) were used as control (n = 4). #P < 0.05 compared with None. *P < 0.05 compared with vehicle. (B) Retinal Müller cells (rMC-1) were pretreated with apocynin (Apo, 0.5 mM) or vehicle (Veh) for 0.5 hour. Cells were then treated with TNF-α (5 ng/mL) for 2 hours. CCL2 mRNA was measured by quantitative PCR (n = 4). #P < 0.05 compared with None. *P < 0.05 compared with vehicle. (C) Bovine retinal ECs were pretreated with apocynin (Apo, 1 mM) or vehicle (Veh) for 0.5 hour. Cells were then treated with TNF-α (10 ng/mL) for 2 hours. CCL2 mRNA was determined by semiquantitative PCR. Representative image is shown (n = 3). (D) Quantitative data of (C). #P < 0.05 compared with None. *P < 0.05 compared with vehicle.
Requirement of NAD(P)H Oxidase/ROS Pathway for CCL2 Production in Human ECs
The vascular endothelium serves as the frontline during inflammation and is critical for leukocyte recruitment through the production of chemokines and adhesion molecules. Thus, the role of NAD(P)H oxidase in CCL2 production was further studied with the use of primary HUVECs. Similar results were achieved in these cells. TNF-α dramatically increased the expression of CCL2. Apocynin dose dependently blocked CCL2 production and achieved maximal inhibition at 1 mM (Fig. 4A). Interestingly, this effect was also observed with VEGF stimulation (Fig. 4B). VEGF is another important mediator of vascular inflammation, and its expression increases in patients with diabetic retinopathy.30 Because human ECs were highly sensitive to TNF-α and expressed little CCL2 under basal conditions, they were studied to further evaluate the effect of NAD(P)H oxidase and to understand the molecular mechanisms by which NAD(P)H oxidase regulates CCL2 production.
Figure 4.
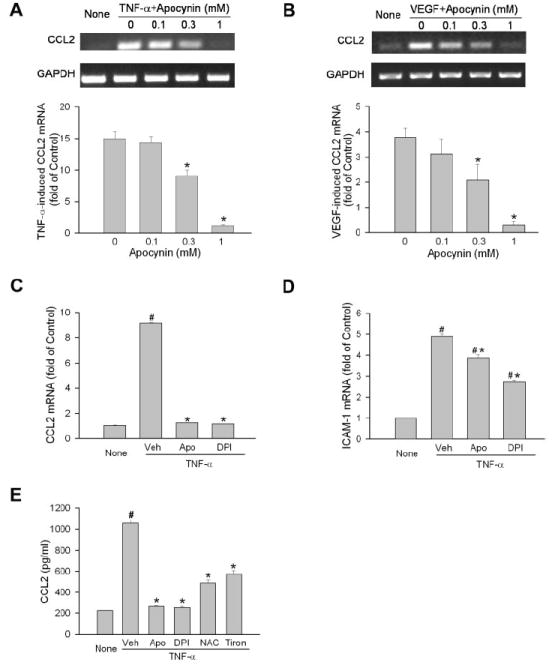
Involvement of NAD(P)H oxidase/ROS pathway in CCL2 production in human ECs. (A) Human ECs were pretreated with apocynin at the concentrations indicated for 0.5 hour. Cells were then stimulated with TNF-α (0.03 ng/mL) for 2 hours. CCL2 mRNA was determined by semiquantitative PCR (n = 3). CCL2 level in cells without treatment (None) is used as reference. *P < 0.05 compared with apocynin at 0 mM. (B) Human ECs were treated and analyzed as in (A) except that VEGF (30 ng/mL) was used as agonist (n = 3). (C) Human ECs were pretreated with apocynin (1 mM), DPI (20 μM), or vehicle for 0.5 hour. Cells were then simulated with TNF-α (0.03 ng/mL) for 2 hours. CCL2 mRNA was measured by quantitative PCR. CCL2 level in cells without treatment (None) is used as reference (n = 4). #P < 0.05 compared with None. *P < 0.05 compared with vehicle. (D) Cells were treated as in (C), and ICAM-1 expression was measured (n = 4). (E) Human ECs were pretreated with vehicle, apocynin (1 mM), DPI (20 μM), N-acetyl-l-cysteine (10 mM), or Tiron (10 mM) for 0.5 hour and were then simulated with TNF-α (0.03 ng/mL) for 3 hours. Culture media were collected, and CCL2 levels were determined by ELISA (n = 4). #P < 0.05 compared with None. *P < 0.05 compared with vehicle.
Complete blockade of CCL2 production by apocynin raised the possibility of a nonspecific effect. Diphenylene iodonium chloride (DPI), another inhibitor for NAD(P)H oxidase, was used to verify the role of NAD(P)H oxidase. The result of this study showed that DPI abolished TNF-α–induced CCL2 production as efficiently as apocynin (Fig. 4C). In addition, TNF-α–induced ICAM-1 expression was only slightly decreased by apocynin and DPI, indicating the specificity of the apocynin and DPI effect on CCL2 (Fig. 4D). Consistent with mRNA expression, the determination of CCL2 production in the conditioned medium after cells were treated with inhibitors and TNF-α revealed that NAD(P)H oxidase inhibitors apocynin and DPI, antioxidant N-acetyl-l-cysteine, and superoxide scavenger Tiron all significantly blocked TNF-α–induced CCL2 protein expression (Fig. 4E), which further supported the role of the NAD(P)H oxidase/ROS pathway in CCL2 production.
CCL2 mRNA Stability Unaffected by NAD(P)H Oxidase Blockade
The effect of apocynin in blocking TNF-α–induced increases in CCL2 mRNA levels may result from an action reducing mRNA stability or inhibiting CCL2 expression. To understand whether the inhibition of NAD(P)H oxidase changed the stability of CCL2 mRNA, CCL2 mRNA half-life was determined after gene transcription was blocked with the use of actinomycin D. As shown in Figure 5, CCL2 mRNA was rapidly degraded and had a short half-life of approximately 27 minutes. In the presence of TNF-α, the degradation of CCL2 mRNA was slightly reduced and had a half-life of approximately 45 minutes. However, TNF-α–increased CCL2 mRNA stability was not reduced by apocynin, suggesting the effect of NAD(P)H oxidase involves the regulation of CCL2 expression.
Figure 5.
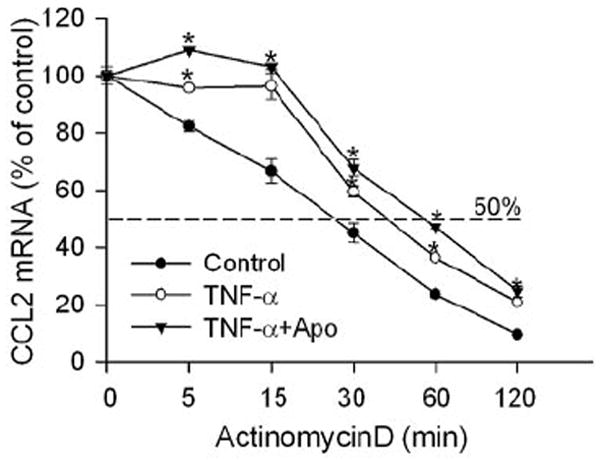
Inhibition of NAD(P)H oxidase does not attenuate CCL2 mRNA degradation. Human ECs were pretreated with TNF-α (0.03 ng/mL) for 1 hour. Cells were then treated with actinomycin D (10 μg/mL) to block gene transcription in the presence or absence of NAD(P)H oxidase inhibitor apocynin (1 mM) for the time indicated. CCL2 mRNA in cells was determined by quantitative PCR. Cells without transcription blockade were used as a reference (n = 3). *P < 0.05 compared with cells without TNF-α treatment for the same period of actinomycin D treatment.
NF-κB and Akt as Downstream Targets for NAD(P)H Oxidase in CCL2 Production
Studies have shown that NF-κB is required in TNF-α–induced CCL2 production in tumor cells.31 To determine whether the NAD(P)H oxidase effect in decreasing CCL2 production involves the blockade of NF-κB, TNF-α–induced NF-κB activity was detected in the presence or absence of apocynin by NF-κB–driven luciferase reporter gene assay. As shown in Figure 6A, stimulation of human ECs resulted in an 8.3-fold increase of luciferase activity. In the presence of apocynin, this increase was significantly reduced to 3.3-fold (a 69% reduction). Given that the commonly used NF-κB inhibitor PDTC is also an antioxidant,32 a sphingosine kinase inhibitor was included as a positive control for the experiment. Sphingosine kinase activity is indispensible for TNF-α–induced NF-κB activation in ECs.33 Inhibition of sphingosine kinase completely blocked luciferase activity induced by TNF-α (Fig. 6A). Unexpectedly, determining CCL2 expression showed that the inhibition of sphingosine kinase only partially blocked TNF-α–induced CCL2 production (a 46% reduction), whereas apocynin achieved 100% inhibition (Fig. 6B), suggesting that molecules other than NF-κB were involved in transducing the TNF-α-NAD(P)H oxidase-CCL2 signal.
Figure 6.
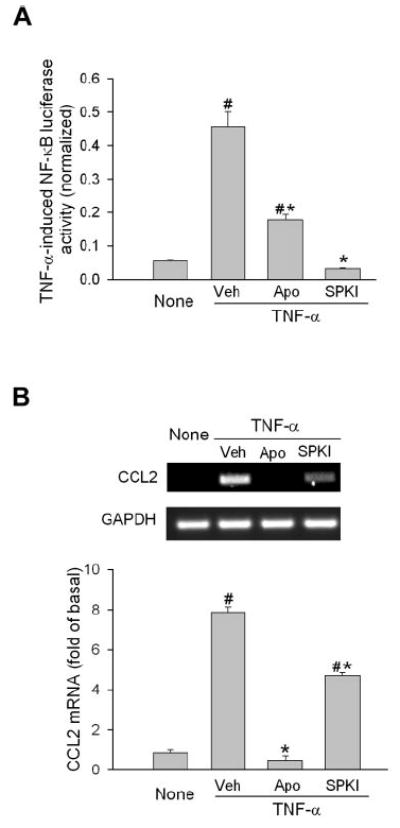
Inhibition of NAD(P)H oxidase partially blocks NF-κB activation. (A) Human ECs were transfected with a plasmid containing the luciferase gene under the control of the NF-κB promoter and an internal control. Then cells were preincubated with NAD(P)H oxidase inhibitor (apocynin, 1 mM), or sphingosine kinase inhibitor (SPKI, 10 μM) and stimulated with TNF-α (1 ng/mL) for 6 hours. Luciferase activity was measured and normalized to internal control to eliminate the transfection variation (n = 8–12). #P < 0.05 compared with None. *P < 0.05 compared with vehicle. (B) ECs were pretreated with apocynin or sphingosine kinase inhibitor as in (A). Cells were then stimulated with TNF-α for 2 hours, and CCL2 mRNA was determined by semiquantitative PCR (n = 3). #P < 0.05 compared with None. *P < 0.05 compared with vehicle.
Some reports indicate that p38MAPK and Akt are involved in TNF-α–induced CCL2 production in ECs.34,35 Experiments were conducted to determine whether they are the downstream targets for NAD(P)H oxidase. Treatment of ECs with TNF-α resulted in a rapid increase in p38MAPK phosphorylation, with maximal effect at 10 minutes, and in Akt phosphorylation, with maximal effect at 30 minutes (Fig. 7A). In the presence of apocynin, TNF-α–induced Akt phosphorylation was completely blocked, whereas the activation of p38MAPK was not changed (Fig. 7A). Interestingly, inhibition of Akt by LY294002 substantially inhibited TNF-α–induced CCL2 production (Fig. 7B). Inhibition of JNK and ERK did not attenuate TNF-α–induced CCL2 production (data not shown). These results indicated that Akt, but not p38MAPK, is involved in the TNF-α–CCL2 signaling pathway.
Figure 7.
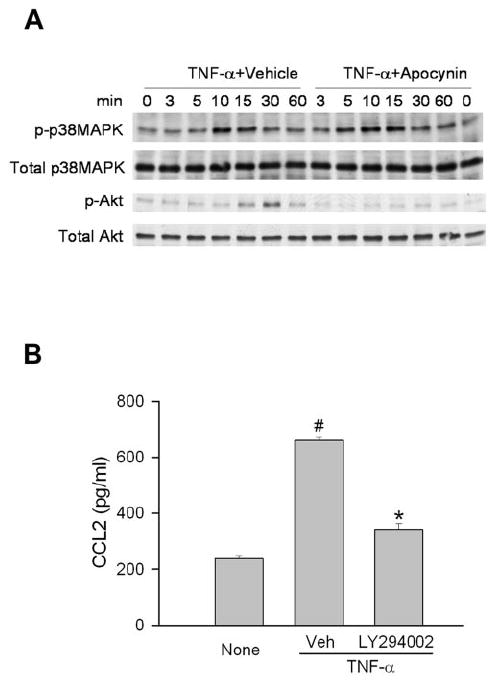
Akt, but not p38MAPK, serves as the downstream target for NAD(P)H oxidase. (A) Human ECs were preincubated with 1 mM apocynin for 30 minutes. Cells were then stimulated with TNF-α (1 ng/mL) for the time indicated, and phosphorylated and total p38MAPK and Akt were detected by immunoblot (n = 3). (B) After human Ecs were pretreated with vehicle or LY294002 (10 μM) for 0.5 hour, they were treated with TNF-α for 3 hours. Culture media were collected, and CCL2 levels were determined by ELISA (n = 3). #P < 0.05 compared with None. *P < 0.05 compared with vehicle control.
Discussion
NAD(P)H oxidase activity has been shown to play a critical role in eye diseases, diabetic complications, and cardiovascular diseases. 17,20,36 Blockade of NAD(P)H oxidase has been shown to reduce retinal vascular inflammation, neovascularization, and vascular hyperpermeability.20,36 Nevertheless, the downstream targets for NAD(P)H oxidase in the inflammatory process are largely unknown. In the present study, we provide the first evidence that NAD(P)H oxidase activity is required for the production of CCL2, an important player in inflammatory responses, during retinal inflammation. Our studies demonstrated that the inhibition of NAD(P)H oxidase significantly attenuated LPS-induced CCL2 production in the EIU model. In vitro studies indicated that NAD(P)H oxidase plays a general role in controlling CCL2 production because the inhibition of NAD(P)H oxidase blocked CCL2 production in different cell types, including retinal microglia, Müller cells, and ECs, in response to a variety of inflammatory stimuli, including LPS, TNF-α, and VEGF. CCL2 is critically involved in vascular inflammation by recruiting leukocytes and is implicated in angiogenesis and EC hyperpermeability by binding to its receptor in ECs.6,37,38 Blockade of CCL2 production has been shown to reduce pathologic signs in uveitis and ischemic retinopathy and to prevent photoreceptor cell damage during retinal detachment by reducing inflammation.13,14,39 Our finding that NAD(P)H oxidase regulates CCL2 production in retinal inflammation further underscores the therapeutic benefit of blocking NAD(P)H oxidase in retinal diseases.
Of note, LPS induces more CCL2 production in EIU than in cultured cells treated with LPS or cytokine alone, which suggests a more complicated response to LPS in vivo. The retina is composed of multiple cell types that regulate each other in physiological and pathologic conditions. LPS may initiate the inflammatory response and induce the production of proinflammatory cytokines in different cell types, such as Müller cells, macrophages/microglia, and astrocytes. As a consequence, CCL2 expression in EIU may arise from a combined or even a synergistic direct effect of LPS on cells and from the indirect impact of other proinflammatory cytokines. However, by showing that the blockade of NAD(P)H oxidase abrogates CCL2 production in vivo and in vitro, our studies clearly demonstrated a critical and general role of NAD(P)H oxidase in regulating CCL2 production.
mRNA level can be regulated by changing expression or by altering mRNA half-life. It has been shown that activated protein C increases CCL2 mRNA in ECs by enhancing the stability of CCL2 mRNA.40 Studies also have shown that TNF-α and ROS have the ability to augment mRNA stability for some proteins. 41,42 This information raises the possibility that the inhibition of NAD(P)H oxidase may accelerate CCL2 mRNA degradation and thus reduce the CCL2 mRNA level. This possibility was excluded by our finding that TNF-α only marginally increased the half-life of CCL2 mRNA and that apocynin treatment did not block this increase.
NF-κB is a redox-sensitive molecule that plays a critical role in inflammatory responses.32 Previous studies with tumor cells have shown that TNF-α–induced CCL2 expression requires NF-κB activity.31 We suspected that NF-κB is a downstream target for NAD(P)H oxidase. Unexpectedly, we found apocynin inhibited TNF-α–induced NF-κB promoter activity by only 69%, whereas it blocked CCL2 production 100%. In contrast, an inhibitor for sphingosine kinase blocked NF-κB activity 100%, whereas it reduced CCL2 production by only 46%. This contradiction raises the possibility that NF-κB is not the only downstream target for NAD(P)H oxidase in the regulation of CCL2 production in human ECs. In contrast with CCL2, ICAM-1 expression in TNF-α–treated cells correlated closely with the activation of NF-κB, and apocynin only partially reduced its expression. Nevertheless, our result does not rule out the possibility that the control of NF-κB activity by NAD(P)H oxidase is important for regulating CCL2 production in other cell types.
Akt has been shown to be activated by ROS through PI3Kdependent mechanisms.43 ROS have been implicated in the PI3K/Akt pathway by the activation of PI3K or the inactivation of PTEN, which dephosphorylates Akt.44,45 Activation of Akt has also been shown to regulate IL-1β–induced CCL2 production by inducing the activation of AP-1 but not NF-κB in RPE cells.46 In our present study, we provide the first evidence that NAD(P)H oxidase activity is critical for TNF-α–induced Akt activation because this activation was completely abolished by the inhibition of NAD(P)H oxidase. In addition, our results also suggest that Akt is involved in the TNF-α-NAD(P)H oxidase CCL2 signal because inhibiting Akt almost completely blocked TNF-α–induced CCL2 production. In contrast to Akt, our study ruled out the role of MAPKs in this human EC because the blockade of ERK and JNK did not reduce TNF-α–induced CCL2 production (data not shown) and the inhibition of NAD(P)H oxidase did not change the activation of p38MAPK. Akt activation is beneficial because it protects cells from death. However, our finding suggests that blocking Akt activation in Ecs may be useful in reducing inflammation by decreasing oxidative stress-elicited CCL2 production. An analogous role has been demonstrated by showing that the blockade of Akt activation prevents VEGF-induced vascular hyperpermeability and consequently blocks brain swelling.47 Further studies are needed to determine how to fine-tune the NAD(P)H oxidase/Akt pathway to prevent inflammation while maintaining the beneficial role of Akt in retinal diseases.
Acknowledgments
The authors thank Sylvia Smith for providing rMC-1 cells and Erhard Bieberich for providing pNF-κB-luc and pRL-CMV plasmids.
Supported by National Eye Institute Grants R01 EY04618 and R01 EY11766; Veterans Administration Career Development Award and VA Merit Review Award (RBC); National Heart, Lung, and Blood Institute Grant R01 HL70215 (RWC); and the Greater Southeast Affiliate’s Postdoctoral Fellowship AHA0725604B (WZ).
Footnotes
Disclosure: W. Zhang, None; M. Rojas, None; B. Lilly, None; N.-T. Tsai, None; T. Lemtalsi, None; G.I. Liou, None; R.W. Caldwell, None; R.B. Caldwell, None
References
- 1.Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86(4):363–365. doi: 10.1136/bjo.86.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198(3):483–489. doi: 10.1084/jem.20022027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishida S, Yamashiro K, Usui T, et al. Leukocytes mediate retinal vascular remodeling during development and vaso-obliteration in disease. Nat Med. 2003;9(6):781–788. doi: 10.1038/nm877. [DOI] [PubMed] [Google Scholar]
- 4.Koizumi K, Poulaki V, Doehmen S, et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Invest Ophthalmol Vis Sci. 2003;44(5):2184–2191. doi: 10.1167/iovs.02-0589. [DOI] [PubMed] [Google Scholar]
- 5.Antonetti DA, Barber AJ, Bronson SK, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55(9):2401–2411. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 6.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95(9):858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 7.Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101(3):234–247. doi: 10.1161/CIRCRESAHA.107.151860b. [DOI] [PubMed] [Google Scholar]
- 8.Randolph GJ, Furie MB. A soluble gradient of endogenous monocyte chemoattractant protein-1 promotes the transendothelial migration of monocytes in vitro. J Immunol. 1995;155(7):3610–3618. [PubMed] [Google Scholar]
- 9.Gerszten RE, Garcia-Zepeda EA, Lim YC, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398(6729):718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 10.Rollins BJ, Walz A, Baggiolini M. Recombinant human MCP-1/JE induces chemotaxis, calcium flux, and the respiratory burst in human monocytes. Blood. 1991;78(4):1112–1116. [PubMed] [Google Scholar]
- 11.Karpus WJ, Lukacs NW, Kennedy KJ, et al. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol. 1997;158(9):4129–4136. [PubMed] [Google Scholar]
- 12.Gosling J, Slaymaker S, Gu L, et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest. 1999;103(6):773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuaillon N, Shen DF, Berger RB, et al. MCP-1 expression in endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 2002;43(5):1493–1498. [PubMed] [Google Scholar]
- 14.Yoshida S, Yoshida A, Ishibashi T, Elner SG, Elner VM. Role of MCP-1 and MIP-1α in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J Leukoc Biol. 2003;73(1):137–144. doi: 10.1189/jlb.0302117. [DOI] [PubMed] [Google Scholar]
- 15.Nagata M. Inflammatory cells and oxygen radicals. Curr Drug Targets Inflamm Allergy. 2005;4(4):503–504. doi: 10.2174/1568010054526322. [DOI] [PubMed] [Google Scholar]
- 16.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98(4):453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 17.Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8(5–6):691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 18.El-Remessy AB, Behzadian MA, Abou-Mohamed G, et al. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol. 2003;162(6):1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis EA, Guberski DL, Somogyi-Mann M, Grant MB. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 2000;28(1):91–101. doi: 10.1016/s0891-5849(99)00216-6. [DOI] [PubMed] [Google Scholar]
- 20.Al-Shabrawey M, Rojas M, Sanders T, et al. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008;49(7):3239–3244. doi: 10.1167/iovs.08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith LE, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35(1):101–111. [PubMed] [Google Scholar]
- 22.Liou GI, Auchampach JA, Hillard CJ, et al. Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Invest Ophthalmol Vis Sci. 2008;49:5526–5531. doi: 10.1167/iovs.08-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarthy VP, Brodjian SJ, Dutt K, et al. Establishment and characterization of a retinal Muller cell line. Invest Ophthalmol Vis Sci. 1998;39(1):212–216. [PubMed] [Google Scholar]
- 24.Behzadian MA, Windsor LJ, Ghaly N, et al. VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB J. 2003;17(6):752–754. doi: 10.1096/fj.02-0484fje. [DOI] [PubMed] [Google Scholar]
- 25.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11(1):95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 26.Yamagishi S, Inagaki Y, Nakamura K, et al. Pigment epithelium-derived factor inhibits TNF-alpha–induced interleukin-6 expression in endothelial cells by suppressing NADPH oxidase-mediated reactive oxygen species generation. J Mol Cell Cardiol. 2004;37(2):497–506. doi: 10.1016/j.yjmcc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25(6):2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardiner TA, Gibson DS, de Gooyer TE, et al. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am J Pathol. 2005;166(2):637–644. doi: 10.1016/s0002-9440(10)62284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mo JS, Matsukawa A, Ohkawara S, Yoshinaga M. Involvement of TNF alpha, IL-1 beta and IL-1 receptor antagonist in LPS-induced rabbit uveitis. Exp Eye Res. 1998;66(5):547–557. doi: 10.1006/exer.1997.0451. [DOI] [PubMed] [Google Scholar]
- 30.Spirin KS, Saghizadeh M, Lewin SL, et al. Basement membrane and growth factor gene expression in normal and diabetic human retinas. Curr Eye Res. 1999;18(6):490–499. doi: 10.1076/ceyr.18.6.490.5267. [DOI] [PubMed] [Google Scholar]
- 31.Ueda A, Okuda K, Ohno S, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153(5):2052–2063. [PubMed] [Google Scholar]
- 32.Haddad JJ. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal. 2002;14(11):879–897. doi: 10.1016/s0898-6568(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 33.Xia P, Gamble JR, Rye KA, et al. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci U S A. 1998;95(24):14196–14201. doi: 10.1073/pnas.95.24.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goebeler M, Kilian K, Gillitzer R, et al. The MKK6/p38 stress kinase cascade is critical for tumor necrosis factor-alpha–induced expression of monocyte-chemoattractant protein-1 in endothelial cells. Blood. 1999;93(3):857–865. [PubMed] [Google Scholar]
- 35.Murao K, Ohyama T, Imachi H, et al. TNF-alpha stimulation of MCP-1 expression is mediated by the Akt/PKB signal transduction pathway in vascular endothelial cells. Biochem Biophys Res Commun. 2000;276(2):791–796. doi: 10.1006/bbrc.2000.3497. [DOI] [PubMed] [Google Scholar]
- 36.Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167(2):599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116(pt 22):4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 38.Salcedo R, Ponce ML, Young HA, et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96(1):34–40. [PubMed] [Google Scholar]
- 39.Nakazawa T, Hisatomi T, Nakazawa C, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A. 2007;104(7):2425–2430. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brueckmann M, Marx A, Weiler HM, et al. Stabilization of monocyte chemoattractant protein-1-mRNA by activated protein C. Thromb Haemost. 2003;89(1):149–160. [PubMed] [Google Scholar]
- 41.Gorospe M, Kumar S, Baglioni C. Tumor necrosis factor increases stability of interleukin-1 mRNA by activating protein kinase C. J Biol Chem. 1993;268(9):6214–6220. [PubMed] [Google Scholar]
- 42.Lin FY, Chen YH, Tasi JS, et al. Endotoxin induces toll-like receptor 4 expression in vascular smooth muscle cells via NADPH oxidase activation and mitogen-activated protein kinase signaling pathways. Arterioscler Thromb Vasc Biol. 2006;26(12):2630–2637. doi: 10.1161/01.ATV.0000247259.01257.b3. [DOI] [PubMed] [Google Scholar]
- 43.Ushio-Fukai M, Alexander RW, Akers M, et al. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274(32):22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 44.Qin S, Chock PB. Implication of phosphatidylinositol 3-kinase membrane recruitment in hydrogen peroxide-induced activation of PI3K and Akt. Biochemistry. 2003;42(10):2995–3003. doi: 10.1021/bi0205911. [DOI] [PubMed] [Google Scholar]
- 45.Cruz CM, Rinna A, Forman HJ, et al. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282(5):2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bian ZM, Elner SG, Yoshida A, Elner VM. Differential involvement of phosphoinositide 3-kinase/Akt in human RPE MCP-1 and IL-8 expression. Invest Ophthalmol Vis Sci. 2004;45(6):1887–1896. doi: 10.1167/iovs.03-0608. [DOI] [PubMed] [Google Scholar]
- 47.Kilic E, Kilic U, Wang Y, et al. The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 2006;20(8):1185–1187. doi: 10.1096/fj.05-4829fje. [DOI] [PubMed] [Google Scholar]