

Abstract
During corticogenesis, the balance between the self-renewal of radial glial stem cells and the production of their descendent progenitor cells is essential in generating the correct size and cell composition of the neocortex. How the stem-to-progenitor cell transition is regulated is poorly understood. FGFs are commonly implicated in promoting proliferation of neural precursor cells, but it is unclear how they exert their effects on stem cells, progenitor cells, or both in vivo. Here, three FGF receptor genes are simultaneously deleted during cortical neurogenesis. In these mutants, radial glia are depleted due to an increased transition from an uncommitted state to a more differentiated one, initially causing an increase in progenitors, but ultimately resulting in a smaller cortex. The proliferation rate of progenitors themselves, however, is unchanged. These results indicate that FGFs normally repress the radial glia to progenitor cell transition during corticogenesis.
Introduction
Achieving the correct size, shape, and cell composition for a tissue is essential for its optimal functioning. The size and complexity of the neocortex, for example, underlies elaborate behaviors. At the onset of neocortical neurogenesis, neuroepithelial cells become radial glial cells (RGCs), a transition that is in part promoted by Fgf10 (Sahara and O'Leary, 2009). Whereas neuroepithelial cells divide symmetrically to expand their numbers, RGCs likely act as stem cells that divide increasingly in an asymmetric manner to self-renew and generate restricted intermediate progenitor cells (IPCs) and neurons (Kriegstein et al., 2006; Merkle and Alvarez-Buylla, 2006). When comparing different mammals, the generation of increased numbers of IPCs underlies increased cortical complexity and size (Kriegstein et al., 2006). Thus the balance between self-renewal and progression to a more restricted state is critical in regulating the number of IPCs and ultimately cortical size. The mechanisms that regulate this progression, however, are poorly understood.
FGF2 is used as a mitogen for cultured cortical precursor cells (Kilpatrick and Bartlett, 1993; Vescovi et al., 1993). However, precursors cultured with growth factors can change their multipotency, masking their original state in vivo (Doetsch et al., 2002). Furthermore, infusion of growth factors in vivo can have the same effect (Doetsch et al., 2002). There are few clear examples of loss-of-function studies demonstrating a role for FGFs in regulating mitotic activity in the developing brain in vivo (Mason, 2007). In Fgf2−/− mice the number of dorsal neuroepithelial cells is reduced to half of the normal number before the start of neurogenesis. Conversely, the injection of FGF2 into embryonic cerebral ventricles produces an increase in cortical size (Vaccarino et al., 1999; Raballo et al., 2000).
These studies demonstrate the importance of FGF2 for the neuroepithelium, but leave key questions unanswered. First, in Fgf2−/− mice the defect in the early neuroepithelium is likely to partly account for the later cortical phenotype, leaving unresolved whether FGFs are needed during neurogenesis to maintain RGCs, IPCs, or both. Second, Fgf2 was deleted from all cells, leaving open the possibility of indirect effects from surrounding non-neural tissue. And third, other FGF ligands are expressed and may function redundantly masking a greater role for FGFs.
Here, to address the issue of indirect effects of surrounding tissues on cortical neurogenesis and to establish a cell-autonomous mode of action, FGF receptor rather than ligand genes are deleted in all cortical precursors or in a mosaic pattern. In both cases a conditional genetic approach is used in which Cre-recombination occurs after neurogenesis has started bypassing the requirement for FGF signaling in cortical patterning and neuroepithelial expansion (Raballo et al., 2000; Fukuchi-Shimogori and Grove, 2001; Garel et al., 2003; Paek et al., 2009). Moreover, due to potential functional redundancy between both FGF ligands and receptors, all three FGF receptor genes that are expressed in cortical precursors, Fgfr1, Fgfr2, and Fgfr3 are deleted. The consequences of abolishing FGF signaling for RGC self-renewal, IPC proliferation, cell survival, and neuronal differentiation are analyzed in vivo and in vitro.
Materials and Methods
Generation of mutant animals.
The mutant Fgfr alleles and the hGFAP-Cre transgenic mice were described previously (Deng et al., 1996; Zhuo et al., 2001; Hébert et al., 2003; Malatesta et al., 2003; Yu et al., 2003) (The Jackson Laboratory, stock 004600). Mice were genotyped by PCR as described previously (Zhuo et al., 2001). Noon on the day of the plug is E0.5. Control embryos were either hGFAP-Cre negative or heterozygous GFAP-Cre;Fgfr1flox/+;Fgfrflox/+;Fgfr3+/−, neither of which exhibit the phenotypes described in this study.
Tissue preparation and histology.
Embryos were either embedded freshly in OCT and frozen or fixed overnight in 4% paraformaldehyde, 4°C, cryoprotected in 20% sucrose, and embedded in OCT. Sections (12 μm) were stained with 0.1% cresyl violet (Nissl staining). E18.5 coronal sections were selected at three points along the anterior–posterior axis: anterior, located at the point where the corpus callosum crosses the hemispheres and the hippocampal commissure appears; middle, located between the hippocampal commissure and where the medial–lateral habenular and the ventral–lateral thalamic nuclei appear; posterior, located posterior to the ventral–lateral thalamic nucleus.
RNA in situ hybridization.
In situ hybridization on frozen sections was performed using 35S-labeled antisense probes as described previously (Frantz et al., 1994). Plasmids used to make probes for Erm (Chotteau-Lelièvre et al., 1997), Er81 (generous gift from J. Rubenstein, University of California, San Francisco, San Francisco, CA), Hes5 (Akazawa et al., 1992), Pax6 (Walther and Gruss, 1991), Svet1 (Tarabykin et al., 2001), and Cux2 (Quaggin et al., 1996) have been described previously.
Immunohistochemistry and fluorescence microscopy.
Immunohistochemistry on cryosections (12 μm) of either fresh-frozen or paraformaldehyde-fixed brain tissue were performed according to standard methods with overnight primary antibody incubations at 4°C and 1 h room temperature secondary antibody incubations (Texas Red or FITC-conjugated, 1:200, Jackson ImmunoResearch). The primary antibodies that were used are as follows: rabbit anti-PH3 (1:200, Millipore), rabbit TuJ1 (1:200, Covance), mouse anti-PCNA (1:500, Sigma), rat anti-BrdU (1:1000, Oxford Biotechnology), rabbit anti-TBR2 (1:1000, Millipore Bioscience Research Reagents), mouse anti-PAX6 (1:500, Hybridoma Bank), mouse anti-Numb (1:1000, generous gift from Weimin Zhong, Yale University, New Haven, CT), rabbit anti-β-catenin (1:200, Millipore), rabbit anti-Caspase3 (1:200, Cell Signaling Technology), mouse anti-N-cadherin (1:2, Hybridoma Bank), mouse anti-NeuN (1:100, Millipore), rabbit anti-TBR1 (1:500, Millipore Bioscience Research Reagents), mouse anti-PAR-3 (1:2, Hybridoma Bank), and rabbit anti-OLIG2 (1:400, Millipore). Sections were analyzed with a Zeiss Axioskop2 Plus microscope, except for BrdU/TuJ1 colabeling experiments, which were analyzed with a confocal fluorescence Zeiss LSM 5 Duo Scanner.
BrdU and TUNEL labeling.
Pregnant dams received an intraperitoneal injection of BrdU, 100 μg/g body weight, 1 h before they were killed. For cell cycle exit assays, pregnant E12.75 or E14.5 females were injected with BrdU, and embryos were collected at E13.5 and E15.5, respectively, and processed for immunohistochemistry. Sections were denatured in 2 N HCl for 1 h at room temperature followed by neutralization in 0.1 m sodium borate, pH 8.5, before incubation with the primary antibody. For double labeling with PCNA or Ki67, before blocking in goat serum, sections were heated in 10 mm sodium citrate at pH 6.0 in a microwave oven for antigen retrieval. Anti-PCNA or anti-Ki67 and the FITC-conjugated secondary antibodies were incubated before labeling with BrdU antibody. For double labeling with TuJ1, TBR2, or PAX6, immunohistochemistry was performed using the standard protocol for the respective antibodies followed by labeling with the anti-BrdU antibody. TUNEL staining was performed following the manufacturer's protocol (Roche).
MAP kinase assay.
E15.5 posterior cortices were dissected. Cells were dissociated and cultured in 37°C/5% CO2 for 60 min in DMEM supplemented with N2. Each sample was divided into two tubes and FGF (50 ng/ml) was added to one of the tubes and incubated for 5 min before collecting cells. Cells were resuspended in SDS loading buffer and boiled before Western blot was performed using anti-Erk1/2 (rabbit 1:500, Cell Signaling Technology) and anti-phospho-Erk1/2 (rabbit 1:500, Cell Signaling Technology) antibodies.
In utero electroporation. The Cre recombinase coding sequence was subcloned in a pCAG expression vector upstream of an IRES-GFP cassette. The pCAG-GFP construct was used as control vector. The vectors were delivered into the lateral ventricle of E13.5 telencephalons in vivo using a glass micropipette. In utero electroporation was performed as previously described (Bultje et al., 2009). Embryos were collected at E15.5 for immunohistochemistry.
Cumulative BrdU labeling.
Cumulative BrdU labeling was performed by repeated intraperitoneal injections of E15.5 pregnant females performed at 3 h intervals with 3 mg BrdU/injection. Mice were killed 30 min after the last BrdU injection (except for the 1 h time point, which was 60 min) and embryos were collected for anti-BrdU immunostaining of sections through the dorsal telencephalon. The labeling index for each time point was determined as the proportion of BrdU+ cells with respect to the precursor pool (PCNA+ cells). Tc (cell cycle length) and Ts (s-phase length) were estimated according to the method previously described (Nowakowski et al., 1989).
Neurosphere assay.
Neurospheres were derived from mechanically dissociated single cells from posterior cortices of E13.5 embryos. Ten thousand cells per milliliter were plated in 96-well (100 μl/well) and 6-well (3 ml/well) uncoated plates in basal serum-free media (DMEM/F12 supplemented with N2, B27, glutamine, and 1% pen/strep) containing heparin (2 μg/ml) and either FGF2 (20 ng/ml), EGF (20 ng/ml), or both. Primary neurospheres were passaged after 4–5 d to form secondary neurospheres and cultured for 5–6 additional days. BrdU (3 μg/ml) was added to the culture 1.5 h before collecting and fixing neurospheres in 4% PFA for 30 min at room temperature. Neurospheres were sunk in 20% sucrose and embedded in OCT for cryosectioning and immunostaining.
Quantification and statistical analysis.
Fluorescent images were captured on a Zeiss Axioskop2 Plus fluorescence microscope with Axiovision version 4.4 software or Zeiss LSM 5 Duo confocal microscope with LSM image browser. All the measurements were done using sections that match the same anterior–posterior level of control and mutant littermate brains. The statistical analysis was performed using Student's t test. Both hemispheres of at least three matching sections from each brain were used for measurement and compared between mutant and control littermates using more than one litter. Except for phosphohistone-H3+ cells (see below), cells were counted within a radial segment containing ∼200 Hoechst+ cells spanning from the ventricular surface to the pia (not included). The radial segments are centered midway between the pallial–subpallial ventricular zone (VZ) boundary and the dorsalmost bend of the cortical VZ at the interhemispheric fissure. PH3+ cells were counted along the whole length of the dorsal ventricle in both hemispheres from four matched sections from the posterior telencephalon and normalized to the length of the ventricular surface. To count the number of BrdU-labeled PAX6+ and TBR2+ cells, cells were counted in an ∼100-μm-wide radial segment of the entire cortical thickness (normalized to 100 μm).
The length of the cortical surface of the lateral ventricle was determined in Nissl-stained sections by measuring the distance as shown between the arrows in Figure 2B. At least five sections spanning from anterior to posterior regions and both hemispheres of each section were measured and compared between control and mutant brains from three pairs of littermates. The thickness of the cortex was measured from a similar set of sections as described for the ventricular length above. The position corresponds to the bars shown in Figure 2B (midway between the pallial–subpallial VZ boundary and the dorsalmost bend of the cortical VZ at the interhemispheric fissure) and the measurement spans from the surface of the ventricle to the pia. The thickness of the proliferative zone was also determined in Nissl-stained coronal brain sections. The measurement was performed again midway between the pallial–subpallial VZ boundary and the dorsal VZ bend. At least three sections and both hemispheres from each of the anterior, medial, and posterior telencephalon regions were used. The thickness of the neuronal layer (TuJ1 immunostaining) was determined in the same position using a similar set of sections.
Figure 2.
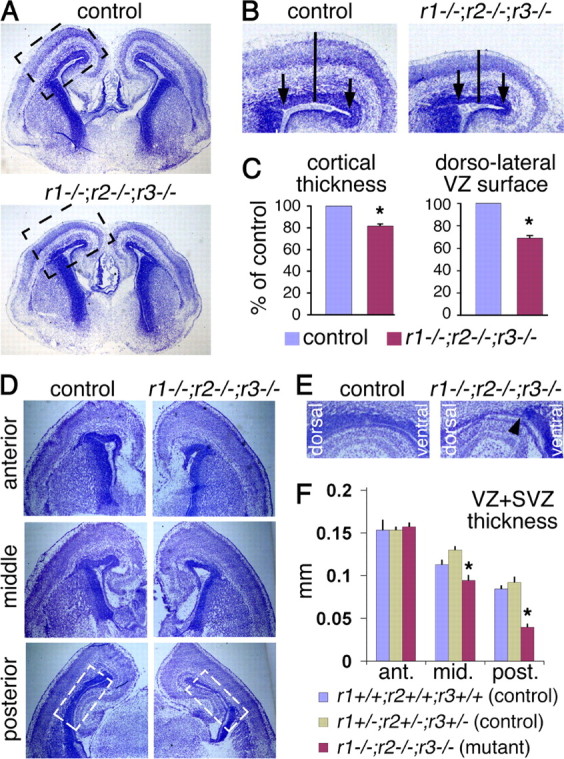
Loss of FGF signaling leads to a thinner and shorter cortex and a thinner cortical proliferative layer at E18.5. A, Cresyl violet stain of coronal brain sections from a control and Fgfr triple mutant. The mutant appears to have a smaller cortex. B, Enlargement of the boxed cortical areas in A. Solid bars illustrate the location where the thickness of the cortex is measured and the distance between the arrows indicates the length of the dorsolateral VZ surface. C, Quantification of cortical thickness and dorsolateral VZ length. D, Coronal sections along the anterior–posterior axis of the telencephalon reveal a decrease in the thickness of the proliferative layer (dark blue). E, Enlargement of the boxed area in the posterior sections shown in D; the arrowhead in the triple mutant marks the border between the dorsal and ventral proliferative zones, suggesting a sharp decrease in proliferating cells specifically in the dorsal telencephalon. F, Quantification of the thickness of the proliferative layer along the anterior–posterior axis in a control that lacks Cre and is essentially wild-type (blue), a control carrying Cre but heterozygous for the floxed alleles of Fgfr1, 2, and the null allele of Fgfr3 (green), and a triple Fgfr mutant (red).
Quantitative RT-PCR.
The dorsal telencephalons from E13.5 embryos were used to extract total RNA. Two mutants and two controls were used in each case. Standard RT-PCR was performed using primers for GAPDH, Tbr2, Cux2, Svet1, and Pax6 mRNA and amplification was performed for 27, 30, 33, 36, and 39 cycles within the same PCR run. Equal amounts of cDNA were used for both control and mutant samples. After electrophoresis, the products were analyzed by ImageJ software. Relative intensity of each band was calculated using the GAPDH band from the same PCR cycle as a standard.
Results
hGFAP-Cre-mediated deletion of FGF receptor genes leads to loss of FGF signaling
FGFs are a family of 22 ligands which signal through four tyrosine kinase receptors (Dono, 2003; Mason, 2007). To directly investigate the roles of FGF signaling in regulating corticogenesis, Fgfr1, Fgfr2, and Fgfr3 were deleted specifically in cortical precursor cells after the onset of neurogenesis in mice. Since Fgfr4 expression is not detected in the neocortical precursors throughout development (Fig. 1D and data not shown), simultaneously knocking out Fgfr1, 2, and 3 should lead to a complete loss of FGF signaling. Mice carrying an hGFAP-Cre transgene were used to conditionally knock out Fgfr1 and Fgfr2 in an Fgfr3-null background (Fig. 1A). The hGFAP promoter directs expression of Cre recombinase specifically in precursor cells of the dorsal telencephalon and cerebellum starting at ∼E13 resulting in recombined floxed alleles in most or all precursor cells and their progeny from this developmental stage forward (Zhuo et al., 2001; Malatesta et al., 2003). This approach allows us to bypass the requirements for FGF signaling in earlier patterning events (Mason, 2007) and focus on its requirement in cortical neurogenesis, specifically in distinguishing the roles of FGF signaling in RGCs versus IPCs.
Figure 1.

Deletion of FGF receptor genes in the developing cortex leads to loss of FGF signaling. A, Cross used to obtain mutants in which three Fgfr genes are simultaneously missing from the developing cortex. B–F, RNA in situ hybridization on coronal sections of control and double Fgfr1;Fgfr2 mutant brains. Expression of Fgfr1 (B, E) and Fgfr2 (C, F) (white grains) is lost specifically in the dorsal telencephalon at E13.5 (B, C) and E15.5 (E, F). D, Fgfr4 is not expressed in the telencephalon. G, H, Expression of Erm and Er81, downstream reporters for FGF signaling, is specifically lost from the E15.5 dorsal telencephalon of Fgfr triple mutants. I, Western blot showing phosphorylation of ERK (P-ERK) in response to FGF2 in control but not Fgfr triple mutant E15.5 cortical cells in culture.
The hGFAP-Cre;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3−/− mutants (henceforth referred to as Fgfr triple mutants) are recovered at the expected Mendelian ratio until E18.5 but die perinatally. To determine the efficiency of recombination, RNA in situ hybridization analysis using Fgfr1 and Fgfr2 probes complementary to the sequences between the loxP sites was performed for E12.5, E13.5, and E15.5 embryos. At E12.5, expression of Fgfr1 and Fgfr2 was unaffected, and no phenotype could be detected (supplemental Fig. 1, available at www.jneurosci.org as supplemental material; and data not shown). At E13.5 and E15.5, however, high expression of Fgfr1 and Fgfr2 was detected in the telencephalic proliferative areas surrounding the ventricles of controls, but not hGFAP-Cre;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3+/+ or hGFAP-Cre;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3+/− double mutants (Fig. 1B,C,E,F). Transcripts for both receptors were lost specifically in the dorsal telencephalon. To confirm loss of FGF signaling in Fgfr triple mutants, expression of Erm and Er81, which are induced by FGF signaling (Minowada et al., 1999; Münchberg and Steinbeisser, 1999; Raible and Brand, 2001), was examined. At E13.5, expression of Erm is specifically absent from the dorsal telencephalon of Fgfr triple mutants (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). At E15.5, expression of both Erm and Er81 is detectable throughout the telencephalic proliferative areas in control embryos, whereas in the Fgfr triple mutants, it is lost from the dorsal telencephalon and maintained ventrally (Fig. 1G,H). Moreover, consistent with a loss of FGF signaling in cortical precursor cells, dissociated E15.5 cortical cells from controls respond to FGF2 by phosphorylating ERK (MAPK) in culture, whereas mutant cells fail to do so (Fig. 1I).
Loss of FGF signaling leads to a smaller cortex and a thinner proliferative area
For the experiments described below, no significant differences could be observed between triple heterozygous hGFAP-Cre;Fgfr1+/fx;Fgfr2+/fx;Fgfr3+/− mice and mice that did not carry hGFAP-Cre, which are functionally wild type (Fig. 2F and data not shown). Therefore, for the analyses described below, “control” refers to either triple heterozygous littermates or littermates that are hGFAP-Cre negative.
Cresyl violet staining of sections reveals that although the gross morphology of the Fgfr triple mutant neocortex is normal at E18.5, the size of the hemispheres appears reduced compared with that of control littermates (Fig. 2A). More specifically, a significant reduction in the length of the ventricular surface along the dorsolateral axis and in the radial thickness of the cortex is observed (Fig. 2B,C). Unlike the Fgfr triple mutant, all three combinations of Fgfr double mutants appeared normal in size (data not shown), suggesting that the three FGF receptors can compensate for each other with respect to regulating cortical size.
One possible cause of the reduced cortical size in the E18.5 Fgfr triple mutants is a reduction in the number of precursor cells. Consistent with this possibility, the combined thickness of the two proliferative layers, the VZ and subventricular zone (SVZ), which cannot be readily distinguished by cresyl violet staining, is significantly reduced chiefly in the posterior cortical regions (Fig. 2D–F). The reason for the more apparent phenotype in the posterior regions is not clear, but may reflect a gradient in Fgfr expression or function or perhaps more likely the normal anterior–posterior developmental gradient of corticogenesis (Bayer and Altman, 1987). Nevertheless, we focused subsequent analyses on mid to posterior regions. Remarkably, a sharp demarcation in the thickness of the proliferative layers of the cortex and of the ventral telencephalon was observed (Fig. 2E, arrowhead), presumably due to the loss of FGF receptors specifically in the dorsal telencephalon.
FGF signaling is required to maintain the number of RGCs but not IPCs
To examine whether the reduced thickness of the cortical proliferative zone observed at E18.5 is due to an earlier reduction in the numbers of RGCs, IPCs, or both, we first examined markers for RGCs and IPCs by 35S-RNA in situ hybridization. The RGCs, which likely act as stem cells during neurogenesis and whose cell bodies form the VZ, are marked by expression of Pax6 and Hes5 (Englund et al., 2005). At E13.5, the radial thickness of Hes5 and Pax6 expression in Fgfr triple mutants is similar to that observed in control embryos (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). In contrast, at E15.5, the thickness of the Hes5 and Pax6 expression domain is markedly reduced in the mutant dorsal telencephalon (Fig. 3A–C). Cux2 and Svet1 expression mark the SVZ, in which reside the cell bodies of the IPCs but not the RGCs (Tarabykin et al., 2001; Zimmer et al., 2004). Cortical expression of these markers is similar at E13.5 in both controls and mutants by in situ hybridization analysis (data not shown), although an increase is detected by semiquantitative PCR (supplemental Fig. 2D,E, available at www.jneurosci.org as supplemental material). At E15.5, the thickness of the expression domains for Cux2 and Svet1 is also similar (Fig. 3D,E), although there may be a slightly thicker domain of expression in the mutant (Fig. 3F). These results suggest that FGF signaling is required to maintain the numbers of RGCs but does not directly affect IPCs.
Figure 3.
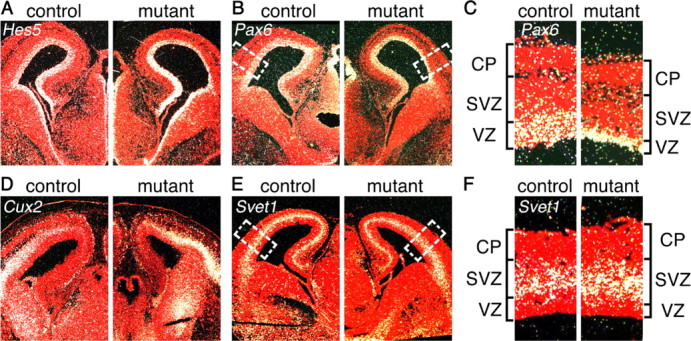
Reduced thickness of Hes5 and Pax6 RNA expression domains suggests a loss of radial glial stem cells in the Fgfr triple mutant. RNA in situ hybridization at E15.5 for Hes5 (A) and Pax6 (B), two VZ markers, and Cux2 (D) and Svet1 (E), two SVZ markers, indicates a smaller VZ in the Fgfr triple mutants compared with controls. C, Enlargement of the boxed areas in B, showing reduced thickness of the Pax6 expression domain. F, Enlargement of the boxed area in E showing only a slight increase, if any, in the thickness of the Svet1 expression domain. CP, Cortical plate.
To quantify and confirm these findings, we first performed immunostaining for RGCs using an anti-PAX6 antibody and for IPCs using an anti-TBR2 antibody (Englund et al., 2005). At E13.5, the number of PAX6+ cells was significantly decreased by ∼30% in the mutant dorsal telencephalon compared with controls (Fig. 4A) (92.0 ± 6.8 SE PAX6+ cells/100 μm of ventricular surface in mutants compared with 125.2 ± 6.0 in controls, p = 0.001). Although RNA in situ hybridization with a Pax6 probe failed to reveal this decrease (above), this is presumably due to the scattered 35S signal and the inability to count Pax6-expressing cells (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). At E15.5, there was an even greater loss of PAX6+ cells in the mutant (∼60% decrease) (Fig. 4C) (56.1 ± 2.5 PAX6+ cells/100 μm in mutants and 146.8 ± 5.0 in controls, p = 0.0001), in accordance with the RNA in situ results (Fig. 3C). Conversely, the number of TBR2+ cells, which are generated from PAX6+ cells, was found to be ∼40% higher at E13.5 and slightly higher at E15.5 in the Fgfr triple mutant compared with controls (Fig. 4B,D) (E13.5: 103.5 ± 5.5 TBR2+ cells/100 μm in mutants compared with 68.5 ± 3.9 in controls, p < 0.0001; E15.5: 108.3 ± 4.7 in mutants and 96.3 ± 3.5 in controls, p = 0.03). Consistent with this, the number of basal PH3+ cells, presumably IPCs undergoing mitoses, is significantly increased at E13.5 and only slightly increased at E15.5 (supplemental Fig. 3, yellow bars, available at www.jneurosci.org as supplemental material) (E13.5, 21% basal/total for control, 33% for mutant, p = 0.005; E15.5, 19% for control, 24% for mutant, not significant). In addition, the percentage of TBR2+ cells incorporating BrdU after a 1 h pulse is the same in controls and mutants at E15.5 (supplemental Fig. 4A–C, available at www.jneurosci.org as supplemental material), suggesting that FGF signaling is not required to maintain the proliferation of IPCs. Second, to further quantify expression levels, we performed semiquantitative RT-PCR for Pax6, Tbr2, Cux2, and Svet1 at E13.5 and found that consistent with the results above, Pax6 mRNA is detectably decreased whereas Tbr2, Cux2, and Svet1 are increased in the mutant compared with control (supplemental Fig. 2D,E, available at www.jneurosci.org as supplemental material). Together, these results demonstrate a requirement for FGF signaling in maintaining RGCs, but not IPCs.
Figure 4.
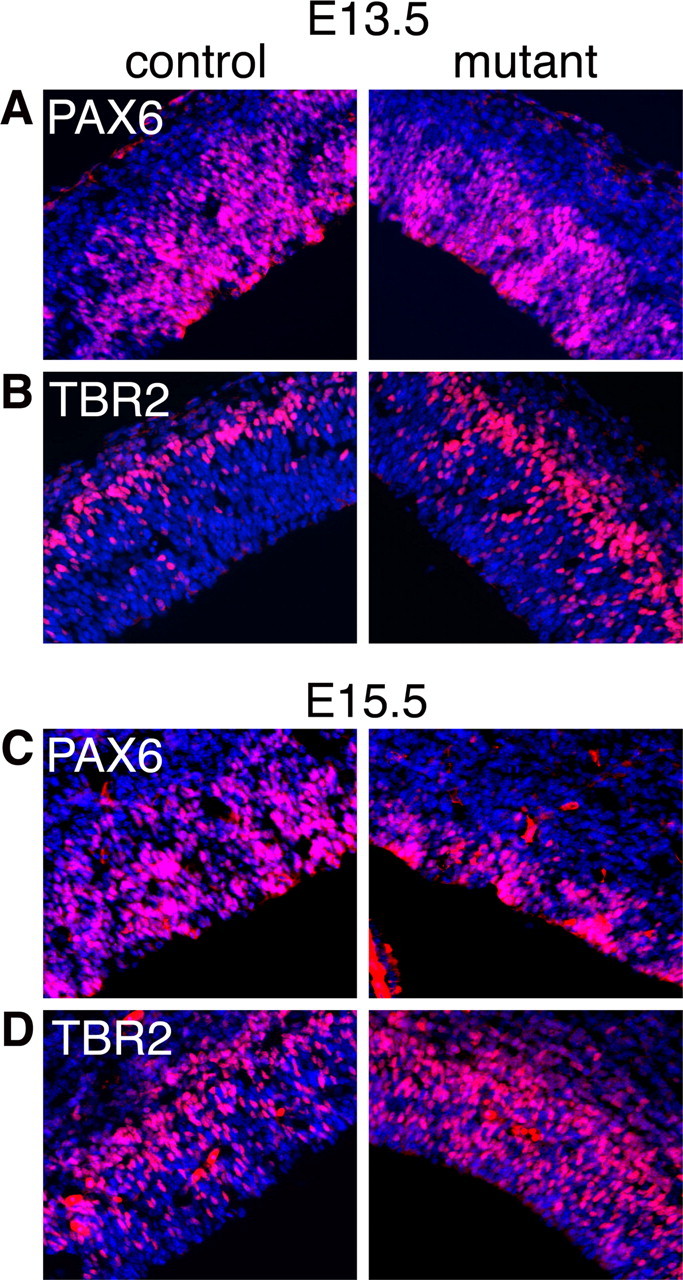
The number of PAX6+, but not TBR2+ cells is reduced in the Fgfr triple mutant. Immunohistochemistry using anti-PAX6 (A, C) or anti-TBR2 (B, D) antibodies on coronal sections of E13.5 (A, B) and E15.5 (C, D) control and mutant cortexes. Cell counts per 100-μm-wide radial segments (see Materials and Methods) reveal significant decreases and increases in the numbers of PAX6+ and TBR2+ cells, respectively, at both ages.
FGF signaling is required to maintain the self-renewal of neurosphere-forming cells
To examine the properties of control and Fgfr triple mutant cells in culture, cells from the posterior regions of E13.5 cortices were dissociated and cultured in the presence of EGF, FGF2, or EGF and FGF2. Control cells gave rise to larger and more primary neurospheres in response to FGF2 than triple Fgfr mutant cells (Fig. 5). Importantly, zero secondary neurospheres could be derived from the mutant primary neurospheres treated with FGF2, whereas these were readily derived from the control primary neurospheres (data not shown), suggesting that FGF signaling is required for self-renewal or maintenance of stem-like cells in culture. The nature of the control and mutant neurospheres treated with FGF2 also differed, with mutant neurospheres containing far fewer BrdU+ cells and more TuJ1+ cells compared with controls (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material), consistent with precursor cells progressing to a more differentiated state. EGF on its own had little effect on control cells, but when it was provided in combination with FGF2 it increased the size, but not the number, of primary neurospheres (Fig. 5). EGF on its own or with FGF2 also had little effect on mutant cells.
Figure 5.

Fgfr triple mutant neurospheres do not grow or propagate. A, FGF2 increases the number and the size of control, but not Fgfr triple mutant, primary neurospheres derived from E13.5 cortices. EGF has little effect on either the control or mutant, whereas EGF + FGF2 increases the size but not the number of neurospheres in controls but not mutants. B, Quantification of neurosphere numbers and size similar to those shown in A. Size refers to the two-dimensional area of pictured neurospheres.
It was not possible to detect the expression of Pax6 or Tbr2 in either control or mutant neurospheres treated with FGF2, perhaps because they are ventralized based on expression of Olig2 (supplemental Fig. 5C, available at www.jneurosci.org as supplemental material) (Hack et al., 2004), a ventral telencephalic marker (Takebayashi et al., 2000). This is consistent with a role for FGFs in ventralizing telencephalic precursor cells in vivo (for review, see Hébert and Fishell, 2008). Nevertheless, the reduced number of mutant primary neurospheres and the absence of any FGF-derived secondary neurospheres suggest that FGFs are required to maintain stem-like cells in vitro, similar to their requirement in maintaining RGCs in vivo. Moreover, the lack of a difference in the size or the number of EGF-treated neurospheres between mutants and controls as well as the increase in the size but not the number of control neurospheres treated with FGF2+EGF compared with FGF alone suggests that, whereas FGF signaling maintains stem (or neurosphere-forming) cells, EGF promotes the proliferation of more restricted progenitors.
RGC survival, organization, and cell cycle length appear normal in Fgfr mutants
To determine the cause of the reduced number of PAX6+ VZ cells in the Fgfr triple mutants, we examined several mechanisms that could account for their loss. The first is that FGF signaling may be required for the survival of RGCs, as is the case for all early (E9) telencephalic precursor cells (Paek et al., 2009). Both TUNEL analysis and Caspase 3 immunostaining revealed no differences between Fgfr triple mutants and controls in the number of apoptotic cells in the E13.5 and E15.5 dorsal telencephalon, which was very low in all cases (data not shown). Therefore, FGF signaling is not required for cell survival at these stages.
A second possibility is that the cell cycle length is increased in Fgfr mutant cells, leading to a slower rate of self-renewal. However, a cumulative BrdU incorporation experiment revealed that the cell cycle and the S-phase lengths for PCNA+ cortical cells are similar in E15.5 controls and mutants (Fig. 6) with cell cycle times of 20.6 and 21.0 h for controls and mutants, respectively, and S-phase lengths of 7.8 and 7.7 h, respectively [note that previous measurements indicated a shorter cell cycle time of 15.1 h and an S-phase length of 3.1 h (Takahashi et al., 1993), which may differ from the results presented here because we used E15.5 rather than E14.5 and the quantification method we used includes all PCNA+ cells in a radial column rather than only cells within a certain distance from the ventricle]. Moreover, despite a decrease in the absolute number of PAX6+ VZ cells (Fig. 4A,C), the percentage of PAX6+ cells that incorporate BrdU at E15.5 is unchanged (supplemental Fig. 4D–F, available at www.jneurosci.org as supplemental material), suggesting that the proliferative index for these precursors is unchanged. The finding that the proliferative kinetics for all precursors and the percentage of PAX6+ cells that incorporate BrdU are both unchanged is consistent with there also being no change in the fraction of TBR2+ cells that incorporate BrdU (supplemental Fig. 4A–C, available at www.jneurosci.org as supplemental material). Therefore, the cell cycle length of RGCs is unlikely to be affected, similar to the lack of any difference in cell cycle kinetics for Fgf2−/− mutant neuroepithelial cells (Vaccarino et al., 1999).
Figure 6.

The overall proliferative kinetics of E15.5 precursors are not affected by loss of FGF signaling. A, Representative coronal sections of control and mutant cortices double-immunolabeled for PCNA (green) and BrdU (red) administered 1 h before sacrifice, which is the first time point in the graph in B. B, Cumulative BrdU incorporation assay indicating that there is no change in the cell cycle length or length of S phase in mutant cells compared with controls (see Materials and Methods for details).
A third possibility is that the organization of RGCs along the ventricle is disrupted in the Fgfr triple mutants, indirectly resulting in a reduction in their numbers. Disruption of the apical–basal polarity or adherens junctions of radial glial stem cells can result in their conversion to IPCs or their delamination into the ventricle, respectively (Li et al., 2003; Machon et al., 2003; Cappello et al., 2006). To examine whether loss of FGF signaling perturbs the structural organization of the VZ, we examined the distribution of the adherens junction molecules N-cadherin and β-catenin, and the polarity determinants Numb and Par3 (Zhong et al., 1996; Lin et al., 2000; Takeichi, 2007) at E13.5 and E15.5 by immunohistochemistry. The distribution of these proteins in the Fgfr triple mutant was similar if not identical to that in controls (supplemental Fig. 6, available at www.jneurosci.org as supplemental material; and data not shown), suggesting that the structure of the VZ upon loss of FGF signaling is intact. Consistent with this, in the mutant telencephalon at E13.5 and E15.5, most M-phase nuclei (phospho-histoneH3+) localized normally to the ventricular surface (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
FGF signaling inhibits RGCs from progressing into IPCs or neurons
Another possible explanation for the observed decrease in the number of RGCs is that FGF signaling is required to maintain these cells in a stem-like state and inhibit their progression toward more differentiated states such as IPCs and neurons. If this is the case, then shortly after loss of FGF signaling we would expect an initial and premature increase in the number of IPCs and/or neurons in the mutants compared with controls. At later stages a decrease in IPCs and neurons would be expected as the decrease in stem cells eventually translates to fewer overall differentiated progeny. Our data confirm this explanation.
At E13.5 there is a ∼40% increase in the number of TBR2+ IPCs in mutants compared with controls (Fig. 4B). To test whether this increase is due to newly generated IPCs, BrdU was administered 16 h before collecting the embryos at E13.5. Colabeling for TBR2 and BrdU revealed a ∼30% increase in the percentage of double-positive TBR2/BrdU cells in mutants (Fig. 7A) (44.9 ± 3.2 SE % BrdU+TBR2+/BrdU+ cells in mutants compared with 34.6 ± 2.9% in controls, p < 0.02), indicative of an initial premature increase in the production of IPCs. This increase probably underrepresents the effect of losing FGF signaling because at the time of BrdU administration (∼E12.75) some cells may not have lost signaling yet. Nevertheless, there is an unambiguous increase in newly born IPCs.
Figure 7.
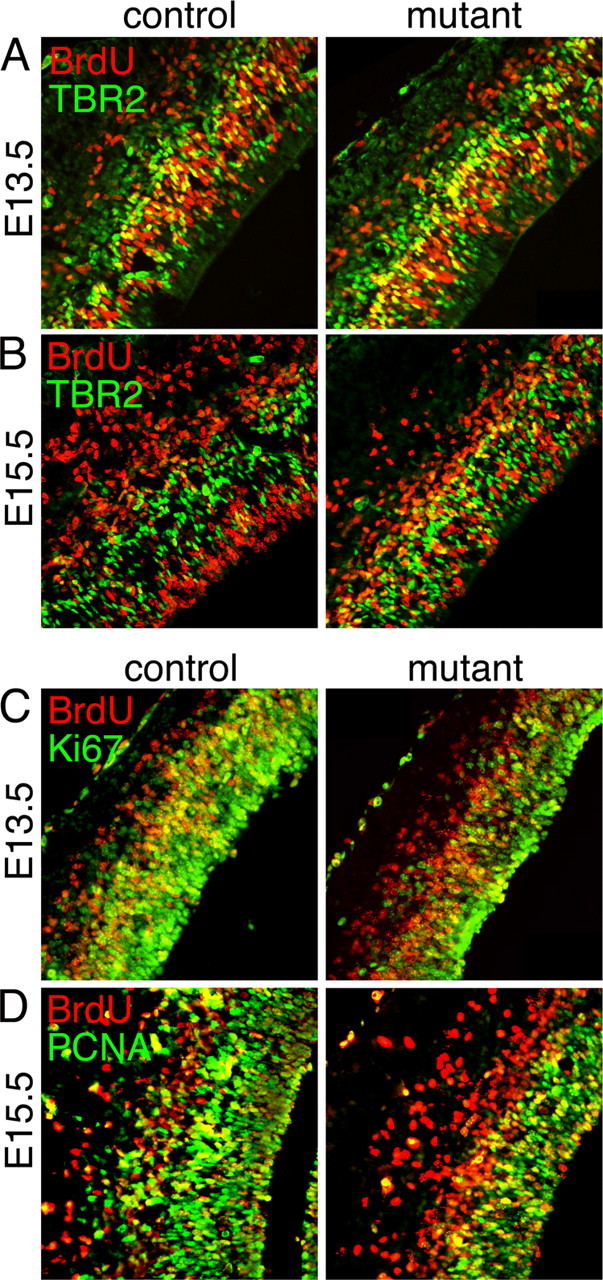
An increase in the percentage of newly generated TBR2+ cells and postmitotic cells is observed in the E13.5 and E15.5 triple mutant cortices. BrdU was administered to pregnant mothers 16 (A, C) or 24 (B, D) hours before collecting E13.5 and E15.5 embryos, respectively. Coronal sections of control and mutant cortices were colabeled for BrdU and TBR2 (A, B), BrdU and Ki67 (C), or BrdU and PCNA (D), revealing an increase in the mutant of the percentage of both newly generated TBR2+BrdU+ cells and cells exiting the cell cycle (BrdU+Ki67− or BrdU+PCNA−).
At E15.5, with an administration of BrdU 24 h before collecting the embryos, the percentage of TBR2+BrdU+ colabeled cells is increased by ∼75% in mutants compared with controls (Fig. 7B) (39.4 ± 2.2% BrdU+TBR2+/BrdU+ cells in mutants compared with 22.8 ± 1.5% in controls, p < 0.0001). The absolute number of TBR2+ cells at this stage, however, is only modestly increased (∼15%) (Fig. 4D), consistent with the idea that although RGCs are increasingly prone to generate IPCs as they lose FGF signaling, the absolute number of IPCs generated is gradually decreasing as PAX6+ RGCs quickly become depleted in the mutant (∼60% depletion by E15.5) (Fig. 4; see also Fig. 3C).
Despite an overall decrease in the size of the cortical plate by E18.5 (Fig. 2), an early increase in the production of postmitotic neurons is observed at E13.5. To quantify the number of postmitotic cells being generated, BrdU was administered 16 and 24 h before collecting E13.5 and E15.5 embryos, respectively. Sections were labeled with BrdU and PCNA or BrdU and Ki67, another marker for cycling cells. The percentage of BrdU+Ki67− or BrdU+PCNA− cells over the total number of BrdU+ cells, which is the fraction of cells exiting the cell cycle, is increased by ∼85% and ∼75% at E13.5 and E15.5, respectively, in the mutants compared with controls (Fig. 7C,D) (E13.5: 27.7 ± 4.5 SE % BrdU+Ki67−/BrdU+ cells in mutants compared with 15.2 ± 2.3% in controls, p < 0.02; E15.5: 40.1 ± 1.9% BrdU+PCNA−/BrdU+ cells in mutants and 23.5 ± 1.6 in controls, p < 0.0001). This increase in cell cycle exit is accompanied by an early increase in neuron production. Both the absolute thickness of the neuronal layer, marked by TuJ1 staining, and the number of neurons, marked by staining for TBR1, are significantly increased in the mutants compared with the controls at E13.5 (supplemental Fig. 7A,B, available at www.jneurosci.org as supplemental material). By E15.5, although the fraction of neurons generated from precursors within the previous 24 h (BrdU+TuJ1+/BrdU+ cells) remains high in the mutant compared with controls (supplemental Fig. 7E, available at www.jneurosci.org as supplemental material), the absolute thickness of the neuronal layer and the number of neurons (NeuN+) in the mutants is already slightly diminished or similar compared with controls (supplemental Fig. 7C,D, available at www.jneurosci.org as supplemental material). Together these data indicate that the decrease in RGCs observed in the Fgfr triple mutants is due to an increase in their progression toward a more differentiated state, resulting in an early increase in IPCs and neurons followed by an eventual overall decrease in their numbers due to stem cell depletion.
FGF signaling acts cell-autonomously to maintain RGCs
To test the hypothesis that the Fgfr genes are required cell-autonomously to maintain RGCs, a mosaic analysis was undertaken. Using in utero electroporation of a Cre-GFP plasmid into E15.5 Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3−/− embryos (that do not carry the hGFAP-Cre transgene), only electroporated GFP+ cells can become mutant for all three Fgfr genes. Surrounding nonelectroporated GFP− cells remain mutant only for Fgfr3. A GFP plasmid without Cre was used as a control.
Two days after E13.5 electroporations, the percentage of GFP+ cells that are also PAX6+ or BrdU+ is significantly reduced with Cre-GFP compared with GFP alone (Fig. 8). Conversely, the percentage of GFP+ cells that are also TBR2+ or TBR1+ is significantly increased with Cre-GFP compared with GFP (Fig. 8). Surrounding GFP− cells, however, are unaffected in all cases. Therefore, only cells which express Cre and are likely mutant for all three receptors show a change in fate, demonstrating a cell-autonomous role for Fgfr genes in maintaining RGCs.
Figure 8.

Mosaic loss of FGF signaling results in a cell-autonomous reduction of VZ cells and precocious neurogenesis. A, Representative coronal sections of E15.5 dorsomedial cortex of control and Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3−/− (no hGFAP-Cre) embryos after electroporation of either GFP or Cre-GFP plasmids at E13.5. BrdU was administered 1 h before collecting embryos. Immunostaining was done for PAX6, BrdU, TBR2 and TBR1. Overall there are consistently fewer GFP+ cells in the VZ in the presence of Cre. B, Quantification of colabeled cells over GFP+ cells (left column) revealed a lower percentage of PAX6+ and BrdU+ cells and a higher percentage of TBR2+ and TBR1+ cells with Cre-GFP. In GFP− cells (unelectroporated; right panel), the percentages of cells labeled with the different markers remains similar.
Discussion
In this report, three FGF receptor genes were conditionally deleted, leading to the loss of FGF signaling specifically in the dorsal telencephalon. Loss of signaling in these mutants occurs once neurogenesis has started allowing us to bypass its essential roles in patterning and expanding the cortical neuroepithelium at earlier ages. Our results provide evidence that, as for cell-intrinsic fate determinants such as CBF1 and Cdc42 for example (Cappello et al., 2006; Mizutani et al., 2007), extracellular signals regulate not simply the proliferation or survival of RGCs as previously shown, but specifically their progression to IPCs during neurogenesis in vivo.
During the development of any tissue, a balance between the self-renewal of stem cells and their progression to more differentiated states is critical in obtaining the correct size and cell composition of the final product. Hence an understanding of the factors that regulate this balance is key. Lineage tracing and real time imaging approaches have shown that cortical neuroepithelial cells divide exclusively in a symmetric mode before neurogenesis, expanding the neuroepithelium, and then become RGCs and divide increasingly in an asymmetric mode once neurogenesis begins, giving rise to another RGC and either a neuron or an IPC (Noctor et al., 2004; Götz and Huttner, 2005). IPCs themselves mainly divide symmetrically giving rise to neurons. Our data indicate that loss of FGF signaling during neurogenesis does not directly affect the proliferation of TBR2+ IPCs, but results specifically in a decrease in the number of PAX6+ RGCs due to an increased rate of progression to more differentiated states. Compared with controls, in the early Fgfr triple mutant (E13.5–E15.5), there is a greater production of IPCs and neurons as the number of RGCs is decreasing, indicating that RGCs are progressing to IPCs in greater numbers. Ultimately, due to the depletion of PAX6+ RGCs, their progeny also become fewer as evidenced by a smaller cortex at E18.5. The higher rate of progression of RGCs to IPCs does not appear to be due to a disruption of the VZ structure. Moreover, a similar result is observed in cultured mutant neurospheres. Together, our data implicate FGF signaling in cell-autonomously maintaining the self-renewal of PAX6+ VZ cells and inhibiting their conversion to a more committed SVZ fate. Since in the Fgfr triple mutants there is an abnormal early increase in the number of both IPCs and neurons concurrently, it remains unclear from our experiments what fraction of the additional neurons results directly from RGCs prematurely exiting the cell cycle to differentiate and what fraction results indirectly from the increased number of IPCs, which then differentiate into neurons.
Previous studies indicated that loss of Fgf2 results in a ∼50% decrease in cortical precursor cells before the onset of neurogenesis, and it was suggested that this decrease may be due to stem cells becoming quiescent (Vaccarino et al., 1999; Raballo et al., 2000). Evidence from our study, however, indicates that once neurogenesis has started the diminished numbers of Fgfr mutant radial glial stem cells result from their premature progression along the differentiation lineage rather than their entrance into quiescence. Namely, upon loss of FGF signaling, rather than a decrease in the number of IPCs and neurons which would occur concurrently with radial glia becoming quiescent, there is an initial increase in the numbers of newly born IPCs and neurons, indicative of premature differentiation. These findings are nevertheless reconcilable if FGF signaling functions to inhibit entrance into quiescence before neurogenesis and inhibits progression along a differentiation pathway once neurogenesis starts. In both cases, there would be an eventual depletion in neuronal numbers later in development and decrease in cortical size, as observed (Fig. 2) (Raballo et al., 2000).
In normal embryos, the FGF-responsive genes Erm and Er81 appear to be specifically expressed in the telencephalic VZ, with little or no expression in the SVZ (Fig. 1). This suggests that FGF signaling may only be active in VZ cells, i.e., radial glial cell bodies, and that the downregulation of FGF signaling may be required for the transition from radial glia to progenitor cell during cortical neurogenesis. This implies that other factors that are yet to be identified must act to counteract FGF signaling in establishing the balance between radial glial self-renewal and progression to an IPC fate.
The function of FGF signaling in maintaining the self-renewal and undifferentiated state of cortical stem cells is reminiscent of its role in the spinal cord stem zone. A balance between opposing FGF and retinoic acid (RA) signals regulates the onset of differentiation in this tissue (Diez del Corral and Storey, 2004). While RA drives differentiation in part by inhibiting Fgf8 expression, FGF signaling in turn inhibits retinoid synthesis to preserve the precursor cell pool in the stem zone (Diez del Corral and Storey, 2004). It is likely that similar mutually inhibitory mechanisms also act in the developing cortex, but whether or not they involve RA or other potential differentiation factors, such as BMPs or neurotrophins (Ghosh and Greenberg, 1995; Li et al., 1998), remains unknown.
Many factors, including transcription factors, miRNAs, chromatin remodeling enzymes, cell polarity determinants, and extracellular factors, have been implicated in maintaining neural stem cell self-renewal or neural precursor cell proliferation [for review, see Doe (2008) and Shi et al. (2008)]. Only a few studies have examined what regulates the stem cell to progenitor cell transition (Cappello et al., 2006; Mizutani et al., 2007; Quinn et al., 2007; Arnold et al., 2008; Sessa et al., 2008; Gauthier-Fisher et al., 2009). Numb, as an asymmetrically localized intracellular fate determinant, is of particular interest and is essential in regulating the balance between self-renewal and differentiation of cortical stem cells by promoting cell cycling (Zhong et al., 1996; Petersen et al., 2002, 2004). During normal development, FGF signaling could convey to stem cells in a permissive manner the potential to remain undifferentiated, while unequal inheritance by daughter cells of intrinsic factors such as Numb or Numbl would play a more instructive role. Conversely, even though FGF signaling does not appear to be required to regulate Numb localization (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), it may still play a more active role in regulating Numb activity.
FGF signaling appears to be continuously required for telencephalic and cortical development. However, its functions change depending on the developmental stage. At the time of initial telencephalon formation, simultaneous deletion of all three FGF receptor genes results in ablation of the telencephalon due to massive cell death (Paek et al., 2009). At this stage, FGF signaling probably also induces a telencephalic cell fate and patterns the telencephalon (Hébert and Fishell, 2008; Paek et al., 2009) and then the neocortex (Fukuchi-Shimogori and Grove, 2001; Garel et al., 2003). Before neurogenesis, FGF2 (and perhaps others) appears to directly maintain the proliferation of telencephalic neuroepithelial cells (Vaccarino et al., 1999; Raballo et al., 2000). Fgf10 then promotes the transition of neuroepithelial cells to RGCs at the onset of neurogenesis, at least in rostral areas (Sahara et al., 2009). The results presented here demonstrate that FGF signaling is subsequently required once neurogenesis starts to inhibit radial glial stem cells from progressing to a more differentiated state without directly affecting intermediate progenitors. Unlike at the stage of initial telencephalon formation, simultaneous deletion of all three FGF receptor genes during neurogenesis does not result in cell death. The different stage-specific requirements for FGF signaling may reflect either changes in the environment of the precursor cells or changes inherent to the cells themselves. It is of interest to identify the underlying causes for the differing stage-specific responses.
Footnotes
This work was supported in part by the James S. McDonnell Foundation for Brain Tumor Research, the Feinberg Foundation for Neurofibromatosis Research, National Institute of Mental Health Grant 70596, and the Alexandrine and Alexander L. Sinsheimer Foundation. We are grateful to our laboratory members for insightful discussions and comments on this manuscript; to Juha Partanen and Janet Rossant (Fgfr1), Kai Yu and David Ornitz (Fgfr2), and Chu-Xia Deng and Philip Leder (Fgfr3) for mice; to Weimin Zhong for his generous gift of the anti-Numb antibody; and to Solen Gokhan and Aldrin Molero for invaluable help with the neurospheres.
References
- Akazawa C, Sasai Y, Nakanishi S, Kageyama R. Molecular characterization of a rat negative regulator with a basic helix-loop-helix structure predominantly expressed in the developing nervous system. J Biol Chem. 1992;267:21879–21885. [PubMed] [Google Scholar]
- Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, Molnár Z, Robertson EJ, Groszer M. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008;22:2479–2484. doi: 10.1101/gad.475408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Directions in neurogenetic gradients and patterns of anatomical connections in the telencephalon. Prog Neurobiol. 1987;29:57–106. doi: 10.1016/0301-0082(87)90015-3. [DOI] [PubMed] [Google Scholar]
- Bultje RS, Castaneda-Castellanos DR, Jan LY, Jan YN, Kriegstein AR, Shi SH. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron. 2009;63:189–202. doi: 10.1016/j.neuron.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, Wilsch-Bräuninger M, Eilken HM, Rieger MA, Schroeder TT, Huttner WB, Brakebusch C, Götz M. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci. 2006;9:1099–1107. doi: 10.1038/nn1744. [DOI] [PubMed] [Google Scholar]
- Chotteau-Lelièvre A, Desbiens X, Pelczar H, Defossez PA, de Launoit Y. Differential expression patterns of the PEA3 group transcription factors through murine embryonic development. Oncogene. 1997;15:937–952. doi: 10.1038/sj.onc.1201261. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Diez del Corral R, Storey KG. Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays. 2004;26:857–869. doi: 10.1002/bies.20080. [DOI] [PubMed] [Google Scholar]
- Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Dono R. Fibroblast growth factors as regulators of central nervous system development and function. Am J Physiol Regul Integr Comp Physiol. 2003;284:R867–R881. doi: 10.1152/ajpregu.00533.2002. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz GD, Weimann JM, Levin ME, McConnell SK. Otx1 and Otx2 define layers and regions in developing cerebral cortex and cerebellum. J Neurosci. 1994;14:5725–5740. doi: 10.1523/JNEUROSCI.14-10-05725.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA. Neocortex patterning by the secreted signaling molecule FGF8. Science. 2001;294:1071–1074. doi: 10.1126/science.1064252. [DOI] [PubMed] [Google Scholar]
- Garel S, Huffman KJ, Rubenstein JL. Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development. 2003;130:1903–1914. doi: 10.1242/dev.00416. [DOI] [PubMed] [Google Scholar]
- Gauthier-Fisher A, Lin DC, Greeve M, Kaplan DR, Rottapel R, Miller FD. Lfc and Tctex-1 regulate the genesis of neurons from cortical precursor cells. Nat Neurosci. 2009;12:735–744. doi: 10.1038/nn.2339. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Hack MA, Sugimori M, Lundberg C, Nakafuku M, Götz M. Regionalization and fate specification in neurospheres: the role of Olig2 and Pax6. Mol Cell Neurosci. 2004;25:664–678. doi: 10.1016/j.mcn.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nat Rev Neurosci. 2008;9:678–685. doi: 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, Lin M, Partanen J, Rossant J, McConnell SK. FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development. 2003;130:1101–1111. doi: 10.1242/dev.00334. [DOI] [PubMed] [Google Scholar]
- Kilpatrick TJ, Bartlett PF. Cloning and growth of multipotential neural precursors: requirements for proliferation and differentiation. Neuron. 1993;10:255–265. doi: 10.1016/0896-6273(93)90316-j. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martínez-Cerdeño V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci. 2006;7:883–890. doi: 10.1038/nrn2008. [DOI] [PubMed] [Google Scholar]
- Li HS, Wang D, Shen Q, Schonemann MD, Gorski JA, Jones KR, Temple S, Jan LY, Jan YN. Inactivation of Numb and Numblike in embryonic dorsal forebrain impairs neurogenesis and disrupts cortical morphogenesis. Neuron. 2003;40:1105–1118. doi: 10.1016/s0896-6273(03)00755-4. [DOI] [PubMed] [Google Scholar]
- Li W, Cogswell CA, LoTurco JJ. Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by BMP. J Neurosci. 1998;18:8853–8862. doi: 10.1523/JNEUROSCI.18-21-08853.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience. 2003;122:129–143. doi: 10.1016/s0306-4522(03)00519-0. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Götz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Mason I. Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci. 2007;8:583–596. doi: 10.1038/nrn2189. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Alvarez-Buylla A. Neural stem cells in mammalian development. Curr Opin Cell Biol. 2006;18:704–709. doi: 10.1016/j.ceb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubüser A, Sun X, Hacohen N, Krasnow MA, Martin GR. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 1999;126:4465–4475. doi: 10.1242/dev.126.20.4465. [DOI] [PubMed] [Google Scholar]
- Mizutani K, Yoon K, Dang L, Tokunaga A, Gaiano N. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature. 2007;449:351–355. doi: 10.1038/nature06090. [DOI] [PubMed] [Google Scholar]
- Münchberg SR, Steinbeisser H. The Xenopus Ets transcription factor XER81 is a target of the FGF signaling pathway. Mech Dev. 1999;80:53–65. doi: 10.1016/s0925-4773(98)00193-2. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol. 1989;18:311–318. doi: 10.1007/BF01190834. [DOI] [PubMed] [Google Scholar]
- Paek H, Gutin G, Hébert JM. FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development. 2009;136:2457–2465. doi: 10.1242/dev.032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Hwang JK, Jan YN, Zhong W. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature. 2002;419:929–934. doi: 10.1038/nature01124. [DOI] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Krauss S, Zhong W. Continuing role for mouse Numb and Numbl in maintaining progenitor cells during cortical neurogenesis. Nat Neurosci. 2004;7:803–811. doi: 10.1038/nn1289. [DOI] [PubMed] [Google Scholar]
- Quaggin SE, Heuvel GB, Golden K, Bodmer R, Igarashi P. Primary structure, neural-specific expression, and chromosomal localization of Cux-2, a second murine homeobox gene related to Drosophila cut. J Biol Chem. 1996;271:22624–22634. doi: 10.1074/jbc.271.37.22624. [DOI] [PubMed] [Google Scholar]
- Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD, Price DJ. Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol. 2007;302:50–65. doi: 10.1016/j.ydbio.2006.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM. Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci. 2000;20:5012–5023. doi: 10.1523/JNEUROSCI.20-13-05012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raible F, Brand M. Tight transcriptional control of the ETS domain factors Erm and Pea3 by Fgf signaling during early zebrafish development. Mech Dev. 2001;107:105–117. doi: 10.1016/s0925-4773(01)00456-7. [DOI] [PubMed] [Google Scholar]
- Sahara S, O'Leary DDM. Fgf10 regulates transition period of cortical stem cell differentiation to radial glia controlling generation of neurons and basal progenitors. Neuron. 2009;63:48–62. doi: 10.1016/j.neuron.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron. 2008;60:56–69. doi: 10.1016/j.neuron.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sun G, Zhao C, Stewart R. Neural stem cell self-renewal. Crit Rev Oncol Hematol. 2008;65:43–53. doi: 10.1016/j.critrevonc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr Cell cycle parameters and patterns of nuclear movements in the neocortical proliferative zone of the fetal mouse. J Neurosci. 1993;13:820–833. doi: 10.1523/JNEUROSCI.13-02-00820.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Yoshida S, Sugimori M, Kosako H, Kominami R, Nakafuku M, Nabeshima Y. Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech Dev. 2000;99:143–148. doi: 10.1016/s0925-4773(00)00466-4. [DOI] [PubMed] [Google Scholar]
- Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. 2007;8:11–20. doi: 10.1038/nrn2043. [DOI] [PubMed] [Google Scholar]
- Tarabykin V, Stoykova A, Usman N, Gruss P. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development. 2001;128:1983–1993. doi: 10.1242/dev.128.11.1983. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:848. doi: 10.1038/12226. [DOI] [PubMed] [Google Scholar]
- Vescovi AL, Reynolds BA, Fraser DD, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron. 1993;11:951–966. doi: 10.1016/0896-6273(93)90124-a. [DOI] [PubMed] [Google Scholar]
- Walther C, Gruss P. Pax-6, a murine paired box gene, is expressed in the developing CNS. Development. 1991;113:1435–1449. doi: 10.1242/dev.113.4.1435. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhong W, Feder JN, Jiang MM, Jan LY, Jan YN. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron. 1996;17:43–53. doi: 10.1016/s0896-6273(00)80279-2. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- Zimmer C, Tiveron MC, Bodmer R, Cremer H. Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb Cortex. 2004;14:1408–1420. doi: 10.1093/cercor/bhh102. [DOI] [PubMed] [Google Scholar]