

Abstract
Effective kinetic resolution of α–aryl-, α–aryloxy-, and α–arylthioalkanoic acids has been achieved via in situ generation of their symmetrical anhydrides and enantioselective alcoholysis in the presence of homobenzotetramisole (HBTM) 3.
Keywords: acylation, anhydrides, carboxylic acids, kinetic resolution
Resolution of racemic carboxylic acids is commonly accomplished via fractional crystallization of their diastereomeric salts with chiral amines.[1] The need for the trial-and-error optimization of conditions, utilization of stoichiometric amounts of chiral resolving agents and repeated recrystallizations often detract from the convenience of this approach. One of the alternatives to this so-called Classical Resolution is Kinetic Resolution[2] (KR), which can be achieved via enantioselective alcoholysis of activated carboxylic acid derivatives in the presence of chiral catalysts. Although examples of this process have been known for some time, high selectivity factors[3] have been obtained in relatively few cases.[4] In contrast to the earlier studies using preformed acyl donors as substrates, two groups recently reported successful KR of carboxylic acids activated in situ. Ishihara et al.[5] utilized their histidine-derived enantioselective acylation catalyst to resolve carboxylic acids bearing a pyrrolidinocarboxy moiety at the α–position. Shiina et al.[6] successfully resolved several 2-arylpropionic acids using BTM 2, previously developed in our laboratory in the context of the KR of benzylic alcohols.[7b]
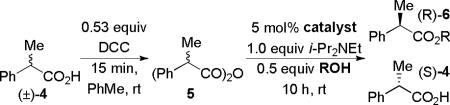
In both previous studies, the free acids were activated using equimolar amounts of condensing agents, such as pivaloyl chloride, DCC (Ishihara) or p-methoxybenzoic anhydride (Shiina). We reasoned that since kinetic resolutions are typically carried out to ca. 50% conversion, it should be sufficient to activate only one half of a substrate acid via in situ generation of its symmetrical anhydride. Treating racemic 2-phenylpropionic acid 4 with 0.53 equiv of DCC in toluene, as expected, resulted in rapid precipitation of dicyclohexylurea and clean formation of the anhydride,[8] presumably as a mixture of d,l- and meso-diastereomers. Alcoholysis of this mixture in the presence of catalysts 1, 2 and 3 was investigated next (Table 1).
Table 1.
Variation of the catalyst and alcohol substrate.
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | ROH | %conv[a] | s |
| 1 | Cl-PIQ 1 | i-PrOH | 20 | 1.9 |
| 2 | BTM 2 | i-PrOH | 5 | 1.7 |
| 3 | HBTM 3 | i-PrOH | 48 | 6.9 |
| 4 | HBTM 3 | PhCH2OH | 50 | 5.2 |
| 5 | HBTM 3 | Ph2CHOH | 53 | 9.3 |
| 6 | HBTM 3 | 1-Np2CHOH | 46 | 33 |
| 7 | Cl-PIQ 1 | 1-Np2CHOH | 49 | 2.6 |
| 8 | BTM 2 | 1-Np2CHOH | 17 | 24 |
Modest enantioselectivities were obtained using isopropanol (entries 1-3).[10] HBTM 3[7c] proved to be clearly superior to Cl-PIQ 1[7a] and BTM 2,[7b] in terms of both the reaction rate and enantioselectivity. Benzyl alcohol produced a lower selectivity factor than isopropanol (entry 4). On the other hand, diarylcarbinols, such as benzhydrol (entry 5) and especially di-(1–naphthyl)-methanol (entry 6) employed in Shiina's work,[6] led to significant improvement. Under the same conditions, Cl-PIQ 1 displayed very low enantioselectivity (entry 7), whereas BTM 2 (entry 8) produced a markedly lower reaction rate than HBTM 3.
Screening different solvents (Table 2, entries 1-5) led us to conclude that the initially chosen toluene was, in fact, optimal. Lowering the temperature to 0 °C (entry 6) increased the selectivity factor by ca. 10 %. Further cooling, however, did not lead to any improvement (entries 7 and 8).
Table 2.
Solvent and temperature effects[a]
| Entry | Solvent | temp | time, h | % conv | s |
|---|---|---|---|---|---|
| 1 | PhMe | 23 | 10 | 46 | 33 |
| 2 | CHCl3 | 23 | 10 | 39 | 24 |
| 3 | CH2Cl2 | 23 | 10 | 42 | 21 |
| 4 | THF | 23 | 10 | 28 | 20 |
| 5 | MeCN | 23 | 10 | 26 | 11 |
| 6 | PhMe | 0 | 24 | 45 | 36 |
| 7 | PhMe | −20 | 24 | 43 | 36 |
| 8 | PhMe | −40 | 24 | 39 | 29 |
Conditions: 1.0 equiv (±)-4 in the specified solvent was treated with 0.53 equiv DCC at rt for 15 min, then with 0.05 equiv of 3, 0.50 equiv 1-Np2CHOH, and 1.0 equiv i-Pr2NEt.
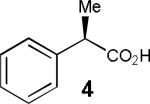
Having thus optimized the reaction conditions, we proceeded to investigate the substrate scope of the new method (Table 3). Racemic forms of nonsteroidal antiinflammatory drugs ibuprofen 7, naproxen 8, and flurbiprofen 9 were successfully resolved using the optimized set of conditions (entries 2-4). Increasing the size of the α-substituent from methyl to ethyl led to lower selectivities (entry 5). A similar observation was made in the case of α–methoxyphenylacetic acid 11 (entry 6). However, much to our surprise, the absolute stereochemistry of the fast-reacting enantiomer in this case proved to be the opposite of that observed in all previous cases. Intrigued by these findings, we investigated substrates 12-14 bearing an aryloxy or an arylthio group and obtained excellent selectivity factors (entries 7-9). It should be noted that the nonenzymatic KR of this type of substrates has not been previously reported.[11] On the other hand, the geometrically similar acid 15 reacted with barely detectable enantioselectivity (entry 10).
Table 3.
Substrate scope[a]
| Entry | (±)-substrate[b] | %eeSM/%eePR | s (% conv) |
|---|---|---|---|
| 1 |
|
72/88 | 36 (45) |
| 2 |
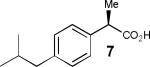
|
67/90 | 37 (43) |
| 3 |
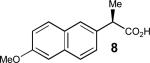
|
76/91 | 48 (46) |
| 4[c] |
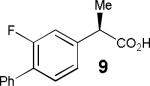
|
80/86 | 33 (48) |
| 5 |
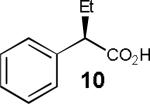
|
54/71 | 10 (43) |
| 6 |
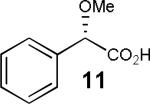
|
75/79 | 19 (49) |
| 7 |
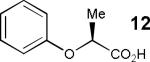
|
76/90 | 42 (45) |
| 8 |
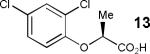
|
61/95 | 74 (39) |
| 9[d] |
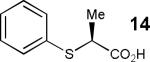
|
41/93 | 39 (31) |
| 10 |
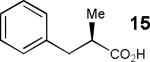
|
5/7 | 1.2 (40) |
Conditions: Same as in Table 2, entry 6, unless specified otherwise.
The absolute configuration of the fast-reacting enantiomer is shown.
Reaction time 2 d.
Reaction time 3 d.
Although the mechanism of chiral recognition in the process described above is not fully understood at present, a preliminary analysis of the experimental results is possible. The enantioselectivity clearly depends on the alcohol employed, which suggests that the first step of the catalytic cycle is rapid and reversible and that the enantiodiscrimination occurs in the second step (Figure 2).
Figure 2.

Proposed catalytic cycle.
By analogy with the previously proposed transition state for the KR of secondary benzylic alcohols,[12] the model shown in Figure 3 may be envisioned. The structure-selectivity trends observed so far are consistent with a Felkin-Ahn-like model (17b).[13] In the case of arylalkanoic acids (4, 7-10), R1 = Ar, R2 = Me or Et. However, when a small, electron-withdrawing substituent is introduced (cf. substrate 11), the situation is reversed: R1 = OMe, R2 = Ph. The reversed selectivity is increased further when R2 becomes smaller and R1 becomes larger and/or more electron-deficient (cf. 11 vs. 12-14).
Figure 3.

Proposed transition state model. The carboxylate anion is omitted for clarity.
In conclusion, we have developed a new protocol for the nonenzymatic KR of carboxylic acids via their symmetrical anhydrides, which compares favorably with previous methods[5,6] in terms of cost, experimental convenience, and enantioselectivity. Further investigations aimed at expanding the scope of this methodology and probing the validity of the proposed TS model are underway and will be reported in due course.
Experimental Section
Optimized procedure
N,N'-dicyclohexylcarbodiimide (22 mg, 0.106 mmol) was added to a stirring solution or suspension of the specified racemic acid substrate (0.20 mmol) in 1mL of toluene at room temperature. In the case of poorly soluble substrates, sonication was used to facilitate the reaction. After 15 min, i-Pr2NEt (35 μL, 0.2 mmol), and di-(1-naphthyl)methanol (28 mg, 0.10 mmol) were added, and the mixture was cooled in an ice bath to 0 °C for 5 min before adding HBTM (50 μL of 0.20 M stock solution in toluene). After stirring for 24 h at 0 °C (except for substrates 9 and 14, requiring 2 and 3 days, respectively), the reaction mixture was quenched by adding 1 mL of saturated aqueous NH4Cl. The aqueous layer was extracted with CH2Cl2 (3×2 mL). The organic layer was extracted with 1 M NaHCO3, dried with Na2SO4, concentrated, and then subjected to flash column chromatography (hexanes/EtOAc 20:1) to give the pure ester product. Acidification of the aqueous NH4Cl layer and/or the NaHCO3 extract to pH 2 gave the unreacted acid substrate. Ee's of the ester and the unreacted acid were obtained by chiral stationary phase HPLC analysis and used to calculate the % conversion and the selectivity factor according to Ref. [2a]. The results reported in each entry in Table 3 are averages of two runs.
Figure 1.

Amidine-based catalysts
Acknowledgements
This study was supported in part by NIGMS (NIH R01 GM072682). Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant No. P41RR0954).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.200######.
References
- 1.Faigl F, Fogassy E, Nógrádi M, Pálovics E, Schindler J. Tetrahedron Asymm. 2008;19:519. [Google Scholar]
- 2.a Kagan HB, Fiaud JC. Top. Stereochem. 1988;18:249. [Google Scholar]; b Vedejs E, Jure M. Angew. Chem., Int. Ed. 2005;44:3974. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 3.Selectivity factor is defined as S = k(fast-reacting enantiomer)/k(slow-reacting enantiomer).[2a] S values ≥ 20 are considered practically useful.[2b]
- 4.a Narasaka K, Kanai F, Okudo M, Miyoshi N. Chem. Lett. 1989:1187. [Google Scholar]; b Tian S-K, Chen Y, Hang J, Tang L, McDaid P, Deng L. Acc. Chem. Res. 2004;37:621. doi: 10.1021/ar030048s. [DOI] [PubMed] [Google Scholar]; c Notte GT, Sammakia T, Steel PJ. J. Am. Chem. Soc. 2005;127:13502. doi: 10.1021/ja053743z. [DOI] [PubMed] [Google Scholar]; d Notte GT, Sammakia T. J. Am. Chem. Soc. 2006;128:4230. doi: 10.1021/ja058010t. [DOI] [PubMed] [Google Scholar]
- 5.Ishihara K, Kosugi Y, Umemura S, Sakakura A. Org. Lett. 2008;10:3191. doi: 10.1021/ol801007m. [DOI] [PubMed] [Google Scholar]
- 6.Shiina I, Nakata K, Onda Y. Eur. J. Org. Chem. 2008:5887. [Google Scholar]
- 7.a Birman VB, Jiang H. Org. Lett. 2005;7:3445. doi: 10.1021/ol051063v. [DOI] [PubMed] [Google Scholar]; b Birman VB, Li X. Org. Lett. 2006;8:1351. doi: 10.1021/ol060065s. [DOI] [PubMed] [Google Scholar]; c Birman VB, Li X. Org. Lett. 2008;10:1115. doi: 10.1021/ol703119n. [DOI] [PubMed] [Google Scholar]
- 8.Chen FMF, Kuroda K, Benoiton NL. Synthesis. 1978:928. [Google Scholar]
- 9.The possibility of racemization/epimerization of intermediates under the reaction conditions has been ruled out by a control experiment. See Supporting Information.
- 10.A modified protocol using 1.2 equiv of DCC to produce the O-acylisourea instead of the anhydride proved to be much less effective. The conversion to the ester was low, presumably as a consequence of the N-acylurea formation (detected in the mixture after the reaction).
- 11.For examples of the enzymatic KR of α–aryloxy- and α–arylthiopropionic acids, see: Cipiciani A, Bellezza F, Fringuelli F, Stillitano M. Tetrahedron: Asymmetry. 1999;10:4599.Koul S, Koul JL, Singh B, Kapoor M, Parshad R, Manhas K, Taneja SC, Qazi GN. Tetrahedron: Asymmetry. 2005;16:2575.
- 12.a Birman VB, Uffman EW, Jiang H, Li X, Kilbane CJ. J. Am. Chem. Soc. 2004;126:12226. doi: 10.1021/ja0491477. [DOI] [PubMed] [Google Scholar]; b Li X, Peng L, Houk KN, Birman VB. J. Am. Chem. Soc. 2008;130:13836. doi: 10.1021/ja805275s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Cherest M, Felkin H, Prudent N. Tetrahedron Lett. 1968;9:2199. [Google Scholar]; b Anh NT, Eisenstein O. Nouv. J. Chim. 1977;1:61. [Google Scholar]; c Anh NT. Top. Curr. Chem. 1980;88:145. [Google Scholar]