

Abstract
The heme-thioether ligand interaction often occurs between heme iron and native methionine ligands, but thioether-based heme-coordinating (type II) inhibitors are uncommon due to the difficulty in stabilizing the Fe-S bond. Here, a thioether-based inhibitor (3) of neuronal nitric oxide synthase (nNOS) was designed, and its binding was characterized by spectrophotometry and crystallography. A crystal structure of inhibitor 3 coordinated to heme iron was obtained, representing, to our knowledge, the first crystal structure of a thioether inhibitor complexed to any heme enzyme. A series of related potential inhibitors (4-8) also were evaluated. Compounds 4-8 were all found to be type I (non-heme-coordinating) inhibitors of ferric nNOS, but 4 and 6-8 were found to switch to type II upon heme reduction to the ferrous state, reflecting the higher affinity of thioethers for ferrous heme than for ferric heme. Contrary to what has been widely thought, thioether-heme ligation was found not to increase inhibitor potency, illustrating the intrinsic weakness of the thioether-ferric heme linkage. Subtle changes in the alkyl groups attached to the thioether sulfur caused drastic changes in binding conformation, indicating that hydrophobic contacts play a crucial role in stabilizing the thioether-heme coordination.
Introduction
Iron-sulfur ligand interactions have been widely studied in heme-based enzymes because of their effect on reduction potential,1 involvement in O-O bond cleavage,2 and mediation of a fluxional process.3 The interaction between the heme iron and the native axial Met present in some heme enzymes is of particular interest because of its rarity in coordination chemistry4 and because of the inherently weak affinity of thioether for ferric iron relative to other iron-ligand interactions.1,4-6 Heme-coordinating (type II) inhibitors based on thiolate or thiol moieties have been reported,7-8 but crystal structures are scarce in the literature of thiol- or thiolate-based type II ligands coordinating to the heme in heme-thiolate proteins. Thioether-based type II inhibitors are far less common, although some have been reported for cytochromes P450.7,9 There are no crystal structures of thioether-based inhibitors coordinating to the heme in heme-thiolate enzymes. Structural information is available only for the natural thioether heme ligands in some heme-containing proteins, such as the axial ligand methionine in cytochrome c10 or the bis-methionine ligands found in the heme transporting protein, Shp.11
Nitric oxide synthases (NOS) are in a family of heme-dependent isozymes that catalyze the NADPH-dependent two-step conversion of L-arginine and two equivalents of molecular oxygen to L-citrulline and nitric oxide (NO),12 an important biological messenger molecule.13 Neuronal NOS (nNOS) is an important medicinal target for inhibitors since overproduction of NO by nNOS has been implicated in strokes,14 septic shock,15 seizures,16 schizophrenia,17 migraine headaches,18 and Alzheimer's disease.19 A few type II inhibitors of nNOS have been reported, but they are imidazole-based,20 and no crystal structure of an inhibitor bound as an axial ligand to nNOS heme has been published. The crystal structure of the “modified type II” inhibitor L-thiocitrulline bound to nNOS was reported,21 but the Fe-S distance of 4.0 Å is nearly twice as long as the linkage typically observed between heme iron and native Met ligands.22,23 Alkylated S-thiocitrullines were found to have no ligand interactions with the nNOS heme iron.24 In this paper, we report our efforts on the design, synthesis, characterization, and crystal structures of thioether based type II inhibitors of nNOS.
Results
Design and synthesis of 3-8
One strategy for designing type II inhibitors of heme enzymes is to attach an iron-binding moiety to a substrate backbone. This approach utilizes favorable contacts with the substrate binding pocket to fix the inhibitor in the active site, thereby positioning the potential heme-binding functionality within reach of the heme iron. This strategy has previously been applied to generate numerous imidazole-based type II inhibitors of heme enzymes.20,25,26 Recently, we employed this strategy to design ten analogues of the substrate L-arginine as potential type II inhibitors of nNOS, two of which were thioether-based (1 and 2).27 Despite the low affinity of thioethers for heme-iron compared to other iron-ligand interactions, it was thought that the Fe-S thioether linkage could increase inhibitor potency more effectively than a hydrogen bond or van der Waals contact.27 Each inhibitor in the series was found by difference spectrometry to be type I—that is, capable of displacing water from its position as a sixth axial heme ligand, thus converting the heme iron to a high-spin state, but unable to coordinate as a sixth axial ligand.
Knowing that 1 and 2 failed to show type II ligation to heme in contrast to what was predicted by the computer docking simulation,27 we analyzed the potential ligand-enzyme interactions according to structural information we had previously obtained on the hydrophobic pocket defined by Pro565, Val567, and Phe584. This pocket was found to accommodate small alkyl groups extending from inhibitors anchored in the active site by hydrogen bonding with the Glu592 carboxylate, thus further stabilizing inhibitor binding.28-29 In a type II binding conformation, the terminal methyl groups of 1 and 2 would be too small to make any direct van der Waals contacts with the protein, which might account for the failure of 1 and 2 to coordinate to the heme iron. We therefore designed compound 3, which has a terminal ethyl group, in order to establish better contact with the active site hydrophobic pocket. Once 3 was determined to be a type II ligand by UV-visible spectrophotometry and further confirmed by crystal structure (see below), a series of potential inhibitors (4-8, Scheme 1) were designed to investigate which features of the thioether tail of 3 were most important for type II binding.
Scheme 1.

Compounds 4 and 6 were designed to establish whether a terminal ethyl or propyl group could stabilize the Fe-S bond even without the flexibility afforded by the two-carbon linker between the amidine group and sulfur atom that 3 possesses. In 7 and 8 the terminal ethyl group of 3 was replaced by n-propyl and isopropyl groups, respectively, to determine whether the hydrophobic pocket formed by Pro565, Val567, and Phe584 could accommodate a slightly larger or bulkier alkyl group. Compound 5 was designed to test whether type II binding could still be achieved by an inhibitor in which the sulfur atom was moved to a position on the thioether tail farther from the amidine group.
Compounds 3-8 were synthesized by our earlier method (Scheme 1).27 The precursor nitriles, which were obtained through either Michael reactions involving acrylonitrile or SN2 reactions involving bromoacetonitrile, were converted to the corresponding ethyl imidic esters using standard Pinner synthesis conditions.30 The imidic esters were coupled with Boc-L-Orn-OH to generate the Boc-protected forms of 3-8, which were purified using cation exchange chromatography. The pure Boc-protected amidines were treated with 4 M aqueous HCl to yield 3-8 quantitatively.
Difference Spectrometry and Determination of Inhibition Constants
Type II inhibitors, such as imidazole, convert the predominantly high-spin heme of H4B –nNOS to the low-spin state, resulting in a red shift in the Soret absorption band and a “type II” visible difference spectrum, characterized by a peak at 430 nm and a trough at 395 nm.31 Conversely, inhibitors that convert the low-spin heme of the imidazole –nNOS to the pentacoordinate high-spin state (type I inhibitors) give rise to a “type I” difference spectrum, marked by a peak at 390 nm and a trough at 430 nm. Optical difference spectrophotometry was employed as described31 to examine the spectral binding constant, Ks, of 3-8 to the nNOS heme domain. Typical difference spectra are characterized by two distinct features: a peak (ΔAmax) and a trough (ΔAmin). A plot of (ΔAmax - ΔAmin) versus inhibitor concentration gives rise to a typical logarithmic binding isotherm, and the corresponding Hanes-Woolf plot forms a straight line with the x-intercept –Ks (see Supporting Information Fig. S1 for all difference spectra and plots). Therefore, Ks is effectively the same as KD, the dissociation constant. Compound 1, the most potent inhibitor in the series we reported earlier,27 was also synthesized and evaluated since the spectral constants (Ks) had not been previously determined. The heme domain of rat nNOS was used for all spectral titrations to eliminate interference from the absorption of flavins in the reductase domain and to directly correlate to the subsequent crystallographic characterization, which used the same heme domain sample. Compound 3 gave rise to a type II difference spectrum toward ferric nNOS (Figure 1), verifying that the thioether of 3 binds, as expected, to the heme iron as a sixth axial ligand. Compound 1 and 4-8 all exhibited type I difference spectra toward ferric nNOS, indicating that none of the inhibitors interacted with the ferric heme iron as an axial thioether ligand.
Figure 1.
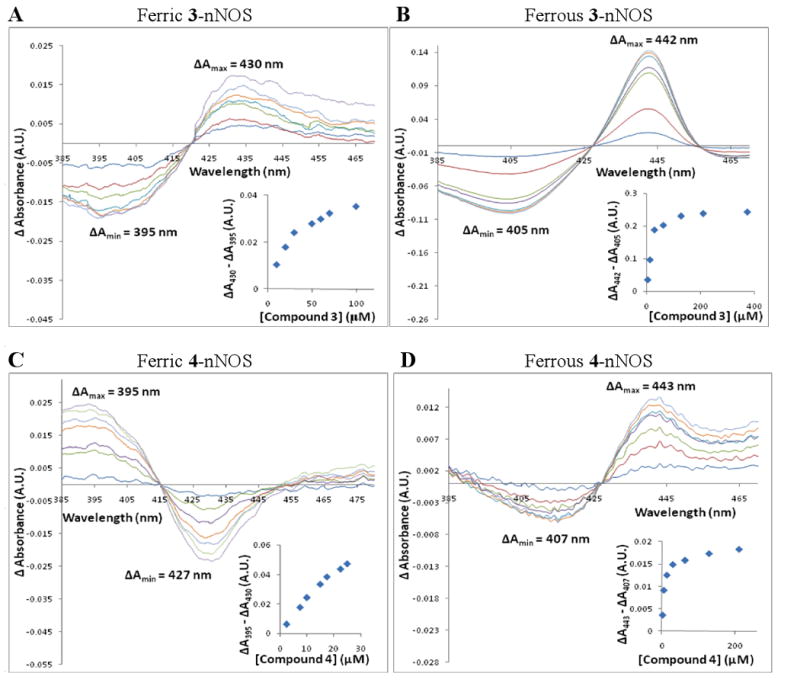
Ferric and ferrous difference spectra for 3 and 4. PANEL A: Titration of ferric nNOS-BH4 with 3. PANEL B: Titration of ferrous nNOS-BH4 with 3. PANEL C: Titration of ferric nNOS-imidazole with 4 (the concentration of imidazole was 300 μM). PANEL D: Titration of ferrous nNOS-BH4 with 4. In each chart, the absorption spectrum of either heme domain nNOS-BH4 (Panels A, B, and D) or heme domain nNOS-imidazole (Panel C) was set as the baseline. Aliquots of inhibitor solution were added sequentially; each curve represents the difference spectrum after addition of a particular aliquot. Spectra were normalized to zero absorbance at 420 (Panel A), 427 (B, D), and 415 nm (C), respectively. In each chart, distinct ΔAmax and ΔAmin values were observed, indicating conversion of the heme iron from high spin to low spin (Panels A, B, and D) or from low spin to high spin (Panel C). The insets are plots of (ΔAmax - ΔAmin) versus inhibitor concentration. The value (ΔAmax - ΔAmin) increased logarithmically with increasing concentration, as expected for binding isotherms. Hanes-Woolf plots were constructed for (inhibitor concentration)/(ΔAmax - ΔAmin) versus inhibitor concentration; the negative x-intercepts of the linear regressions were taken as the spectral constants (Ks).31 Based on the data shown above and data from replicate experiments, Ks values of 34 ± 2 (Panel A), 15.5 ± 0.1 (B), 15 ± 3 (C), and 8 ± 1 μM (D) were calculated. For ferric 4-nNOS, the Ks value was calculated from the apparent Ks value as described.32 Ferrous difference spectra were obtained under anaerobic conditions, and nNOS was reduced using 2-10 mM sodium dithionite. All titrations were performed in 100 mM Hepes buffer at pH 7.5. The concentration of nNOS heme domain was 3.75 μM in the ferric titrations and 2.4 μM in the ferrous titrations. For each titration, the total volume change was < 2%. The data shown are representative of at least two replicate experiments.
The Ks value of 3 was significantly lower than the Ks values of the other inhibitors except for 1 and 4, for which the Ks values were in the low μM range. Apparent Ks values for type I inhibitors were converted to actual Ks values as described.32 No spectral change occurred when the low-spin imidazole-nNOS complex was titrated with 3, and the predominantly high-spin H4B-nNOS became even more high-spin when titrated with 4-7, as evidenced by a blue shift in the Soret band (data not shown). Interestingly, 8 was found to give rise to a type II difference spectrum when added at very high concentrations to the predominantly high-spin H4B-nNOS (see Supporting Information Fig. S3). This observation suggests that when 8 binds to the nNOS active site, it produces a mixture of high-spin and low-spin states, as was reported for L-thiocitrulline.33
To investigate whether low Ks values correlated with high potency, IC50 values were determined as described34 for 3-8 against rat nNOS, and Ki values were calculated from the IC50 values (Table 1).35 In general, low spectral constants corresponded to low Ki values. Compound 4 was found to be significantly more potent (Ki = 8.6 ± 0.9 μM) than the other inhibitors, while 3, the only type II ligand in the series, was found to be moderately potent (Ki = 33 ± 2 μM). Compound 5 displayed potency comparable to that of 3 while 6-8 were considerably less potent. None of the inhibitors was found to be selective for nNOS over the other NOS isoforms, endothelial NOS (eNOS) and inducible NOS (iNOS) (data not shown). This lack of selectivity was not surprising since the NOS isozymes have very high sequence identity in the active site, and no features of 3-8 were expected to reach far outside the substrate binding pocket. These results are in line with the earlier finding that substrate analogues often fail to exhibit NOS isoform selectivity.36
Table 1.
Summary of Spectral and Inhibition Data obtained for 1 and 3-8
| R | Ki (μM) | Fe3+ type | Fe3+ Ks (μM)a | Fe2+ type | Fe2+ Ks (μM) | |
|---|---|---|---|---|---|---|
| Imidazole | -- | 29.7 ± 0.8 | II | 120 ± 20 | I | N/D |
| L-Nitroarginine | NHNO2 | 0.9 ± 0.2 | I | 1.07 ± 0.01 | I | N/D |
| 1 | CH2SCH3 | 5.9 ± 0.4 | I | 2.2 ± 0.2 | I | N/D |
| 3 | (CH2)2SCH2CH3 | 33 ± 2 | II | 34 ± 2 | II | 15.5 ± 0.1 |
| 4 | CH2SCH2CH3 | 8.6 ± 0.9 | I | 15 ± 3 | II | 8 ± 1 |
| 5 | (CH2)3SCH3 | 32 ± 4 | I | 170 ± 10 | N/D | N/D |
| 6 | CH3S(CH2)2CH3 | 59 ± 4 | I | 550 ± 50 | II | 22 ± 2 |
| 7 | (CH2)2S(CH2)2CH3 | >500 | I | 1940 ± 90 | II | 110. ± 9 |
| 8 | (CH2)2SCH(CH3)2 | >500 | I / II | 1010 ± 40b | II | 77 ± 8 |
For type I inhibitors, “actual Ks” values are shown, converted from apparent Ks based on the concentration of imidazole used.32
This is the type I Ks value, i.e., from titration of imidazole—nNOS.
All values are reported as the mean ± the standard deviation of 2-3 experiments.
Since the ferrous heme-thioether linkage is known to be considerably stronger than the corresponding ferric interaction,1 all spectrophotometric assays were repeated with ferrous nNOS to test whether the oxidation state of the heme iron affected inhibitor binding. Sodium dithionite was added in excess to reduce ferric heme to the ferrous state. Toward ferrous nNOS, 3, 7, and 8 generated typical type II spectral shifts with a peak at ∼443 nm and a trough around 405 nm (Figs. 1 and S1). The latter is the Soret peak position known for reduced ferrous iron in nNOS.
Compounds 4 and 6 also produced type II spectral shifts with ferrous nNOS, but unlike 3, 7, and 8, they failed to exhibit a clear isosbestic point. The peak position of 6 is around 430 nm rather than 443 nm, which is distinct from 3-4 and 7-8. Titration of ferrous nNOS with 1 gave only a trough at 423 nm while 5 showed an altered peak position at 424 nm and a trough at 402 nm. Apparently, the greater affinity of the thioether sulfur for ferrous heme caused 4 and 6-8 to switch from type I to type II ligands while 1 and 5 most likely do not have direct interaction from the sulfur to the ferrous heme iron. Ferrous Ks values were calculated for 3-4 and 6-8; in each case, the ferrous Ks was lower than its corresponding ferric value, indicating that tighter binding occurs when Fe2+-S bonds are formed. Compound 4 was found to have the lowest ferrous Ks (8 ± 1 μM), but the most substantial decreases in Ks from ferric to ferrous were observed for 6 (25-fold), 7 (18-fold), and 8 (13-fold).
Additionally, absolute absorption spectra were obtained over the entire visible range for 1 and 3-8, and for nNOS alone and L-arginine as controls (see Supporting Information Fig. S2 for all absolute absorption spectra). Ferrous thioether-heme complexes are known to give rise to split Q bands, 9,37-38 and this spectral feature was observed in the absolute absorption spectrum of the ferrous 3-nNOS complex from 530-580 nm (Figure 2). The sharp Soret band observed around 450 nm also resembles that of the CO bound, low-spin ferrous heme in a heme-thiolate protein. In fact, several heme-thiolate protein ligands, including CO, NO, thiocyanite, phosphine, pyridine, thiol, and thioether share the “hyperporphyrin” spectral features with intense double Soret peaks around 450 nm and 350-380 nm (data not shown). 9,38-39 Compounds 4 and 6-8 also exhibited the type II feature, but only as a shoulder to the high-spin Soret peak, which occurred around 412 nm, while for 3 the entire Soret peak shifted to 443nm with only a small shoulder at the shorter wavelength side. The type II shoulder for compounds 4 and 6-8 indicates that a mixture of high-spin and low-spin ferrous heme exists. Inhibitors inducing larger low-spin heme population exhibited larger shoulders at 443 nm. However, there seemed to be no direct correlation between the low-spin population and binding affinity. For instance, 6 had a rather small 443 nm shoulder (Fig. S2) and generated weak absorbance changes in the ferrous titration (Fig. S1), but its Ks value was much lower than those of 7-8, even though the latter showed more pronounced absorbance changes (Fig. S1).
Figure 2.
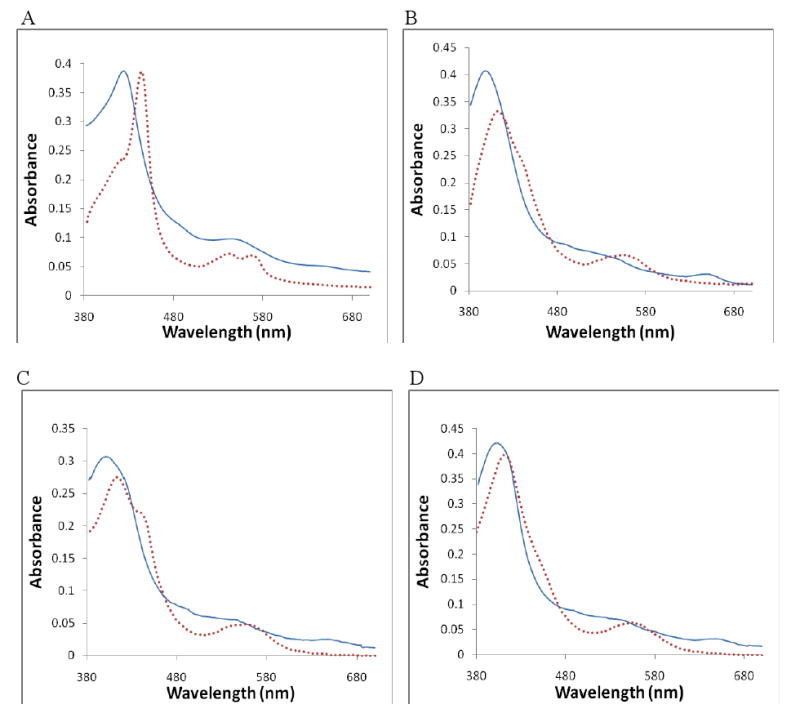
Absolute absorption spectra in the ferric and ferrous oxidation states for Panel A) 3 -nNOS; B) 4 -nNOS; C) 8 -nNOS; and D) H4B -nNOS with no inhibitor present as a control. In each panel, the solid line represents the ferric spectrum, and the dotted line represents the ferrous spectrum.
Crystal structures of nNOS inhibitor complexes
Crystal structures were obtained for 1 and 3-8 bound to the nNOS active site. Compounds 3, 4, 6 and 8 all exhibited continuous 2Fo-Fc density contoured at 1.0 σ between inhibitor and iron, even though 4, 6, and 8 were all found to generate type I high-spin spectra with ferric nNOS. Compounds 1, 5, and 7 exhibited no density that reflected interaction with the heme iron. Clear density was present for the arginine-like backbone of each inhibitor, indicating that they all made favorable contacts to the substrate binding site through extensive hydrogen bonds, but the density varied for the thioether alkyl tails.
Structural refinements were carried out for the nNOS-inhibitor complexes of 1, 3-6, and 8. The crystallographic statistics are summarized in Table S1 in Supporting Information. The structure for 3 was refined to 1.8 Å resolution. As expected by manually modeling of the inhibitor in the nNOS active site, the refined crystal structure of 3 revealed that the two methylene groups separating the amidine from the sulfur afforded the flexibility needed to place the sulfur directly over the heme iron (Figure 3A). The terminal ethyl group fits into the hydrophobic pocket formed by Pro565, Val567, and Phe584. The Fe-S distance is about 2.51Å in one molecule of the dimer and 2.59 Å in the second, slightly longer than the 2.35-2.42 Å Fe-S distance observed for methionine ligands in cytochrome c systems,22 yet significantly shorter than the 4.0 Å Fe-S non-bonded distance reported for L-thiocitrulline.21 Compounds 7 and 8 have even bulkier n-propyl and isopropyl groups, respectively, after the sulfur atom, which resulted in disorder of the tail end of the inhibitors. The 7-nNOS complex exhibited rather poor density for the inhibitor and was therefore not refined. Presumably, the linear three carbon tail is too long to fit into the hydrophobic pocket. Although an accurate Fe-S distance could not be determined because of poor density quality, our best estimate is greater than 3.0 Å (data not shown). Compared to the n-propyl group in 7, the branched, shorter isopropyl tail in 8 fits slightly better, although it was also disordered with no clear density for a single conformation. The inhibitor, therefore, was modeled and structure refined with two alternate conformations with equal occupancy (50% each), as shown in Fig. S4. Without good density, the isopropyl tail was modeled only tentatively at positions where it was not clashing with the protein. The closest Fe-S distance is 2.8 Å, while the alternate conformation gives a longer Fe-S distance of ∼4.9 Å.
Figure 3.
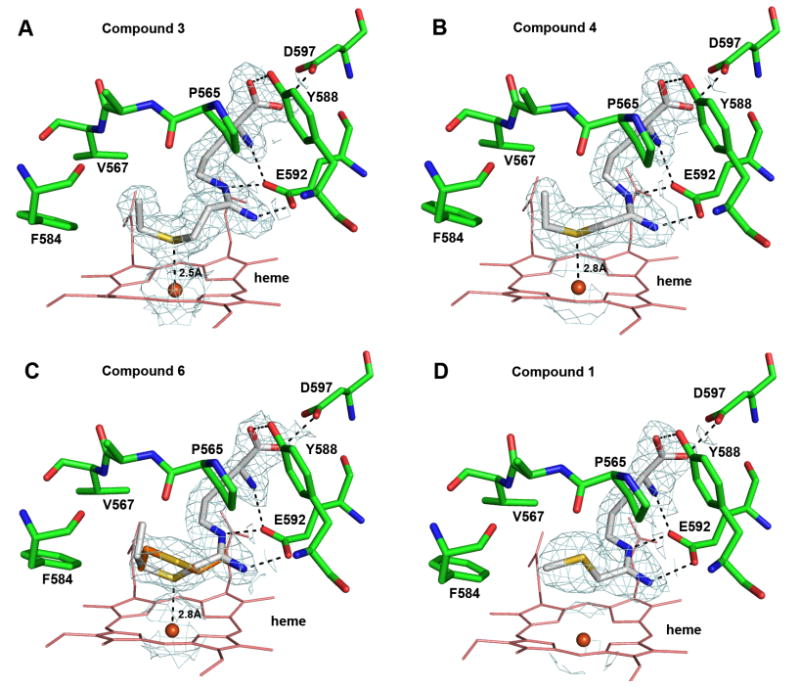
Active site binding conformations for 1 (panel D), 3 (A), 4 (B), and 6 (C) based on the refined X-ray crystal structures. Note that for 3 and 6 alternate conformations were observed, shown with two different colors. For 1 and 4, a single conformation was observed. The hydrogen bonds are depicted with dashed lines. The mean Fe-S distance from the two subunits for 3, 4, and 6 is labeled. The 2Fo-Fc electron density map contoured at 1 σ is also displayed around the bound inhibitor and the heme iron.
The structure at 2.10 Å resolution for 4 (Figure 3B) revealed that, as with 3, the sulfur atom is positioned directly over the heme iron, and the Fe-S distance of 2.8 Å is longer than that of 3. Furthermore, the density for the terminal ethyl group is well defined, as for 3, and can be modeled in a single conformation. The good fit of this inhibitor results from its short one carbon linker from the amidine to sulfur and a small ethyl tail after the sulfur atom that does not make any unfavorable contacts with the protein that would promote disordering. The longer propyl tail in 6 did not fit as well. The refined structure for the 6-nNOS complex at 2.10 Å resolution showed the sulfur atom positioned directly over the heme iron (Figure 3 C) with an Fe-S distance of 2.8 Å. The terminal n-propyl group was disordered, and the two alternate conformations with equal occupancy depicted in Figure 3C were based on partial density.
Structural refinement for 1 at 2.15 Å revealed that the entire thioether tail swings away from the heme in a type I conformation (Figure 3D), and the Fe-S distance is 5.1 Å. The density is solid for the entire ligand, making 1, 3, and 4 the compounds whose thioether tail exhibited no disorder. Apparently, it is the longer ethyl tail and favorable hydrophobic interactions in 3 and 4 that help to stabilize the Fe-S bond while the smaller methyl tail in 1 or 2 is not long enough to support Fe-S ligation. Another compound that did not ligate to the heme iron is 5. The 2.20 Å structure of the 5-nNOS complex exhibited good density for the arginine-like backbone, although the remainder of the inhibitor exhibited poor density. The density of the terminal alkyl group was incomplete with the sulfur atom and its methyl tail positioned beyond the existing electron density boundary (Fig. S4). It appears, therefore, that 5 binds via the arginine backbone, but the three-carbon linker between the amidine and the sulfur atom is apparently too long to allow sulfur ligation to the heme iron. According to the available partial density, one conformation was built with an estimated Fe-S distance of 4.5 Å, as shown in Fig. S4. Judging from the negative residual density around the sulfur atom, an alternate conformation with a shorter Fe-S distance was possible, but no consistent model could be refined because of the limited density.
Dithionite-reduced crystal structures
An apparent paradox in what has been described so far is that type I ligands, like 4 and 8, showed substantial Fe-S interaction in the structures using crystals prepared with nNOS heme domain in its ferric state, even though the spectral data indicate that these do not form low-spin complexes when the iron is in the ferric state. However, 4 and 8 do form low-spin type II complexes with the heme iron in the ferrous state. It is well known that metal centers can undergo reduction in the X-ray beam,40 so it could be that our structures of 4 and 8 complexed to nNOS are really in the ferrous state owing to X-ray-induced reduction. To test this hypothesis, dithionite reduced nNOS-inhibitor crystals were prepared for 3-5 in the hope that these crystal structures would strictly reflect the ferrous inhibitor conformation. As it has previously been reported that the Fe-S bond length for native Met ligands can decrease upon heme reduction,22 we thought there might be a decrease in Fe-S distance in our dithionite-reduced crystal structures. The structure for the 4-nNOS complex in the reduced state at 2.10 Å resolution revealed the Fe-S distance to be 2.7 Å (Fig. S5), which is not significantly different from the Fe-S distance observed for the structure from the crystal not treated by dithionite. The data for 3 with the dithionite reduced crystal at 2.40Å was not fully refined because of marginal data quality, although the electron density and Fe-S distance were very similar to data obtained from crystals that were not treated with dithionite. There was also no change found for 5 in the dithionite reduced structure. This observation indicates that the three carbon amidine-sulfur linker still could not align the sulfur atom close to the heme iron, even in its ferrous state (Fig. S5). The fact that pretreatment of crystals with dithionite had very little effect on the structures of these three inhibitors suggests that reduction may have occurred rapidly in the X-ray beam.
Discussion
Coordination of thioether to heme
All of the thioether inhibitors reported here exhibit binding to the nNOS active site similar to that of the substrate L-arginine. The extensive hydrogen bonding interactions with the protein from the inhibitor amino acid and amidine groups anchor the inhibitors above the heme plane. However, there are two factors that determine whether or not each thioether inhibitor can behave as a heme ligand: 1) the length of the linker from the amidine to the sulfur atom, and 2) the size of the tail after the sulfur atom. From the structural data reported here, it is obvious that both one-carbon and two-carbon linkers can bring the sulfur atom to the vicinity of the heme iron, while a three carbon linker, as in 5, is too long. Whether or not the inhibitor can bind directly to the heme is also controlled by the size of its terminal alkyl tail; the tail must fit into the hydrophobic pocket formed by Pro565, Val567, and Phe584. The terminal methyl groups of 1 and 2, which we reported earlier,27 are not large enough to interact favorably with the hydrophobic pocket, and 1 exhibits no type II conformations in the crystal structure. Compounds 3, 4, 6, and 8, on the other hand, all exhibit conformations that hold the thioether sulfur in close proximity to the heme iron. The best fits are the ethyl groups in 3 and 4, giving no disorder or alternate conformations in their crystal structures.
The bulkier three-carbon terminal groups of 6-8 can barely be accommodated by the hydrophobic pocket, which enables conformations that hold the sulfur in close proximity to the heme iron; however, the interactions are not as favorable as those of 3 and 4, leading to high levels of disorder in the entire thioether tail and a higher population of type I conformation, which can account for the loss of type II binding toward ferric nNOS. Although both 6 and 7 have an identical n-propyl tail, the fit is better with 6. Again, this superior fit results from the shorter linker in 6, which orients the terminal propyl group differently, restrained by bond angles along the alkyl chain. The branched isopropyl group in 8 is also disordered, but it fits better into the pocket than does the n-propyl group of 7, thereby giving an Fe-S distance of 2.8 Å compared to >3.0Å for 7.
Potency and Fe-S bond
Unexpectedly, coordination to the heme iron has no effect on potency. Compound 3 is essentially equal in potency to 5, even though the latter displays no type II binding. Furthermore, both 1 and 4 fail to ligate to ferric heme, with the Fe-S distance for 1 being more than 5 Å, yet both 1 and 4 are more potent than 3. Therefore, the Fe-S ligand interaction contributes little, if any, to inhibitor binding affinity. Considering the extensive hydrogen bonding interactions from the amino acid and amidine groups to the surrounding protein residues, it is understandable that the thioether tail has only a small influence on inhibitor binding. Nevertheless, our results imply that the Fe-S bond formed from an axial thioether ligand to the heme of a thiolate protein is exceptionally weak, which is reflected by the rather long distances in the range of 2.5 – 2.8 Å, as seen in structures reported here. This type of Fe-S bonding is much weaker than that for the natural thioether-heme ligation in cytochrome c, for which the Fe-S distance is ∼ 2.3 Å. The Fe-S interaction with our NOS inhibitors is so weak that without proper hydrophobic contacts from the terminal alkyl group to the protein, the sulfur atom cannot ligate to the heme, as seen in the cases of 1 and 2. These observations underscore the previously documented low intrinsic affinity of thioethers for ferric heme1,4-6 and imply that, contrary to what has been widely thought,9,27,41 coordination of thioethers to the heme iron does not generally lead to increased potency, at least not in heme-thiolate proteins. Furthermore, our results suggest that while hydrogen bonding42,43 and electrostatic interactions44 of native Cys ligands with nearby residues stabilize heme-thiolate ligation, hydrophobic interactions may play an important role in facilitating the thioether-iron coordination of native Met heme ligands in cytochrome c.
There are two possible explanations for why the thioether to heme interaction seen in our nNOS inhibitor complexes is so weak, exhibiting a rather long Fe-S distance of 2.5–2.8 Å. The first explanation is that hydrophobic contacts between the protein and the terminal alkyl group dominate the binding of the thioether tail, preventing the sulfur atom from getting closer than 2.5–2.8 Å from the heme iron. This seems to be the case for the ethyl group in 3 and 4. The second possibilityis that the Fe-S bond is intrinsically weak, perhaps because of the Cys thiolate ligand. Compared to the proximal heme His ligand seen in cytochrome c, the Cys ligand in heme-thiolate proteins is a stronger electron donor. Therefore, a proximal Cys heme ligand can exert a strong σ-trans effect to the distal ligand more effectively than a proximal His ligand. This trans effect has been illustrated45,46 by a bent NO bound to a synthetic ferric thiolate porphyrin, in contrast to the linear geometry observed for NO bound to a porphyrin trans to a N-donor ligand.47 An axial thiolate ligand effect was also observed during the oxidation of hydrocarbons by iron porphyrins with various axial ligands.48 Porphyrins with the axial thiolate ligand catalyzed alkane hydroxylation more efficiently than did porphyrins coordinated with imidazole or chloride, which preferentially catalyzed alkene epoxidation.
The structures reported here provide the first example of a thioether distal ligand bound to a heme-thiolate protein. In addition, there are no crystal structures reported for a synthetic porphyrin compound with both a thiolate and a thioether as the axial ligands. Therefore, although the observed long Fe-S distance of 2.5-2.8 Å from the thioether ligand to the heme iron could be a special case for nNOS in complex with a substrate analogue thioether inhibitor, we cannot rule out the possibility that this long Fe-S distance might be intrinsic to heme-thiolate proteins.
Type I or type II: Dependence of heme coordination on iron oxidation state
All of the Fe-S distances in our structures are 2.5-2.8 Å, raising the question of whether or not the crystal structures reflect true heme iron type II ligation. Nevertheless, the crystal structure of the 3-nNOS complex should be classified as type II because its Fe-S distance is the shortest among this series of thioether compounds. In good agreement with the Fe-S interaction observed in the structure, spectrophotometry shows that 3 causes a red spectral shift regardless of the heme oxidation state, normally an indication of coordination to the heme iron. The spectral features of 3-nNOS are consistent with those of other thioether compounds that have been classified as low-spin type II ligands based on their EPR signals.7,9,38 Therefore, to the best of our knowledge, we have obtained the first crystal structure of a thioether-based type II inhibitor for any heme enzyme. Compounds 4 and 6-8, however, which show a type II shift in ferrous, but not in ferric spectral assays, cannot be definitively classified as type II. The thioether ligands for P450 previously reported7,38 behaved consistently as type II heme binders. In contrast, thioether compounds 4 and 6-8 show a switch from type I to type II upon heme reduction. One possible explanation for the switching we observed is that in the ferrous state, the sulfur atom is drawn closer to the heme iron due to increased binding affinity. A decrease in the Fe-S distance from 2.42 Å to 2.35 Å upon heme reduction was previously observed in S. cerevisiae iso-1-Cyt c, an enzyme in which the loop domain donating the axial Met ligand to the heme iron possesses high plasticity.22 There is a similar subtle decrease (< 0.1 Å) in Fe-S bond length for the dithionite-reduced crystal structure in the case of 4. It must also be kept in mind that what was observed in the crystal structures is the ferrous complex owing to x-ray induced reduction. Even if this is the case and the Fe-S distance of 2.7-2.8 Å observed for 4 and 6-8 is for the ferrous complexes, this distance would not be expected to increase much (∼0.1Å) in the ferric state. Therefore, the balance between low-spin ferrous and high-spin ferric may be quite subtle, involving Fe-S distance changes barely detectable at our resolution limits.
In summary, our work has produced a crystal structure of a type II thioether-based inhibitor bound to nNOS and demonstrated the crucial role of hydrophobic contacts in stabilizing the thioether-heme coordination. The ability to switch from type I to type II upon heme reduction makes 4 and 6-8 a set of novel thioether-based heme enzyme ligands. A number of issues remain to be resolved. The lack of much difference in Fe-S distances between some type I and II inhibitors observed in the crystal structures was unexpected. However, very subtle differences in Fe-S bond distance, on the order of 0.1Å, and not detectable in our structures, could tip the balance between low- and high-spin complexes. Additional experiments and computational approaches are required. For example, EPR can more accurately probe spin-state while high level density functional theory calculations can provide insights into the relationship between spin-state and Fe-S bond distance. In addition, monitoring heme iron spin state before, during, and after data collection now is possible, although far from routine, using single crystal spectroscopy. These additional approaches should provide further insights into the nature of the thioether Fe-S bond in these and future nNOS inhibitors.
Experimental Section
Protein Preparation and Crystallization
The heme domain of rat nNOS was generated by limited trypsinolysis of the partially purified full length nNOS and further purified through a Superdex 200 gel filtration column (GE Healthcare) as described previously.29 An absorbance ratio of A280nm over A395nm at 1.9 or lower was found to be the quality needed for crystallization. The vapor diffusion sitting drop setup reported29 was used for the crystal growth except that 20 mM histidine was directly added to the nNOS heme domain sample (7-9 mg/ml) before setting up the sitting drops. For the ferric heme preparation, crystals grown freshly were passed through the cryo-protectant solutions described previously29 and then the thioether inhibitors at 10 mM were soaked into the crystals for 4-6 hr at 4°C. For the ferrous heme preparation, cryo-protectant solution along with the crystals was degassed by alternate evacuation and purging with pure argon for 1 hr. These crystals were reduced with 10 mM dithionite and then soaked with 10 mM thioether inhibitor for 4 hr at room temperature in a glovebox. Crystals were flash cooled and stored in liquid nitrogen until data collection.
X-ray Data Collection and Structure determination
All the X-ray diffraction data collections were carried out at 100K at various beamlines of Stanford Synchrotron Radiation Lightsource (SSRL), via remotely controlled robotic operation using Blu Ice data collection control software.49 Raw data frames were processed with HKL2000.50 The bound thioether inhibitors were detected by difference Fourier synthesis using CNS.51 Models of ligands were built in O 52 or COOT53 and complex structures refined with CNS. The final round of refinement was done with REFMAC54 so that the TLS protocol55 could be implemented. Water molecules were added automatically with CNS or REFMAC and then validated visually in COOT. The crystallographic data collection and refinement statistics are summarized in Table S1 in Supporting Information. Coordinates of the eight refined structures were deposited with RCSB protein data bank with accession codes listed in the table.
Enzyme and Assay for IC50 Value Determination
Murine macrophage iNOS,56 rat nNOS,32 and bovine eNOS57 were prepared according to the literature procedures. Nitric oxide formation from NOS was monitored by the hemoglobin capture assay at 22 °C as described.34 A typical assay mixture for nNOS and eNOS contained 10 μM L-arginine, 1 mM CaCl2, 600 unit/mL calmodulin (Sigma, P-2277), 100 μM NADPH, 0.125 mg/mL hemoglobin-A0 (ferrous form, Sigma, H0267), 10 μM H4B, in 100 mM HEPES (pH 7.5). A typical assay mixture for iNOS contained 10 μM L-arginine, 100 μM NADPH, 0.125 mg/mL hemoglobin-A0 (ferrous form), 10 μM H4B, in 100 mM HEPES (pH 7.5). All assays were performed in a final volume of 600 μL and were initiated by addition of enzyme. Hemoglobin absorption was monitored at 401 nm on a Perkin-Elmer Lambda 10 UV-visible spectrophotometer.
Determination of Ki Values
The apparent Ki values were obtained by measuring the percent enzyme inhibition in the presence of 10 μM L-arginine with at least five concentrations of inhibitor. The parameters of the following inhibition equation35 were fitted to the initial velocity data: % inhibition = 100[I]/([I] + Ki(1 + [S]/Km)). Km values for L-arginine were 1.3 μM (nNOS), 8.2 μM (iNOS), and 1.7 μM (eNOS). The selectivity of an inhibitor was defined as the ratio of the respective Ki values.
Ferric Difference Spectrometry
To a quartz cuvette were added 3.75 μM nNOS heme domain (100 μL), 10 μM H4B (1 μL), and 100 mM Hepes buffer (90 μL) at pH 7.5. This solution was scanned against a blank containing Hepes buffer (198 μL) and H4B (2 μL) from 381 nm to 500 nm. To the same cuvette were added 0.8 μL-aliquots of inhibitor solution. The concentration of the inhibitor stock solutions varied from 1.25 mM to 50 mM. After each addition the solution was scanned against the blank. From each spectrum was subtracted the nNOS-only spectrum. In cases when a type II difference spectrum was not observed, a separate cuvette was prepared containing 3.75 μM nNOS (100 μL), 10 μM H4B (1 μL), Hepes buffer (98 μL), and 1 μL of an imidazole solution (the final concentration of imidazole varied from 100 μM to 300 μM). The nNOS-imidazole solution was scanned. Inhibitor solution was then added in 0.8 μL aliquots (the concentration of the inhibitor stock solutions varied from 1 to 50 mM), and from the resulting spectrum was subtracted the nNOS-imidazole spectrum. The total volume of the aliquots did not exceed 5% of the total volume.
Ferrous Difference Spectrometry
The spectral titrations of inhibitor binding to the ferrous nNOS were carried out under anaerobic conditions on a Cary 4 (Varian) spectrophotometer. The buffer used for titration was 100 mM Hepes, pH 7.5, with 5 μM H4B. The buffer was sealed in the serum vials and degassed by purging into solution with the ultrapure argon and pulling vacuum in the head space of vial for a few alternate cycles in 2 hours. All the inhibitor and dithionite stock solutions were made with the degassed buffer inside a glovebox (COY Laboratory Products, Inc.). To each of the sample and reference septum-sealed cuvettes was added 600 μL of buffer containing ∼2.4 μM of nNOS and an excess amount of dithionite (∼2-10 mM). The baseline was setup with both cuvettes in place using a double beam mode of the instrument. For each scan from 360 to 600 nm, small aliquots (1-2 μL) of the inhibitor stock solution with concentrations ranging from 1 to 200 mM were added only to the sample cuvette inside the glovebox. The total volume of the aliquots was less than 2% of the 600 μL buffer in the sample cuvette.
Absolute Absorption Spectra in Ferrous State
The absolute spectra were taken under the anaerobic condition using a degassed Hepes as described in the previous section. Both septum sealed cuvettes were filled with the Hepes buffer containing dithionite and 1-6.5 mM of inhibitor for a baseline correction. The reason for including inhibitor in the baseline was because of their strong absorption in the UV region, which could cause the baseline tilting at high concentration. The nNOS protein sample was then added only to the sample cuvette inside the glovebox. The spectral scan was limited to the 360-700 nm range due to the noisy baseline in the near UV region caused by dithionite.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from NIH with grants GM57353 (T.L.P) and GM49725 (R.B.S.). J.D.M. thanks Jianguo Fang for overexpressing and purifying nNOS; Stephanie Choing for help in preparing 1; and Fengtian Xue, Guoyao Xia, Haitao Ji, and Walter Fast for helpful discussions. H.L. thanks the excellent technical assistance provided by Cynthia Edirisuriya. We thank the beamline staff at SSRL for their excellent support during the remote data collections.
Footnotes
Supporting Information Available: All difference spectra, Lineweaver Burk plots, and full absorption spectra over the visible range for 1 and 3-8; type II difference spectrum obtained upon titration of H4B—nNOS with 8; full experimental details for 3-8 and their precursors; 1H-NMR and 13C-NMR spectra of 3-8, 10, 12, 14, 16, 18, and 20; figures for the structures of nNOS with inhibitors 5 and 8 bound in the ferris state and those with 4-5 bound in the ferrous state; and table of crystallographic data collection and refinement statistics for all the structures reported here.
References
- 1.Tezcan FA, Winkler JR, Gray HB. J Am Chem Soc. 1998;120:13383–13388. [Google Scholar]
- 2.Voegtle, et al. Biochemistry. 2003;42:2475–2484. doi: 10.1021/bi0271502. [DOI] [PubMed] [Google Scholar]
- 3.Zhong L, Wen X, Rabinowitz TM, Russell BS, Karan EF, Bren KL. Proc Natl Acad Sci. 2004;101:8637–8642. doi: 10.1073/pnas.0402033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray SG, Hartley FR. Chem Rev. 1981;81:365–414. [Google Scholar]
- 5.Smith M, McLendon G. J Am Chem Soc. 1981;103:4912–4921. [Google Scholar]
- 6.Schejter A. In: Cytochrome c: A Multidisciplinary Approach. Scott RA, Mauk AG, editors. Univ. Sci. Books; Mill Valley, CA: 1996. pp. 335–345. [Google Scholar]
- 7.Sono M, Andersson LA, Dawson JH. J Biol Chem. 1982;14:8308–8320. [PubMed] [Google Scholar]
- 8.Sono M, Hager LP, Dawson JH. J Biol Chem. 1984;21:13209–13216. [PubMed] [Google Scholar]
- 9.Nastainzcyk W, Ruf HH, Ullrich V. Eur J Biochem. 1975;60:615–620. doi: 10.1111/j.1432-1033.1975.tb21039.x. [DOI] [PubMed] [Google Scholar]
- 10.Bushnell GW, Louie GV, Brayer GD. J Mol Biol. 1990;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 11.Aranda R, IV, Worley CE, Liu M, Bitto E, Cates MS, Olson JS, Lei B, Phillips GN., Jr J Mol Biol. 2007;374:374–383. doi: 10.1016/j.jmb.2007.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffith OW, Steuhr DJ. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 13.Kerwin JE, Jr, Heller M. Med Res Rev. 1994;14:23–74. doi: 10.1002/med.2610140103. [DOI] [PubMed] [Google Scholar]
- 14.Choi DW, Rothman SM. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 15.Crossin KL. Trends Biochem Sci. 1991;16:81–2. doi: 10.1016/0968-0004(91)90036-u. [DOI] [PubMed] [Google Scholar]
- 16.Ferrendellia JA, Blank AC, Gross RA. Brain Res. 1980;200:93–103. doi: 10.1016/0006-8993(80)91097-5. [DOI] [PubMed] [Google Scholar]
- 17.Das I, Khan NS, Puri BK, Sooranna SR, de Belleroche J, Hirsch SR. Biochem Biophys Res Commun. 1995;212:375–80. doi: 10.1006/bbrc.1995.1980. [DOI] [PubMed] [Google Scholar]
- 18.Thomsen LL, Iversen HK, Lassen LH, Olesen J. CNS Drugs. 1994;2:417–22. [Google Scholar]
- 19.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Biochem Biophys Res Commun. 1994;205:659–65. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Martasek P, Roman LJ, Masters BSS, Silverman RB. Bioorg Med Chem. 1999;7:1941–1951. doi: 10.1016/s0968-0896(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 21.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 22.Berghuis AM, Brayer GD. J Mol Biol. 1992;223:959–976. doi: 10.1016/0022-2836(92)90255-i. [DOI] [PubMed] [Google Scholar]
- 23.Matsuura Y, Takano T, Dickerson RE. J Mol Biol. 1982;56:389–409. doi: 10.1016/0022-2836(82)90335-7. [DOI] [PubMed] [Google Scholar]
- 24.Narayanan K, Spack L, McMillan K, Kilbourn RG, Hayward MA, Masters BS, Griffith OW. J Biol Chem. 1995;270:11103–11110. doi: 10.1074/jbc.270.19.11103. [DOI] [PubMed] [Google Scholar]
- 25.Haines DC, Chen B, Tomchick DR, Bondlela M, Hedge A, Machius M, Peterson JA. Biochemistry. 2008;47:3662–3670. doi: 10.1021/bi7023964. [DOI] [PubMed] [Google Scholar]
- 26.Rahman MN, Vlahakis JZ, Szarek WA, Nakatsu K, Jia Z. J Med Chem. 2008;51:5943–5952. doi: 10.1021/jm800505m. [DOI] [PubMed] [Google Scholar]
- 27.Litzinger EA, Martasek P, Roman LJ, Silverman RB. Bioorg Med Chem. 2006;14:3185–3198. doi: 10.1016/j.bmc.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Raman CS, Martásek P, Král V, Masters BSS, Poulos TL. J Inorg Biochem. 2000;81:133–139. doi: 10.1016/s0162-0134(00)00099-4. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 30.Pinner A, Klein F. Umhandlung der Nitrile in Imide Ber. 1877;10:1889–1897. [Google Scholar]
- 31.McMillan K, Masters BSS. Biochemistry. 1993;32:9875–9880. doi: 10.1021/bi00089a001. [DOI] [PubMed] [Google Scholar]
- 32.Roman LJ, Sheta EA, Martasek P, Gross SS, Liu Q, Masters BSS. Proc Natl Acad Sci USA. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frey C, Narayana K, McMillan K, Spack L, Gross SS, Masters BS, Griffith OW. J Bio Chem. 1994;269:26083–26091. [PubMed] [Google Scholar]
- 34.Hevel JM, Marletta MA. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 35.Segel IH. Enzyme Kinetics. John Wiley and Sons; NewYork: 1975. p. 105. [Google Scholar]
- 36.Babu BR, Griffith OW. Curr Opin Chem Biol. 1998;2:491–500. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 37.Cowan JA, Gray HB. Inorg Chem. 1989;28:4554–4556. [Google Scholar]
- 38.Dawson JH, Andersson LA, Sono M. J Biol Chem. 1983;258:13637–13645. [PubMed] [Google Scholar]
- 39.Nastainczyk W, Ruf HH, Ullrich V. Chem-Biol Interactions. 1976;14:251–263. doi: 10.1016/0009-2797(76)90105-8. [DOI] [PubMed] [Google Scholar]
- 40.Beitlich T, Kuhnel K, Schulze-Briese C, Shoeman RL, Schlichting I. J Synchrotron Radiat. 2007;14:11–23. doi: 10.1107/S0909049506049806. [DOI] [PubMed] [Google Scholar]
- 41.Narayanan K, Griffith OW. J Med Chem. 1994;37:885–887. doi: 10.1021/jm00033a004. [DOI] [PubMed] [Google Scholar]
- 42.a) Suzuki N, Higuchi T, Urano Y, Kikuchi K, Uekusa H, Ohashi Y, Uchida T, Kitagawa T, Nagano T. J Am Chem Soc. 1999;121:11571–11572. [Google Scholar]; b) Ueyama N, Nishikawa N, Yamada Y, Okamura T, Nakamura A. J Am Chem Soc. 1996;118:12826–12827. [Google Scholar]
- 43.Yoshioka S, Takahashi S, Ishimori K, Morishima I. J Inorg Biochem. 2000;81:141–151. doi: 10.1016/s0162-0134(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 44.a) Goodin DB. J Biol Inorg Chem. 1996;1:360–363. [Google Scholar]; b) Poulos TL. J Biol Inorg Chem. 1996;1:356–359. [Google Scholar]
- 45.Paulat F, Lehnert N. Inorg Chem. 2007;46:1547–1549. doi: 10.1021/ic070023f. [DOI] [PubMed] [Google Scholar]
- 46.Xu N, Powell DR, Cheng L, Richter-Addo GB. Chem Commun. 2006;19:2030–2032. doi: 10.1039/b602611g. [DOI] [PubMed] [Google Scholar]
- 47.Ellison MK, Scheidt WR. J Am Chem Soc. 1999;121:5210–5219. [Google Scholar]
- 48.Ohno T, Suzuki N, Dokoh T, Urano Y, Kikuchi K, Hirobe M, Higuchi T, Nagano T. Inorg Biochem. 2000;82:123–125. doi: 10.1016/s0162-0134(00)00141-0. [DOI] [PubMed] [Google Scholar]
- 49.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J Synchrotron Rad. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 50.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 51.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 52.Jones TA, Zou JY, Cowan SW, Kjeldgaarrd M. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 55.Winn MD, Isupov MN, Murshudov GN. Acta Cryst. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 56.Hevel JM, White KA, Marletta M. J Biol Chem. 1991;266:22789–22791. [PubMed] [Google Scholar]
- 57.Martasek P, Liu Q, Roman LJ, Gross SS, Sessa WC, Masters BSS. Biochem Biophys Res Commun. 1996;219:359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.