

Abstract
Cigarette smoking is strongly correlated with the onset of non-small cell lung cancer (NSCLC). Nicotine, an active component of cigarettes, has been found to induce proliferation of lung cancer cell lines. In addition, nicotine can induce angiogenesis and confer resistance to apoptosis. All these events are mediated through the nicotinic acetylcholine receptors (nAChRs) on lung cancer cells. In the present study we demonstrate that nicotine can promote anchorage-independent growth in NSCLCs. In addition, nicotine also induced morphological changes characteristic of a migratory, invasive phenotype in NSCLCs on collagen gel. These morphological changes were similar to those induced by the pro-migratory growth factor VEGF. The pro-invasive effects of nicotine were mediated by α7-nAChRs on NSCLCs. RT-PCR analysis showed that the α7-nAChRs were also expressed on human breast cancer and pancreatic cancer cell lines. Nicotine was found to promote proliferation and invasion in human breast cancer. The pro-invasive effects of nicotine were mediated via a nAChR, Src and calcium dependent signaling pathway in breast cancer cells. In a similar fashion nicotine could also induce proliferation and invasion of Aspc1 pancreatic cancer cells. Most importantly, nicotine could induce changes in gene expression consistent with epithelial to mesenchymal transition, characterized by reduction of epithelial markers like E-cadherin expression, ZO-1 staining and concomitant increase in levels of mesenchymal proteins like vimentin and fibronectin in human breast and lung cancer cells. Therefore, it is probable that the ability of nicotine to induce invasion and EMT may contribute to the progression of breast and lung cancers.
Keywords: lung cancer, nicotine, epithelial-mesenchymal transition, tumor progression, metastasis
Introduction
Cigarette smoke is the strongest documented risk factor for the development of lung cancer, accounting for about 157,000 deaths every year in the US 1. A significant proportion of non-small cell lung cancer cases are detected only in the advanced stage after the onset of metastasis, leading to a remarkably low 5-year survival rate (about 15%) in patients 1. Tobacco derived carcinogens like 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N’-nitrosonornicotine (NNN) are known to form DNA adducts, mutating vital growth regulatory genes like p53 and Ras, initiating oncogenesis2–4. In addition, several lines of evidence indicate that cigarette smoking correlates with increased metastasis of lung, pancreatic, breast and bladder cancers 5–7. Although cigarette smoke is a complex mixture of over 4000 compounds, nicotine has been shown the major addictive component of cigarettes 8–10. Nicotine, while not carcinogenic by itself, has been shown to induce proliferation and angiogenesis in several experimental models 11–14; these effects occurred at concentrations normally found in the blood stream of smokers (10−8 M to 10−7M) 12. These levels can vary greatly in smokers, and urine cotinine levels have been reported to range from 1500ng/ml to 8000ng/ml 15–17.
The pathophysiological effects of nicotine are mediated by nicotinic acetylcholine receptors (nAChRs), which are expressed on neurons and neuromuscular junctions 8, 9, 18, 19; in addition, they have been found to be expressed in a variety of non-neuronal cells including endothelial cells and several histological types of lung tissue 20. Several convergent studies show that nicotine induces angiogenesis by upregulation of COX-2, prostacyclin, VEGFR-2, MMPs, uPA, e-NOS activity. Additionally, nicotine can exert its pro-angiogenic activity by increasing the cellular levels of VEGF, bFGF and PDGF. The administration of nicotine increased the severity of choroidal neovascularization and reversed VEGF-induced suppression of MMP-2 activity in mice 7, 21, 22. While there are insights into the molecular mechanisms by which nicotine induces angiogenenesis, however, the molecular mechanisms underlying its role in tumor invasion and metastasis is not yet clear.
Clinical studies have shown an association between smoking and the development of pancreatic as well as breast cancers; further, patients who were smokers showed worse survival profile relative to non-smokers 1, 23, 24. In the present study we show that nicotine can induce invasion and migration of NSCLC cells in a α7-receptor dependent and Src-dependent manner. Whereas the pro-invasive effects of nicotine were mediated by α7-nAChRs in lung cancer cells, α7-nAChRs as well as DhβE-sensitive nAChRs mediated invasion of breast cancer cells. Nicotine was also found to inhibit anoikis (loss of cell-substratum adhesion) in lung airway epithelial cells. Further, we demonstrate that nicotine induces changes in gene expression consistent with epithelial-mesenchymal transition (EMT)25–27; a signature of more advanced and less differentiated cancer. Long-term treatment of lung cancer and breast cancer cells with 1µM nicotine was found to diminish levels of epithelial markers namely β-catenin and E-cadherin, and upregulate mesenchymal proteins like fibronectin and vimentin, indicative of disruption of cell-cell contacts and increased motility. Our results suggest that nicotine plays a key role in the regulation of the complex cellular cascades that modulate cell adhesion, invasion and migration, leading to metastasis.
Materials and Methods
Cell culture
A549, MDA-MB-468 and MCF-7 cells were cultured in F12K or DMEM (Mediatech Cellgro, Virginia) containing 10% FBS (Hyclone). Aspc1 cells were cultured in RPMI supplemented with 10% FBS. Small airway epithelial cells (SAECs) were purchased from Clonetics and cultured in SAGM. The studies using anti-cancer drugs or signal transduction inhibitors were done on cells that were rendered quiescent by serum starvation for 36 hours, following which cells were treated with indicated concentrations of the drugs for 30 minutes. Thereafter, cells were stimulated with 1µM nicotine (Sigma Chemical Company) in the presence or absence of the inhibitors for 18 hours. The concentrations of inhibitors used for the various experiments were 10µM PP2, 1µM atropine, 10µM methylallylaconitine, 1µM α-lobeline, 10µM nifedipine, 1µM DhβE, 10µM α-bungarotoxin and 20µM hexamethonium bromide.
Anoikis Assay
Small airway epithelial cells were grown to 70–80% confluency in SAGM containing growth factors. Cells were harvested by trypsinization and 105 cells were plated on 35mm culture dishes which were either uncoated or coated with polyhema (10mg/ml in ethanol). Subsequently one set of the uncoated or polyhema coated plates were stimulated with Nicotine (1µM) for 18 hours 28. Cells were collected from uncoated plates by trypsinization while in polyhema coated plates non-adherent floating cells were collected using a pipet. After washing with PBS, cells were suspended in 200µl PBS and Cytospin was used to collect cells on the slide by spinning at 500rpm for 15min. TUNEL assay was performed on the fixed cells using Promega’s Dead End Colorimetric TUNEL system. TUNEL positive cells were counted in 4 different fields and % positive cells were plotted.
Soft Agar Growth Assay
Anchorage independent growth was assayed by the soft agar growth assay as described elsewhere 29. The first step involves plating a bottom layer of 0.6% agar in serum-free media in 12-well plates. The plates were incubated at room temperature for 30 minutes to solidify the agar. Cells were harvested by trypsinization and 2000 cells were mixed with 0.3% agarose (made in serum-free media) in the presence of the indicated nicotine concentration and layered carefully on the top of existing 0.6% agarose. Each treatment was done in triplicate. The plates were covered with 1mL of medium supplemented with 10% FBS, in the presence or absence of the indicated concentrations of nicotine and incubated at 37°C in a 5% CO2 incubator for 3 weeks. Nicotine was added every 2 days to the test wells. The covering medium was replaced every week. At the end of 3 weeks, cell colonies were stained with 10mg/ml MTT and colonies with >0.1 mm in diameter were counted under a microscopic field at 40× magnification. Mean colony count was based on numbers from triplicate wells for each treatment condition and was analyzed using two-sided Student's t test.
Collagen gel culture
3D collagen-1 gels were prepared on ice using equal volumes of 3mg/ml collagen solution and 2× HEPES-buffered salt solution [50.4mM HEPES, pH 7.4, 162.6mM NaCl, 10.6mM KCl, 88.2mM NaHCO3, 1.6 mM Na2HPO4, and 11mM D(+)-glucose] yielding a concentration of 1.3 mg/ml following addition of culture medium 30, 31. The collagen gel solution (0.2 ml) was added to each well of the 96-well plate and allowed to set at 37°C for 30 minutes. A549 cells were collected in a single cell suspension and added on the preformed collagen gel. The plates were incubated at 37°C for 3 days, at which time complex structures had formed. The 3-D structures were visualized by phase contrast microscopy.
Invasion Assay
The invasive ability of Aspc-1, MCF-7, A549 and MDA-MB-468 cells was assayed according to the method reported before 32. Briefly, the upper surface of the filters were precoated with collagen (100µg/filter). Matrigel was applied to the upper surface of the filters (50µg/filter) and dried in a hood. These filters were placed in Boyden chambers. Cells were grown to 70% confluency in respective media and were rendered quiescent by serum starvation, then treated with 1µM nicotine in the presence or absence of indicated inhibitors for 18 hours. The inhibitors were added to the cells 30 minutes before the addition of nicotine. For some experiments cells treated with 10% serum were used as the positive control. Following treatment, cells were trypsinized and 7000 cells were plated in the upper chamber of the filter in media containing 0.1% bovine serum albumin (Sigma), indicated inhibitors and nicotine. Media containing 20% fetal bovine serum was placed in the lower well as a chemoattractant and the chambers were incubated at 37°C. After 18 hours, non-migrating cells on the upper surface of the filters were removed by wiping with cotton swabs. The filters were processed first by fixing in methanol followed by staining with hematoxylin. The cells migrating on the other side of the filters were quantitated by counting three different fields under 40× magnification. Data presented is a mean of three independent experiments.
Proliferation Assays
Bromodeoxyuridine (BrdU) labeling kits were obtained from Roche Biochemicals. Cells were plated in poly-D-lysine coated chamber slides at a density of 10,000 cells per well and rendered quiescent by serum starvation for 36 hours. Cells were then re-stimulated with 1µM nicotine or 10% FBS for 18 hours. S-phase cells were visualized by microscopy and quantitated by counting 3 fields of 100 cells in quadruplicate. Data is presented as the percentage of BrdU positive cells out of the 100 cells counted.
Lysate preparation and Western blotting
Lysates from cells treated with different agents were prepared by NP-40 lysis as described earlier 11, 33 and 100µg protein was run on polyacrylamide-SDS gel. The proteins were transferred to a nitrocellulose membrane and immunoblotted with antibodies corresponding to various EMT markers. Polyclonal β-catenin and fibronectin, monoclonal E-cadherin and Vimentin antibodies were obtained from Santa Cruz Biotechnology. Monoclonal antibody to Actin was purchased from Sigma Chemical Co. and polyclonal N-cadherin antibody from Abcam. Western blots are representative of three independent experiments.
Wound Healing Assay
10,000 Aspc-1 cells were plated and grown asynchronously to 90% confluency in a 6-well plate (Falcon Becton Dickinson). These cells were starved in 0.1% fetal bovine serum for 24 hours and then washed with 1× Dulbecco’s phosphate-buffered saline (MediaTech). The cells were scratched with a sterile 200µl pipet tip in three separate places in each well and medium containing 1µM nicotine or 10% fetal bovine serum was added in addition to the two wells that remained serum starved. After 24 hours, the wounds were observed and images were taken in 20× magnification. Similar experiments were performed with A549, MCF-7 and MDA-MB-468 cells, except that cells were not serum starved prior to inflicting the scratch. The cells were treated with 0.01µM, 0.1µM or 1µM of nicotine in serum free media; media containing 10% serum was used as a positive control. The data is representative of three independent experiments.
Immunofluorescence and confocal microscopy
A549, MCF-7 and MDA-MB-468 cells were plated onto poly-D-lysine-coated eight well glass chamber slides (7,000 cells per well) for immunostaining. Cells were rendered quiescent, followed by treatment with 1µM Nicotine for the indicated time points. The cells were fixed with 10% buffered-formalin and double immunofluorescence was performed as per the protocol published previously 34. Primary antibodies used were monoclonal E-cadherin, ZO-1 (Abcam) and polyclonal Crm-1 (Santa Cruz) at 1:100 dilution. Secondary antibodies were goat anti-rabbit Alexa Fluor-546 and anti-mouse Alexa Fluor-488 (Molecular Probes) respectively. Control experiments demonstrated no cross-reactivity between anti-mouse secondary and anti-rabbit primary antibodies and vice-versa; nor were there any detectable staining by secondary antibodies alone (data not shown). DAPI (Vector labs) was used to stain the nuclei. Cells were visualized with a DM16000 inverted Leica TCS SP5 tandem scanning confocal microscope with a 63×/1.40NA oil immersion objective. 405 diode, 488 Argon and 546 HeNe laser lines were applied to excite the cells using AOBS line switching to minimize crosstalk between fluorochromes. Images were produced with three cooled photomultiplier detectors and analyzed with the LAS AF software version 1.6.0 build 1016 (Leica Microsystems, Germany).
Results
Nicotine induces proliferation, migration and invasion of lung cancer cells
The proliferative effects of nicotine were examined in the bronchioalveolar lung carcinoma cell line A549. Nicotine induced a dose-dependent increase in S-phase entry of quiescent A549 cells, with the maximal proliferation being observed at 1µM nicotine (Figure 1A).12. Ability of cells to grow independent of adhesion is a feature of cancer cells and enhanced capability of adherence-independent growth is a feature of advanced tumors. Since nicotine was able to increase the proliferation of A549 cells grown on plastic, efforts were made to assess whether it promoted adherence-independent growth as well. As shown in Figure 1B, treatment of cells with nicotine promoted their growth in soft agar; nicotine treatment increased the number as well as the size of the colonies. Given these results, Boyden Chamber assays were carried out to assess whether nicotine affects the invasive as well as migratory properties of A549 cells. It was found that nicotine could enhance the migration and invasion of A549 cells in a dose-dependent manner; the maximum effect was observed at 1µM and the effect was reduced at 10µM (Figure 1C).
Figure 1.

Nicotine can promote proliferation, invasion and survival pathways in A549 NSCLC cells. (A) Nicotine can induce S-phase entry in A549 NSCLC cells in a dose-dependent manner, as measured by BrdU assays. (B) Nicotine can induce anchorage-independent growth of A549 cells in soft-agar assay. Nicotine increased the size of the individual colonies but had little effect on the total number of colonies. (C) Nicotine can promote migration and invasion of A549 cells in Boyden Chamber assays, in a dose dependent manner, the maximal effect being observed at 1µM nicotine. (D) Nicotine can confer resistance to anoikis of SAECs. SAECs were treated with 1µM nicotine and plated on polyhema coated slides. Apoptosis was measured by TUNEL assays. (E) Treatment of A549 cells with 1µM nicotine caused morphological changes, similar to VEGF on 3D collagen gel assays.
Several lines of evidence show that nicotine not only promotes tumor growth and neovascularization, but plays a role in tumorigenesis as well. Normal adherent cells undergo apoptosis shortly after loss of adhesion to substratum, a phenomenon known as anoikis 35. In-vitro-transformed cells and cancer-derived cells are able to survive and grow in the absence of anchorage to the extracellular matrix (ECM) and their neighboring cells. This represents one of the most important hallmarks of oncogenic transformation of normal cells 36, 37. Since nicotine could enhance adherence-independent growth of cells in soft agar, experiments were carried out to determine whether nicotine could promote anchorage-independent survival of normal lung epithelial cells. Primary small airway epithelial cells (SAECs) were grown on polyhema coated plates to reduce adherence; these cells showed a significant increase in apoptosis compared to cells grown on regular plates. The treatment of the small airway epithelial cells with 1µM nicotine induced resistance to anoikis when grown on polyhema coated plates, showing reduced apoptosis (Figure 1D). This suggests that nicotine could not only promote the proliferation of cells, but can also provide a survival advantage. Based on these findings, attempts were made to assess whether nicotine altered the morphology of A549 cells indicative of a more mesenchymal and migratory potential. Quiescent A549 cells were grown as 3-D cultures on collagen and treated with 1µM nicotine or 100ng/ml (equivalent to 2.38nM) VEGF. Figure 1E shows that the cells treated with nicotine acquired elongated migratory morphology relative to control cells. The angiogenic factor VEGF was taken as the positive control for the assay and similar morphological changes were observed in both the cases. These results clearly show that nicotine can alter the proliferative, migratory and survival properties of cells.
Nicotine induces invasion of lung and breast cancer cells via nAChRs
Ability to invade through a basement membrane and adjacent tissues is critical for metastasis of cancer cells. Since nicotine could promote the invasive properties of cells, we wanted to determine the signaling pathways underlying the pro-invasive activity of nicotine. Towards this purpose, Boyden Chamber assays were done in the presence or absence of various receptor antagonists or signaling inhibitors. Nicotine-induced tumor cell invasion was completely ablated by the generalized nAChR antagonist hexamethonium, but unaffected by atropine, a muscarinic receptor antagonist. In particular, α7-nAChR antagonists α-bungarotoxin and MAA significantly abrogated the nicotine-stimulated invasion of A549 cells, whereas α-lobeline and DhβE did not have any effect (Figure 2A). Thus, the pro-invasive effects of nicotine in A549 cells are primarily mediated by α7-nAChRs. Further, the stimulatory effects of nicotine on A549 invasion were suppressed by 1µM Iressa (gefitinib, EGFR inhibitor), 1µM PP2 (Src inhibitor) and 1µM nifedipine (calcium channel inhibitor). These results show that the pro-invasive effects of nicotine on A549 cells involve α7-nAChR subunits, Src, EGFR activity as well as calcium channel activity.
Figure 2.

Nicotine induces proliferation and invasion in human lung cancer and breast cancer cells. (A) The pro-invasive effects of nicotine require α7-nAChR, Src activity, EGFR and intracellular calcium, in A549 cells as seen in a Boyden Chamber assay. The pro-invasive effects of nicotine were reversed by the general nAChR antagonist hexamethonium, the α7-nAChR antagonists α-bungarotoxin and MAA, the Src inhibitor PP2, calcium channel blocker nifedipine and the EGFR inhibitor Iressa (gefitinib). (B) The α7-nAChR subunit is expressed by multiple human cancer cell lines including breast cancer cell lines MCF-7, MDA-MB-468 and pancreatic cancer cell lines Aspc-1, Panc-1 and CAPAN-2, as examined by RT-PCR. RT-PCR for actin was used as the control. (C) BrdU assays show that nicotine induces dose-dependent proliferation of human breast cancer cell lines MCF-7 and MDA-MB-468, the maximal effect being observed at 1µM. (D) Nicotine promotes invasion of MCF-7 and MDA-MB-468 breast cancer cells at a concentration of 1µM, as measured by Boyden chamber assays. A549 NSCLC cells were used as the positive control for the assay. (E) Nicotine stimulates the anchorage-independent growth of MCF-7 and MDA-MB-468 human breast cancer cells in soft agar assays. MCF-7 and MDA-MB-468 cells treated with 1µM nicotine formed larger colonies in soft agar relative to untreated controls. (F) Nicotine-mediated invasion of MCF-7 and MDA-MB-468 human breast cancer cells is dependent on Src activity and intracellular calcium pathways. The pro-invasive effects on nicotine in these breast cancer cells were mediated by both α7-nAChR and DhβE-sensitive nAChRs.
Since tobacco use has been implicated in the pathogenesis of several non-lung cancers like breast cancer and pancreatic cancer 1, we examined the effect of nicotine on two human breast cancer cell lines. RT-PCR analysis shows the presence of α7 subunit of nAChRs in a variety of breast and pancreatic cancer cell lines (Figure 2B). Given this result, we examined whether nicotine could cause S-phase entry in MCF-7 and MDA-MB-468 cells. BrdU assays demonstrate that the treatment of MCF-7 and MDA-MB-468 cells with varying doses of nicotine induced a dose-dependent cell proliferation, the maximal proliferative effect being observed at 1µM nicotine in both cell lines (Figure 2C). Furthermore, 1µM nicotine could induce invasion of MCF-7 and MDA-MB-468 cells in Boyden chamber assays (Figure 2D) and promote colony formation on soft agar (Figure 2E). Invasion assays using various inhibitors show that the invasion of breast cancer cells in response to nicotine was mediated partially by α7-nAChR and DhβE-sensitive nAChRs (Figure 2F); this is probably due to the reduced amounts of α7 expression in these cells compared to A549 cells (Figure 2B). It can be imagined that DhβE-sensitive α3β4 subunits, which transmit survival signals 28,33, might be contributing to the invasion of the breast cancer cells.
Given that pancreatic cancer also shows correlation with smoking, experiments were carried out to examine whether nicotine had similar effects on pancreatic cancer cells. It was found that nicotine induces proliferation of Aspc1 pancreatic cancer cells, similar to lung cancer and breast cancer cell lines (Figure 3A); this induction was reduced by the Src kinase inhibitor PP2. This is similar to our earlier results on A549 cells 11, suggesting that the same pathways mediate nicotine-mediated proliferation of pancreatic cancer cells as well. Nicotine was also found to promote invasion of pancreatic cancer cells as measured by wound healing assays (Figure 3B). Boyden chamber assays showed that the pro-invasive activity of nicotine was abrogated by the Src inhibitor PP2, suggesting an additional role of Src in both these processes (Figure 3C). These results show that multiple tumor types can respond to physiological concentrations of nicotine by undergoing proliferation and that nicotine can enhance their invasive properties.
Figure 3.

Nicotine induces proliferation and invasion of pancreatic cancer cells. (A). Quiescent Aspc1 cells were stimulated with 1µM nicotine for 18 hours and S-phase entry was measured by BrdU assays. The proliferative effects of nicotine in Aspc1 cells were abrogated in the presence of 1µM of Src inhibitor PP2, indicating that Src function is required for the proliferative effects of nicotine. (B) Wound healing assays show that 1µM nicotine can promote migration and invasion of Aspc1 cells. Cells treated with media containing 10% FBS were used as the positive control for the assay. (C) Nicotine was able to potently promote invasion of Aspc1 pancreatic cancer cells at a concentration of 1µM as seen in a Boyden-chamber assay. The pro-invasive activity of nicotine was abrogated by PP2 demonstrating a requirement for Src function.
Nicotine promotes wound healing in vitro in a dose-dependent manner
Given the results of the Boyden chamber assays and the observation that nicotine can promote migration and wound-healing in pancreatic cancer cells, attempts were made to see whether similar effects are observed in NSCLC and breast cancer cells. Towards this purpose, A549, MCF-7 and MDA-MB-468 cells were grown to 90% confluency and the plates were scratched with a pipet tip. Cells were treated with 0.01µM, 0.1µM or 1µM nicotine for 24 hours; treatment with 10% serum was used as a positive control. It was found that nicotine could induce the migration of cells to the scratched area in a dose-dependent manner (Figure 4). While there was only marginal invasion at 0.01µM nicotine, there were significantly more cells migrating to the wound when exposed to 0.1µM and 1µM nicotine. This result is in agreement with the data obtained in Boyden chamber assays (Figure 2F).
Figure 4.
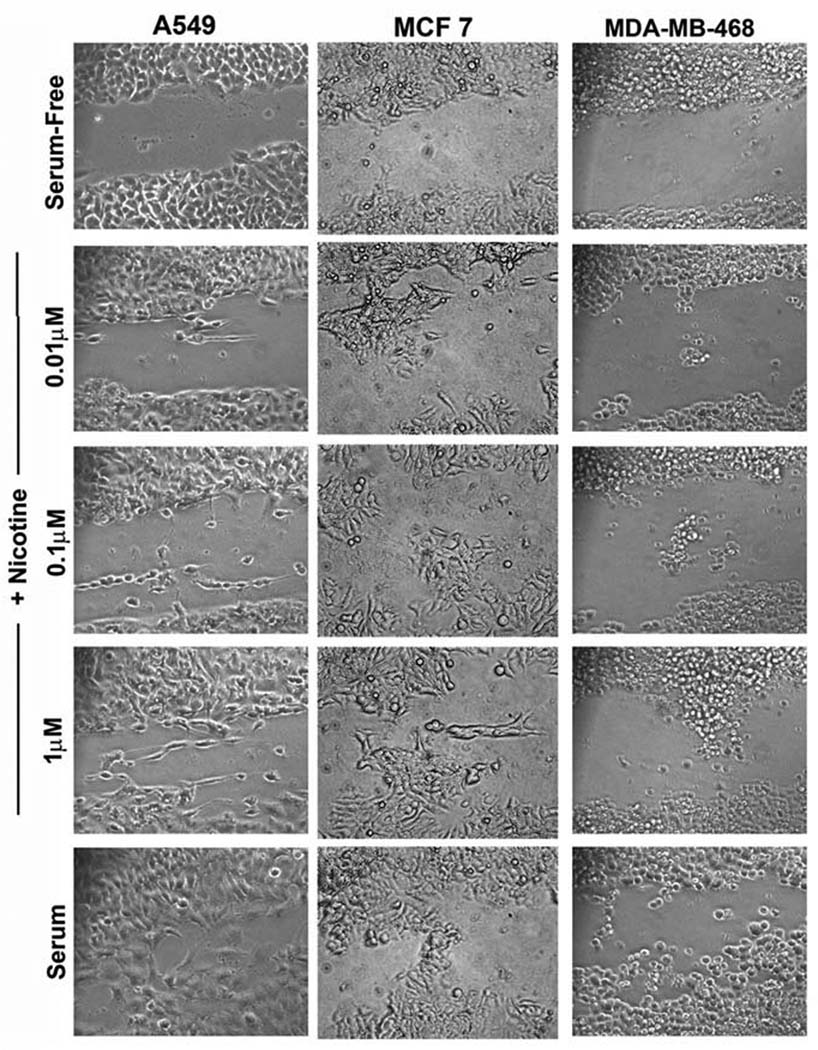
Nicotine can promote invasion of A549, MCF-7 and MDA-MB-468 cells as seen in a wound healing assay. Cells were scratched and treated with the indicated amounts of nicotine; cells treated with 10% serum were used as the positive control. Nicotine induced the migration of the cells in a dose-dependent manner.
Nicotine modulates the levels of multiple proteins involved in epithelial-to mesenchymal transition
Epithelial to mesenchymal transition (EMT) is known to be one of the vital steps for the acquisition of malignant phenotype 25. This transition allows the cell to acquire migratory properties and allows metastasis to a new location. Epithelial-mesenchymal transition involves the repression of epithelial-specific adhesion molecules like E-cadherin and β-catenin with a concomitant expression of proteins like fibronectin and vimentin 30. Since nicotine was found to alter the invasive and migratory properties of cells, we wanted to examine whether long-term treatment of cancer cells with nicotine could induce changes characteristic of the EMT process. Figure 5A shows that treatment of A549 cells with nicotine causes an increase in the levels of mesenchymal proteins like fibronectin and vimentin with a concomitant downregulation of epithelial markers E-cadherin and β-catenin. Interestingly, nicotine induced EMT in breast cancer cell lines like MCF-7 and MDA-MB-468; this is significant since smoking has been correlated with increased metastasis of breast cancers. Experiments were done to assess whether nicotine induced EMT in a dose-dependent fashion. Towards this purpose, A549, MCF-7 or MDA-MB-468 cells were rendered quiescent by growing in 0.1% serum for 24 hours and stimulated with 0.01µM, 0.1µM or 1µM nicotine for 72 hours. It was found that the maximal effect of nicotine was observed at 1µM concentration while 0.1µM nicotine also induced EMT like changes (Figure 5B). The changes were less marginal at the lowest concentration tested. All the three cell lines exposed to nicotine showed a reduction in E-cadherin and β-catenin levels while levels of fibronectin, vimentin and N-cadherin were elevated. This result shows that exposure to nicotine can induce EMT in a variety of human cancer cells.
Figure 5.
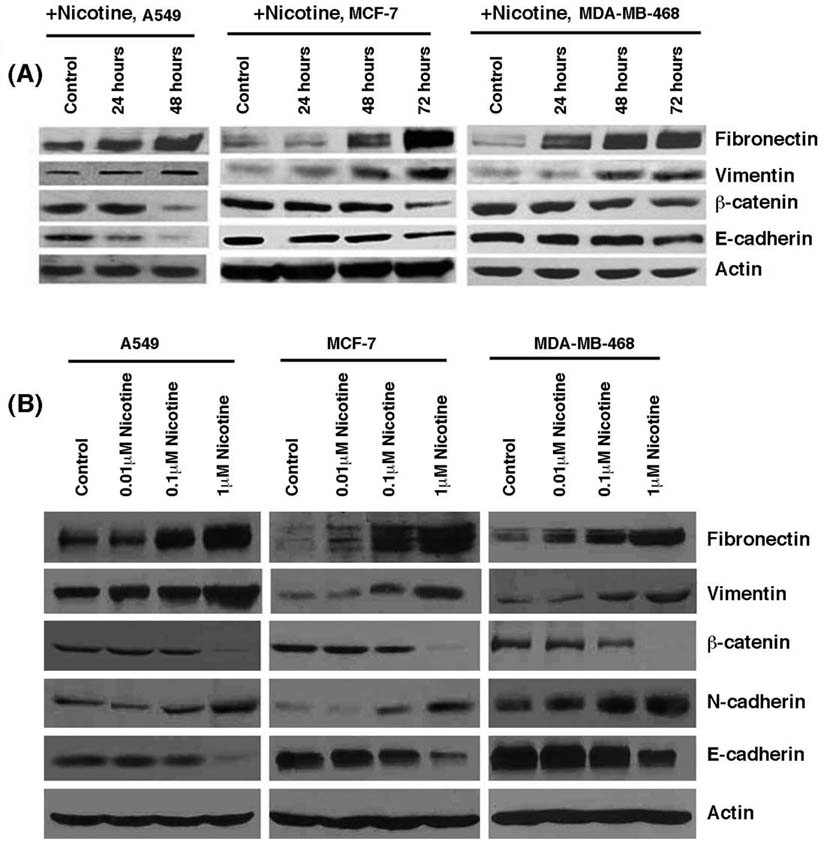
Nicotine induced epithelial to mesenchymal transition in human lung cancer cells and breast cancer cells. (A) Treatment with 1µM nicotine induced downregulation of epithelial markers E-cadherin and β-catenin, whereas it caused concomitant increase of mesenchymal proteins fibronectin and vimentin in A549 human NSCLC as well as MCF-7 and MDA-MB-468 human breast cancer cells. Quiescent cells were treated with 1µM nicotine for the indicated time-points and the expression of epithelial and mesenchymal markers was examined by immunoblotting. Western blotting for β-actin was used as the control for the assay. (B) Induction of EMT by nicotine in a dose-dependent manner. Quiescent cells were treated with the indicated amounts of nicotine for 72 hours and EMT markers examined by western blotting.
These results were further confirmed by immunofluorescence experiments in A549, MCF-7 and MDA-MB-468 cells (Figure 6). E-cadherin was localized on the outer cell membrane in all three cell lines (Figure 6, top panels); DAPI was used to stain the nuclei of the cells. It was observed that treatment with nicotine for 48 or 72 hours led to a significant reduction in the levels of E-cadherin, in all three cell lines (Figure 6, middle and lower panels). These results confirm that the epithelial adhesion molecule E-cadherin is indeed downregulated by nicotine, a molecular signature consistent with EMT.
Figure 6.

Nicotine treatment reduces the expression of E-cadherin as seen in immunofluorescence assays. Treatment with 1µM nicotine causes the downregulation and translocation of epithelial marker E-cadherin from the cell membrane in A549 (NSCLC), MCF-7 and MDA-MB-468 (breast cancer) cells in a time dependent manner. DAPI was used to visualize the nuclei.
Since induction of EMT facilitates detachment of contacts with adjacent cells, attempts were made to assess whether nicotine affected the levels and localization of the tight junction protein, ZO-1. Towards this purpose, an immunofluorescence experiment was conducted on A549, MCF-7 and MDA-MB468 cells. Quiescent cells displayed membranous staining of ZO-1 characteristic of epithelial tight junctions (Figure 7, top panel); Crm-1 was used as a marker for protein levels. However, when the cells were treated with 1µM nicotine for 48 or 72 hours, diffuse ZO-1 staining was observed, characteristic of a migratory mesenchymal phenotype (Figure 7, middle and bottom panel) 26, 27. Further, the overall intensity of the ZO-1 staining was reduced. Taken together, our data indicates that exposure of lung cancer and breast carcinoma cells to nicotine induce EMT like changes, which facilitate disruption of cell-cell contacts and subsequently metastasis.
Figure 7.

Nicotine induces disruption of tight junctions to facilitate EMT and invasion. (A) Nicotine downregulates the membrane localization of the tight junction protein ZO-1 in human A549 lung cancer cells and MCF-7, MDA-MB-468 breast cancer cells. Quiescent cells were treated with 1µM nicotine for the indicated time-points. Immunofluorescence analysis demonstrates the reduction in membrane localization of ZO-1 upon nicotine treatment, indicating that nicotine reduces the presence of membrane-bound ZO-1.
Discussion
The present study shows that nicotine induces a dose-dependent increase in proliferation of lung cancer cells, breast cancer cells and pancreatic cancer cells via α7-nAChRs-mediated signal transduction pathways. These results are in agreement with our earlier studies as well as recent studies from other labs that show the importance of the α7-receptor subunit in nicotine-induced cell proliferation. Indeed, a recent study has shown that proliferation of transplanted A549 cells in nude mice can be inhibited by α-cobratoxin, an α7 receptor antagonist10, 38. Nicotinic acetylcholine receptors particularly α7-nAChRs have been detected in primary endothelial cells as well as human NSCLC cell lines A549, NCI-H23 and H1299; similarly, other labs have shown that these receptor subunits are expressed on human lung cancers 11, 20, 33, 39–42. Thus it appears that the presence of these receptor subunits and their ability to promote cell proliferation might contribute to the growth of tumors already initiated by tobacco-specific carcinogens. The importance of α7-nAChRs in mediating the pathophysiological effects of nicotine is further reinforced by the fact that it is overexpressed on human NSCLC tumors relative to adjacent normal tissue. At the same time, recent studies from the Minna lab had shown that the expression of α6 and β4 subunits is different on tumors from smokers and non-smokers 43; this raises the possibility that other subunits might also be contributing to the growth of lung cancers in vivo. In addition, it has been reported that variations in chromosomal loci 15q24 and 15q25, which harbor genes for nicotinic acetylcholine receptors, correlate with nicotine dependence, lung cancer and peripheral arterial disease 44–46. These and other studies lend support to the idea that exposure to nicotine and enhanced activation of nicotinic acetylcholine receptors contribute to lung cancer.
Apart from non-small cell lung cancer, cigarette smoking has been implicated in the pathogenesis of breast, gastric, colon and cervical cancers 1. Our results show that nicotine can induce proliferation in a variety of cancer cell lines apart from NSCLC; furthermore, this induction appears to be through α7-nAChR subunits, which are expressed on these cell lines. Our findings seem to suggest that nAChRs may be an autocrine mitogenic signaling pathway facilitating the growth of several types of cancers in addition to lung cancer. Several lines of evidence show that β-adrenergic receptors also mediate the proliferative, pro-angiogenic and anti-apoptotic effects of nicotine, mainly in non-lung cancer cells. The mitogenic and proangiogenic effects of nicotine have been found to be mediated by β-adrenergic receptors in colon and gastric cancer cells 47, 48. Studies by Shin et al. (2007) indicate that nicotine promotes growth of gastric cancers via PKC, ERK1/2 phosphorylation, and COX-2 activation in a β-adrenergic receptor-dependent fashion 49. Further, data by Wong et al., (2007) demonstrate that nicotine-induced proliferation of HT-29 colon cancer cells are mediated by both α7-nAChRs and β-adrenergic receptors 47. Their findings show that nicotine binds to α7-nAChRs nicotine on the membranes of HT-29 colon cancer cells and thereby facilitates downstream production of adrenaline and β-adrenergic activation; thus β-adrenergic receptors contribute to the proliferation indirectly. These data reveal that the both α7-nAChRs and β-adrenergic receptors contribute to the mitogenic effects of nicotine in colon cancer cells. The proliferative effects of the tobacco carcinogen NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone] have been found to be mediated by β-adrenergic receptors on human adenocarcinomas of the lungs, pancreas, and breast 40, 50. Thus it is possible that β-adrenergic receptors are also contributing to the observed changes, either directly or indirectly.
Multiple lines of evidence indicate that cigarette smoking not only facilitates proliferation of cancer cells but might also promote their metastatic spread as well 51–53. Invasion and metastasis are complex, multi-step processes that involve alteration of cell adhesion to extracellular matrix proteins as well as disruption of cell-cell junctions. Nicotine has been shown to promote phosphorylation of calpains, upregulation of COX2, VEGF and VEGFR2; these molecules are known to affect the metastasic process 54–56. Our findings shed light on additional pathways that contribute to the pro-invasive effects of nicotine. These include α7-nAChR-mediated activation of Src, calcium channels and upregulation of EGFR. These findings raise the possibility that inhibition of α7 nAChR activity by non-toxic agents or inhibition of its downstream mediators might open novel avenues for the therapy of cancers promoted by smoking 57.
Our data further shows that sustained exposure to nicotine promotes anchorage-independent growth by downregulation of anoikis. Ability to survive independent of a substratum is a feature of cancer cells that is indispensable for metastasis 58. Anoikis prevents normal cells from detaching from their substratum and migrating to different locations; the ability of nicotine to enhance the survival of cancer cells independent of a substratum might be contributing significantly to the ability of these cells to detach and find alternate sites of attachment. Indeed, nicotine could enhance adherence-independent proliferation of tumor cells lines, showing that the proliferative and survival advantages it provides allows the cells to grow robustly independent of a substratum.
Clinical and epidemiological studies have suggested that smokers tend to have more progressed and metastatic cancer than non-smokers 23. Further, smokers had enhanced metastasis of breast cancers to the lung 51, 53. Our findings suggest that nicotine induces changes consistent with EMT is highly relevant in this context. Indeed, earlier studies had suggested that long-term exposure to nicotine could alter the phenotype of epithelial and endothelial cells 12, 28. We provide the molecular changes that facilitate these morphological changes. Results presented here show that chronic treatment with nicotine resulted in the downregulation of ECM proteins E-cadherin and β-catenin, with concomitant increase of fibronectin and vimentin. Several studies show that the decrease in E-cadherin and β-catenin with a concurrent increase in fibronectin and vimentin levels is one of the hallmarks of EMT in lung cancer cells. Further, clinical studies also report that smoking decreases levels of E-cadherin in lung tumors and might be contributing to resistance to chemotherapeutic agents 33. It is probable that apart from its pro-invasive activity, nicotine plays a role in promoting EMT via downregulation of ECM proteins.
The results presented here suggest that while nicotine is not a carcinogen by itself , it has the potential to promote the growth and progression of tumors. The ability of nicotine to promote proliferation, angiogenesis, adherence-independent growth, and EMT while inhibiting anoikis might be contributing significantly to the growth and metastasis of tumors that are sensitive to nicotinic acetylcholine receptors. Further, these results also show that exposure to nicotine can affect tumors of tissues other than that of the lung. Development of agents that can disrupt the nAChR signaling pathway might prove beneficial in the treatment of such cancers.
Acknowledgements
We thank the Analytical Microscopy and Histopathology Core Facilities at Moffitt Cancer Center for their assistance and George Simon for helpful discussions. These studies were supported by the Bankhead-Coley Grant # 06BB-04-9587 to SC. RK is a recipient of a pre-doctoral fellowship from the American Heart Association.
References
- 1.ACS. Cancer facts and figures. American Cancer Society. 2002 [Google Scholar]
- 2.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3(8):461–469. doi: 10.1016/s1470-2045(02)00815-x. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12147432. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS, Abbaspour A, Hoffman D. A study of tobacco carcinogenesis. XLII. Bioassay in A/J mice of some structural analogues of tobacco-specific nitrosamines. Cancer Lett. 1988;42(1–2):141–145. doi: 10.1016/0304-3835(88)90251-0. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=3180033. [DOI] [PubMed] [Google Scholar]
- 4.Sekido Y, Fong KM, Minna JD. Molecular genetics of lung cancer. Annu Rev Med. 2003;54:73–87. doi: 10.1146/annurev.med.54.101601.152202. [DOI] [PubMed] [Google Scholar]
- 5.Arredondo J, Chernyavsky AI, Grando SA. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol Ther. 2006;5(5):511–517. doi: 10.4161/cbt.5.5.2601. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16582591. [DOI] [PubMed] [Google Scholar]
- 6.Bose C, Zhang H, Udupa KB, Chowdhury P. Activation of p-ERK1/2 by nicotine in pancreatic tumor cell line AR42J: effects on proliferation and secretion. Am J Physiol Gastrointest Liver Physiol. 2005;289(5):G926–G934. doi: 10.1152/ajpgi.00138.2005. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16051920. [DOI] [PubMed] [Google Scholar]
- 7.Shin VY, Wu WK, Chu KM, Wong HP, Lam EK, Tai EK, Koo MW, Cho CH. Nicotine induces cyclooxygenase-2 and vascular endothelial growth factor receptor-2 in association with tumor-associated invasion and angiogenesis in gastric cancer. Mol Cancer Res. 2005;3(11):607–615. doi: 10.1158/1541-7786.MCR-05-0106. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16317086. [DOI] [PubMed] [Google Scholar]
- 8.Lindstrom J. Neuronal nicotinic acetylcholine receptors. Ion Channels. 1996;4:377–450. doi: 10.1007/978-1-4899-1775-1_10. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8744214. [DOI] [PubMed] [Google Scholar]
- 9.Lindstrom J. Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol. 1997;15(2):193–222. doi: 10.1007/BF02740634. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9396010. [DOI] [PubMed] [Google Scholar]
- 10.Trombino S, Bisio A, Catassi A, Cesario A, Falugi C, Russo P. Role of the non-neuronal human cholinergic system in lung cancer and mesothelioma: possibility of new therapeutic strategies. Curr Med Chem Anticancer Agents. 2004;4(6):535–542. doi: 10.2174/1568011043352687. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15579018. [DOI] [PubMed] [Google Scholar]
- 11.Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, Chellappan S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116(8):2208–2217. doi: 10.1172/JCI28164. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16862215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nature Medicine. 2001;7(7):833–839. doi: 10.1038/89961. [DOI] [PubMed] [Google Scholar]
- 13.Heeschen C, Weis M, Aicher A, Dimmler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Invest. 2002;110:527–536. doi: 10.1172/JCI14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gazdar AF. Environmental tobacco smoke, carcinogenesis, and angiogenesis: a double whammy? Cancer Cell. 2003;4(3):159–160. doi: 10.1016/s1535-6108(03)00222-8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14522247. [DOI] [PubMed] [Google Scholar]
- 15.Mathai M, Skinner A, Lawton K, Weindling AM. Maternal smoking, urinary cotinine levels and birth-weight. Aust N Z J Obstet Gynaecol. 1990;30(1):33–36. doi: 10.1111/j.1479-828x.1990.tb03192.x. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2346450. [DOI] [PubMed] [Google Scholar]
- 16.Matsukura S, Taminato T, Kitano N, Seino Y, Hamada H, Uchihashi M, Nakajima H, Hirata Y. Effects of environmental tobacco smoke on urinary cotinine excretion in nonsmokers. Evidence for passive smoking. N Engl J Med. 1984;311(13):828–832. doi: 10.1056/NEJM198409273111305. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=6472384. [DOI] [PubMed] [Google Scholar]
- 17.Thompson SG, Stone R, Nanchahal K, Wald NJ. Relation of urinary cotinine concentrations to cigarette smoking and to exposure to other people's smoke. Thorax. 1990;45(5):356–361. doi: 10.1136/thx.45.5.356. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2382242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itier V, Bertrand D. Neuronal nicotinic receptors: from protein structure to function. FEBS Lett. 2001;504(3):118–125. doi: 10.1016/s0014-5793(01)02702-8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11532443. [DOI] [PubMed] [Google Scholar]
- 19.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74(6):363–396. doi: 10.1016/j.pneurobio.2004.09.006. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15649582. [DOI] [PubMed] [Google Scholar]
- 20.Schuller HM, Plummer HK, 3rd, Jull BA. Receptor-mediated effects of nicotine and its nitrosated derivative NNK on pulmonary neuroendocrine cells. Anat Rec. 2003;270A(1):51–58. doi: 10.1002/ar.a.10019. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12494489. [DOI] [PubMed] [Google Scholar]
- 21.Cook JP, Bitterman H. Nicotine and angiogenesis: a new paradigm for tobacco-related diseases. Ann Med. 2004;36:33–40. doi: 10.1080/07853890310017576. [DOI] [PubMed] [Google Scholar]
- 22.Shin VY, Wu WK, Ye YN, So WH, Koo MW, Liu ES, Luo JC, Cho CH. Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal-regulated kinase and cyclooxygenase-2. Carcinogenesis. 2004;25(12):2487–2495. doi: 10.1093/carcin/bgh266. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15319299. [DOI] [PubMed] [Google Scholar]
- 23.Richardson GE, Tucker MA, Venzon DJ, Linnoila RI, Phelps R, Phares JC, Edison M, Ihde DC, Johnson BE. Smoking cessation after successful treatment of small-cell lung cancer is associated with fewer smoking-related second primary cancers. Ann Intern Med. 1993;119(5):383–390. doi: 10.7326/0003-4819-119-5-199309010-00006. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8393311. [DOI] [PubMed] [Google Scholar]
- 24.Vander Ark W, DiNardo LJ, Oliver DS. Factors affecting smoking cessation in patients with head and neck cancer. Laryngoscope. 1997;107(7):888–892. doi: 10.1097/00005537-199707000-00010. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9217125. [DOI] [PubMed] [Google Scholar]
- 25.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–558. doi: 10.1016/j.ceb.2005.08.001. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16098727. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15210113. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res. 2006;66(9):4549–4552. doi: 10.1158/0008-5472.CAN-05-3850. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16651402. [DOI] [PubMed] [Google Scholar]
- 28.West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, Harris C, Belinsky S, Dennis PA. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111(1):81–90. doi: 10.1172/JCI16147. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12511591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaskovich MA, Lin Q, Delarue FL, Sun J, Park HS, Coppola D, Hamilton AD, Sebti SM. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat Biotechnol. 2000;18(10):1065–1070. doi: 10.1038/80257. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11017044. [DOI] [PubMed] [Google Scholar]
- 30.Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156(2):299–313. doi: 10.1083/jcb.200109037. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11790801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janda E, Litos G, Grunert S, Downward J, Beug H. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene. 2002;21(33):5148–5159. doi: 10.1038/sj.onc.1205661. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12140765. [DOI] [PubMed] [Google Scholar]
- 32.Vukanovic J, Passaniti A, Hirata T, Traystman RJ, Hartley-Asp B, Isaacs JT. Antiangiogenic effects of the quinoline-3-carboxamide linomide. Cancer Res. 1993;53(8):1833–1837. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7682157. [PubMed] [Google Scholar]
- 33.Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A. 2006;103(16):6332–6337. doi: 10.1073/pnas.0509313103. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rastogi S, Joshi B, Dasgupta P, Morris M, Wright K, Chellappan S. Prohibitin facilitates cellular senescence by recruiting specific corepressors to inhibit E2F target genes. Mol Cell Biol. 2006;26(11):4161–4171. doi: 10.1128/MCB.02142-05. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16705168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilmore AP. Anoikis. Cell Death Differ. 2005;12 Suppl 2:1473–1477. doi: 10.1038/sj.cdd.4401723. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16247493. [DOI] [PubMed] [Google Scholar]
- 36.Frisch SM. Anoikis. Methods Enzymol. 2000;322:472–479. doi: 10.1016/s0076-6879(00)22043-0. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10914040. [DOI] [PubMed] [Google Scholar]
- 37.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555–562. doi: 10.1016/s0955-0674(00)00251-9. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11544023. [DOI] [PubMed] [Google Scholar]
- 38.Trombino S, Cesario A, Margaritora S, Granone P, Motta G, Falugi C, Russo P. Alpha7-nicotinic acetylcholine receptors affect growth regulation of human mesothelioma cells: role of mitogen-activated protein kinase pathway. Cancer Res. 2004;64(1):135–145. doi: 10.1158/0008-5472.can-03-1672. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14729617. [DOI] [PubMed] [Google Scholar]
- 39.Schuller HM, Orloff M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem Pharmacol. 1998;55(9):1377–1384. doi: 10.1016/s0006-2952(97)00651-5. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10076528. [DOI] [PubMed] [Google Scholar]
- 40.Schuller HM, Tithof PK, Williams M, Plummer H., 3rd The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999;59(18):4510–4515. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10493497. [PubMed] [Google Scholar]
- 41.Minna JD. The molecular biology of lung cancer pathogenesis. Chest. 1993;103(4 Suppl):449S–456S. doi: 10.1378/chest.103.4_supplement.449s. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8462339. [DOI] [PubMed] [Google Scholar]
- 42.Minna JD. Nicotine exposure and bronchial epithelial cell nicotinic acetylcholine receptor expression in the pathogenesis of lung cancer. J Clin Invest. 2003;111(1):31–33. doi: 10.1172/JCI17492. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12511585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam DC, Girard L, Ramirez R, Chau WS, Suen WS, Sheridan S, Tin VP, Chung LP, Wong MP, Shay JW, Gazdar AF, Lam WK, et al. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007;67(10):4638–4647. doi: 10.1158/0008-5472.CAN-06-4628. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17510389. [DOI] [PubMed] [Google Scholar]
- 44.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–642. doi: 10.1038/nature06846. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18385739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452(7187):633–637. doi: 10.1038/nature06885. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18385738. [DOI] [PubMed] [Google Scholar]
- 46.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40(5):616–622. doi: 10.1038/ng.109. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18385676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol Appl Pharmacol. 2007;221(3):261–267. doi: 10.1016/j.taap.2007.04.002. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17498763. [DOI] [PubMed] [Google Scholar]
- 48.Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes colon tumor growth and angiogenesis through beta-adrenergic activation. Toxicol Sci. 2007;97(2):279–287. doi: 10.1093/toxsci/kfm060. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17369603. [DOI] [PubMed] [Google Scholar]
- 49.Shin VY, Wu WK, Chu KM, Koo MW, Wong HP, Lam EK, Tai EK, Cho CH. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Toxicol Sci. 2007;96(1):21–29. doi: 10.1093/toxsci/kfl118. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17003101. [DOI] [PubMed] [Google Scholar]
- 50.Schuller HM. Neurotransmitter receptor-mediated signaling pathways as modulators of carcinogenesis. Prog Exp Tumor Res. 2007;39:45–63. doi: 10.1159/000100045. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17314500. [DOI] [PubMed] [Google Scholar]
- 51.Murin S, Pinkerton KE, Hubbard NE, Erickson K. The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer. Chest. 2004;125(4):1467–1471. doi: 10.1378/chest.125.4.1467. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15078760. [DOI] [PubMed] [Google Scholar]
- 52.Daniell HW. Increased lymph node metastases at mastectomy for breast cancer associated with host obesity, cigarette smoking, age, and large tumor size. Cancer. 1988;62(2):429–435. doi: 10.1002/1097-0142(19880715)62:2<429::aid-cncr2820620230>3.0.co;2-4. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=3383142. [DOI] [PubMed] [Google Scholar]
- 53.Murin S, Inciardi J. Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest. 2001;119(6):1635–1640. doi: 10.1378/chest.119.6.1635. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11399684. [DOI] [PubMed] [Google Scholar]
- 54.Xu L, Deng X. Protein kinase Ciota promotes nicotine-induced migration and invasion of cancer cells via phosphorylation of micro- and m-calpains. J Biol Chem. 2006;281(7):4457–4466. doi: 10.1074/jbc.M510721200. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16361262. [DOI] [PubMed] [Google Scholar]
- 55.Cooke JP. Angiogenesis and the role of the endothelial nicotinic acetylcholine receptor. Life Sci. 2007;80(24–25):2347–2351. doi: 10.1016/j.lfs.2007.01.061. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17383685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanda Y, Watanabe Y. Nicotine-induced vascular endothelial growth factor release via the EGFR-ERK pathway in rat vascular smooth muscle cells. Life Sci. 2007;80(15):1409–1414. doi: 10.1016/j.lfs.2006.12.033. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17286987. [DOI] [PubMed] [Google Scholar]
- 57.Grozio A, Catassi A, Cavalieri Z, Paleari L, Cesario A, Russo P. Nicotine, lung and cancer. Anticancer Agents Med Chem. 2007;7(4):461–466. doi: 10.2174/187152007781058587. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17630920. [DOI] [PubMed] [Google Scholar]
- 58.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10647931. [DOI] [PubMed] [Google Scholar]