

Abstract
Recent findings suggest that the neurexin-neuroligin link promotes both GABAergic and glutamatergic synaptogenesis, but the mechanism by which neurexins influence the clustering of appropriate neuroligins and postsynaptic differentiation remains unclear. Previous studies suggested that the presence or absence of alternatively spliced residues at splice site 4 (S4) in the neurexin LNS domain may regulate neurexin function. We demonstrate that addition of the S4 insert selectively reduces the ability of neurexin-1β to cluster neuroligin-1/3/4 and glutamatergic postsynaptic proteins, although clustering of neuroligin-2 and GABAergic postsynaptic proteins remain strong. Furthermore, addition of the S4 insert decreases the binding affinity of neurexin-1β to neuroligins-1 and -4 but has little effect on binding to neuroligins-2 and -3. Additional structure-function studies reveal the neurexin binding interface mediating synaptogenic activity to be composed primarily of residues in the β2β3, β6β7, and β10β11 loops on one rim of the LNS domain β sandwich. Mutation of two predicted Ca2+-binding residues disrupts postsynaptic protein clustering and binding to neuroligins, consistent with previous findings that neurexin-neuroligin binding is Ca2+ dependent. Glutamatergic postsynaptic clustering was more readily disrupted by the mutagenesis than GABAergic postsynaptic protein clustering. Perhaps neurexins-neuroligins, or neurexin-1β at least, is most important for GABA synapse formation or controlling the balance of GABA and glutamate synapses. These results suggest that differential neurexin-neuroligin binding affinities and splice variations may play an instructive role in postsynaptic differentiation.
Keywords: synaptogenesis, neuroligin, glutamate, GABA, gephyrin, PSD-95, postsynaptic
Introduction
During synaptogenesis, a neuron must cluster the proper postsynaptic proteins opposite the correct presynaptic axon terminals. Recent work suggests that neurexins and neuroligins are core components of both glutamatergic and GABAergic synapses (Brose, 1999; Graf et al., 2004; Prange et al., 2004; Chih et al., 2005). Presynaptic neurexins and postsynaptic neuroligins likely work together with other synaptic cell adhesion molecules of the cadherin and ephrin families, and secreted proteins such as pentraxins, to mediate CNS synaptogenesis (Garner et al., 2002; Li and Sheng, 2003; Biederer, 2005; Craig et al., 2006). We have shown previously that neurexin-1β expressed in a fibroblast or attached to a bead is able to cluster both GABAergic and glutamatergic postsynaptic receptors and scaffolding proteins in contacting dendrites (Graf et al., 2004). Furthermore, our data suggested that neurexins may mediate synaptic differentiation by binding to the proper neuroligin isoforms, thereby causing the clustering of appropriate postsynaptic proteins. Whereas neuroligin-1 localizes to glutamatergic synapses (Song et al., 1999), neuroligin-2 localizes primarily to GABAergic synapses (Graf et al., 2004; Varoqueaux et al., 2004). In addition, whereas direct aggregation of neuroligins-1/3/4 coaggregates the glutamatergic scaffolding protein postsynaptic density-95 (PSD-95) but not the GABAergic scaffolding protein gephryin, aggregation of neuroligin-2 coaggregates both PSD-95 and gephryin, gephryin to a greater extent than PSD-95 (Graf et al., 2004). Thus, the differential binding affinities of the various neurexin isoforms and splice variants to the four neuroligin isoforms may be a source of instruction for postsynaptic differentiation.
Neurexins are a family of presynaptic transmembrane proteins composed of six main isoforms and multiple splice variants (Ushkaryov et al., 1992; Tabuchi and Sudhof, 2002). In situ hybridization studies have shown that the six major neurexin isoforms are expressed in both excitatory and inhibitory neurons (Ullrich et al., 1995), suggesting that postsynaptic differentiation may not be dependent on the general isoform expressed by the presynaptic axon. All neurexins are expressed in multiple splice variants (Ushkaryov et al., 1992), and β-neurexins have one splice site that is of particular interest, because it is located in the LNS domain (named after laminin, neurexin, sex hormone-binding protein), the only domain of neurexin-1β that is both necessary and sufficient for synaptogenic activity (Graf et al., 2004). Addition of the 30 aa insert at this site, called splice site 4 (S4), was reported previously by biochemical assays to completely abolish binding of neurexin-1β to neuroligin (Ichtchenko et al., 1995, 1996) (but see Boucard et al., 2005) and to abolish neurexin-neuroligin-mediated cell adhesion (Nguyen and Sudhof, 1997; Dean et al., 2003).
Thus, we wished to determine whether insertion or deletion of the alternatively spliced residues at S4 affected the synaptogenic activity of neurexin-1β. We show here that rather than destroying the ability of neurexin to bind to the various neuroligins, addition of the S4 insert alters binding and synaptogenic activity in a more complex way. In addition, we used a structure-function assay to determine the loops and individual residues that constitute the binding interface mediating synaptogenic activity of the neurexin-1β LNS domain.
Materials and Methods
Cell culture.
Cultures were prepared from hippocampal neurons using methods described previously (Goslin et al., 1998). Hippocampi were dissected from embryonic day 18 (E18) rat embryos and dissociated by trypsinization and trituration. Neurons were plated at a final concentration of 300,000 cells/dish on poly-l-lysine-coated coverslips in 60 mm culture dishes in MEM supplemented with 10% horse serum. After 2–4 h, coverslips were inverted over an astroglia feeder layer in serum-free MEM with N2 supplements, 0.1% ovalbumin, and 1 mm pyruvate. After 2 d, neuron cultures were treated with cytosine arabinoside (5 μm; Calbiochem, La Jolla, CA) to prevent the overgrowth of glia. Cultures were maintained in N2.1 media, feeding the cells once per week by replacing one-third of the media per dish. Neurons were treated with either 100 μm APV (Research Biochemicals, Natick, MA) or 10 μm (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (Alexis Corporation, San Diego, CA) beginning on day 7. All analyses were performed on neurons that were 8–10 d in vitro.
COS-7 cells were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For protein expression in COS cells, cells were transfected with Effectene Transfection Reagent (Qiagen, Hilden, Germany) and fixed 24 h later. For fibroblast-neuron cocultures, COS-7 cells were transfected with Effectene or Lipofectamine 2000 (Invitrogen, San Diego, CA) and trypsinized 24 h later. Cells were washed twice with FBS supplemented DMEM and plated onto neurons. Fibroblast cells were allowed to adhere and grow on neurons for 24 h before fixation.
DNA constructs.
The expression vector for soluble neurexin-1β-Fc containing amino acids 1–262 of mouse neurexin-1β was received from P. Scheiffele (Columbia University, New York, NY) (Scheiffele et al., 2000). Neurexin-1β-cyan fluorescent protein (CFP) was made as described previously (Graf et al., 2004). This form of neurexin-1β lacks any insert at splice site 4. To add the insert at splice site 4 to both soluble neurexin-Fc and membrane bound neurexin-CFP, amino acids GNNDNERLAIARQRIPYRLGRVVDEWLLDK were inserted between residues 200 and 201 by PCR and triple ligation using silent restriction sites. Mutations of the individual or three to five tandem residues to alanine were created with the Stratagene (La Jolla, CA) Site-Directed Mutagenesis kit. All neurexin variants were verified by sequencing. To create myc-tagged neurexins, the myc tag sequence EQKLISEEDL was added to the neurexin mature N terminus. The hemagglutinin (HA)-tagged neuroligins-1 and -2 (gift from P. Scheiffele) (Scheiffele et al., 2000) both contain the neuroligin-1 signal sequence followed by the HA tag (YPYDVPDYA) and the mature N terminus of each of the neuroligins inserted into the pNice expression vector. HA-neuroligin-3 and -4 were created by replacing the neuroligin-2 sequence following HA with the mature neuroligin-3 and -4 sequences from human neuroligin-3 (BC051715; Open Biosystems, Huntsville, AL) and human neuroligin-4 (BC034018; Open Biosystems). These forms correspond to splice variants neuroligin-1 A2+ B+, neuroligin-2 A2+, neuroligin-3 A2+, and neuroligin-4 lacking inserts.
Antibodies.
The following mouse monoclonal antibodies were used: gephyrin (mAb7a; IgG1; 1:500; Synaptic Systems, Goettingen, Germany); PSD-95 [although the term PSD-95 is used for simplicity, this antibody recognizes PSD-95, PSD-93, SAP102, and SAP97 (Sugiyama et al., 2005); 6G6–1C9; IgG2a; 1:500; Affinity Bioreagents, Golden, CO], neuroligin-1/3/4 (this antibody recognizes neuroligins-1, -3, and -4 but not -2 (Song et al., 1999; Bolliger et al., 2001); 4F9; IgG2a; 1:1000; Synaptic Systems); synapsin (46.1; IgG1; 1:100; Synaptic Systems); HA (IgG2b; 1:500; Roche Products, Welwyn Garden City, UK); myc (9E10; IgG1; 1:2000; Upstate Biotechnology, Lake Placid, NY). Rabbit antibodies were used against synapsin (1:1000; Chemicon, Temecula, CA). A rabbit antibody was generated against a unique peptide of neuroligin-2 RGGGVGADPAEALRPACP conjugated to keyhole limpet hemocyanin via the cysteine (custom generated through Zymed, San Francisco, CA). The antibody was affinity purified against the peptide antigen coupled to Sepharose. This neuroligin-2 antibody specifically recognizes neuroligin-2 expressed in COS cells and gives the same pattern of immunoreactivity at GABA synapses of neurons as previously reported for other neuroligin-2 antibodies (Graf et al., 2004; Varoqueaux et al., 2004). Secondary antibodies were: Alexa488, Alexa568, and Alexa647 conjugated anti-mouse IgG1, anti-mouse IgG2a, anti-mouse IgG2b, and anti-rabbit (Invitrogen); and FITC anti-human (Jackson ImmunoResearch, West Grove, PA).
Immunocytochemistry.
Generally, neurons were fixed for 15 min in warm PBS with 4% paraformaldehyde and 4% sucrose and permeabilized with 0.25% Triton X-100. For experiments involving immunocytochemistry for neuroligin-1/3/4 or -2 in the cocultures, cells were fixed in −20°C methanol for 10 min. Fixed cells were incubated in 10% BSA (30 min; 37°C), appropriate primary antibody (Ab) in PBS with 3% BSA (overnight; 20°C), and secondary Ab (45 min; 37°C). Coverslips were mounted in elvanol (Tris-HCl, glycerol, and polyvinyl alcohol with 2% 1,4-diazabicyclo[2,2,2]octane). Fibroblast-neuron cocultures were generally incubated in mouse IgG1 anti-gephyrin, mouse IgG2a anti-PSD-95, and rabbit anti-synapsin followed by secondary antibodies conjugated to Alexa 488, Alexa 568, and Alexa 647, respectively. Alternately, cocultures were labeled for neuroligin-1/3/4 or neuroligin-2 in the Alexa 568 channel and mouse IgG1 anti-synapsin in the Alexa 647 channel. To determine surface localization of mutated myc-neurexin-CFP proteins in transfected COS cells, cells were stained live with myc antibody for 15 min at room temperature before being fixed and incubated with secondary antibody. Fluorescence and phase-contrast images were captured with a Photometrics (Huntington Beach, CA) Sensys cooled CCD camera mounted on a Zeiss (Thornwood, NY)Axioskop microscope with a 63× 1.4 numerical aperture oil objective using MetaMorph imaging software (Molecular Devices, Sunnyvale, CA). Images were prepared using Adobe Photoshop 5.5 software (Adobe Systems, San Jose, CA).
Neurexin-neuroligin binding.
Both splice variants (−S4 and +S4) and selected point mutant forms of soluble neurexin-Fc (human IgG) were expressed in COS cells. Protein was collected in serum-free media, purified with protein A columns from an ImmunoPure IgG Purification Kit (Pierce, Rockford, IL), and concentrated in PBS with Centricon filters (Millipore, Molsheim, France). To determine neurexin binding to neuroligin, neuroligin-transfected COS cells were costained live with HA antibody and 50 ng/μl soluble neurexin-Fc protein for 15 min at room temperature. Cells were then fixed and incubated with Alexa568-conjugated anti-mouse IgG2b to detect surface HA-neuroligins and FITC-conjugated anti-human Ig to detect bound neurexin-Fc.
Image analysis.
Sets of cells used for quantification were stained simultaneously. Images were randomized before quantification so that the experimenter was blind to the treatment group. For the neuron-fibroblast coculture assay, transfected COS cells were chosen randomly based on phase contrast showing significant contact of COS cells with dendrites. Images were taken of the postsynaptic proteins, the presynaptic protein, and the transfected COS cell [neurexin-CFP or membrane-associated CFP (mCFP)] using the same exposure time for both experimental and control conditions. Images of the presynaptic and postsynaptic proteins were thresholded, and the area for measuring was defined by the perimeter of the transfected COS cell. For each postsynaptic protein cluster, the area and total gray value was measured. A region was drawn around each cluster, and thresholded synapsin was measured through these regions to determine which clusters were synaptic. Only values from postsynaptic protein clusters that were not apposed to synapsin were used in the final quantification.
To determine the binding affinity of soluble neurexin-Fc variants to neuroligins, regions were drawn around the exact perimeter of each cell, and the average gray values of neurexin-Fc and HA-neuroligin were measured within the region. The average gray value of the off-cell background was subtracted, and the ratio of average bound neurexin-Fc intensity to average surface HA-neuroligin intensity was determined.
Analysis was performed using MetaMorph, Microsoft (Seattle, WA) Excel, StatView, and Cricket-Graph. Statistical comparisons of immunofluorescence were made using Student’s unpaired t test. All data are reported as mean ± SEM. Three-dimensional (3D) structure images of the neurexin LNS domain were generated using visual molecular dynamics (Humphrey et al., 1996).
Results
Effect of alternative splicing at S4 on synaptogenic activity
We have shown previously that full-length neurexin-1β-CFP without the insert at splice site 4 (−S4) induces clustering of gephyrin and PSD-95 in a fibroblast-neuron coculture assay (Graf et al., 2004). These studies suggested that neurexin clusters gephyrin and PSD-95 by binding to the various neuroligin isoforms. Because it had been reported previously that addition of the insert at splice site 4 (+S4) abolishes neurexin-neuroligin binding (Ichtchenko et al., 1995, 1996), we determined whether the S4 insert also affected clustering of gephyrin and PSD-95 in our coculture assay. Therefore, we inserted the amino acid sequence GNNDNERLAIARQRIPYRLGRVVDEWLLDK into the LNS domain between residues 200 and 201 of the original neurexin-1β-CFP construct with CFP attached to the C terminus, creating nrx(+S4)-CFP. We then cultured COS fibroblast cells transfected with either nrx(+S4)-CFP or neurexin-CFP with no splice site 4 insert, notated nrx(−S4)-CFP, onto 1-week-old hippocampal neuron cultures grown by the Banker method (Goslin et al., 1998). The cocultures were immunostained for gephyrin and PSD-95. As reported previously (Graf et al., 2004), nrx(−S4)-CFP induced clusters of the postsynaptic scaffolding proteins PSD-95 and gephyrin in dendrites at contact sites with the transfected fibroblasts (Fig. 1A). Absence of immunoreactivity of the presynaptic antigen synapsin distinguished induced clusters of gephyrin and PSD-95 from endogenous synaptic clusters that may happen to underlie transfected fibroblasts. Surprisingly, nrx(+S4)-CFP retained the ability to cluster both gephyrin and PSD-95 (Fig. 1B). For both nrx(+S4)-CFP and nrx(−S4)-CFP, clustering of gephyrin and PSD-95 was strong and often occurred at the edges of the COS cells where the fibroblasts were likely in greatest contact with their environment. Such clustering of gephyrin and PSD-95 was never seen at dendritic contacts of fibroblasts transfected with mCFP (Fig. 1C) or with N-cadherin or NgCAM (Graf et al., 2004).
Figure 1.

Addition of the splice insert at S4 reduces induction of PSD-95 but not gephyrin clusters. A, B, In a fibroblast-neuron coculture assay, COS cells expressing CFP-tagged neurexin-1β without the S4 insert (A) and with the addition of the S4 insert (B) induced clustering of both gephyrin and PSD-95 in contacting dendrites. Induced clusters (arrowheads) were negative for the presynaptic antigen synapsin unlike endogenous synapses (arrows). C, Only synapsin-positive gephyrin and PSD-95 clusters (arrows) were observed under mCFP transfected fibroblasts. D–F, Quantification of the number (D), area (E), and total integrated intensity (F) of synapsin-negative gephyrin and PSD-95 clusters underlying transfected fibroblasts revealed reduced induction of PSD-95 clusters by splice site 4-positive neurexin but no change in induction of gephyrin clusters compared with splice site 4-negative neurexin (∗p < 0.05; n = 20 cells each). Scale bar, 10 μm.
To determine whether the degree of gephyrin and PSD-95 clustering was altered because of addition of the S4 insert, we quantified the number, area, and total integrated intensity of synapsin-immunonegative gephyrin and PSD-95 clusters underlying COS cells transfected with either nrx(−S4)-CFP or nrx (+S4)-CFP (Fig. 1D–F). The presence or absence of the S4 insert had no effect on the number, area, or total intensity of induced gephyrin clusters. However, nonsynaptic PSD-95 clusters were significantly reduced in number, area, and total intensity by 28–44% under COS cells transfected with nrx(+S4)-CFP compared with cells transfected with nrx(−S4)-CFP. Thus, whereas +S4 did not abolish the ability of neurexin to induce gephyrin and PSD-95 clustering, it specifically decreased PSD-95 clustering while leaving gephyrin clustering unaffected. Addition of an extracellular myc tag to both nrx(−S4)-CFP and nrx(+S4)-CFP and subsequent live staining with anti-myc antibody showed that both constructs reached the cell surface with equal efficiency, suggesting that the above results are not a result of disrupted protein processing (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Effect of alternative splicing at S4 on recruitment and binding to neuroligins
The synaptogenic activity of neurexin-1β containing the S4 insert could occur by recruitment of neuroligins or by a novel mechanism perhaps independent of neuroligins. We thus determined whether nrx(+S4)-CFP could recruit endogenous neuroligins to contact sites in the coculture assay, using antibodies specific either for neuroligin-2 or for neuroligins-1, -3, and -4 but not -2. Indeed, like nrx(−S4)-CFP, nrx(+S4)-CFP induced clusters of neuroligin-1/3/4 and -2 on dendrites at sites of contact with the expressing fibroblasts (Fig. 2A,B,D,E). Unlike the endogenous synaptic clusters of neuroligin, the induced clusters lacked apposed synapsin. Such extrasynaptic clusters of neuroligin-1/3/4 and -2 were not seen on dendrites in contact with cells expressing neurexin-CFP with the LNS domain deleted (Fig. 2C,F). Furthermore, quantitatively, compared with the form lacking the S4 insert, addition of the S4 insert to neurexin-1β did not affect clustering of neuroligin-2 but reduced clustering of neuroligin-1/3/4 by 32% (Fig. 2G–I) (∗p < 0.05).
Figure 2.

Addition of the splice insert at S4 reduces recruitment of neuroligin-1/3/4 but not neuroligin-2. A–F, In a fibroblast-neuron coculture assay, COS cells expressing CFP-tagged neurexin-1β without the S4 insert (A, D) and with the addition of the S4 insert (B, E) induced clustering of both neuroligin-1/3/4 (A, B), and neuroligin-2 (D, E) in contacting dendrites. Induced clusters (arrowheads) were negative for the presynaptic antigen synapsin unlike endogenous synapses (arrows). C, F, Only synapsin-positive neuroligin clusters (arrows) were observed under COS cells expressing CFP-tagged neurexin-1β deleted for the LNS domain. G–I, Quantification of the number (G), area (H), and total integrated intensity (I) of synapsin-negative neuroligin (NLG)-1, -3, -4, and -2 clusters underlying transfected fibroblasts revealed reduced induction of neuroligin-1/3/4 clusters by splice site 4-positive neurexin but no change in induction of neuroligin-2 clusters compared with splice site 4-negative neurexin (∗p < 0.05; n = 40 cells each). Scale bar, 10 μm.
We next determined directly how addition of the S4 insert affects binding of purified neurexin-1β to neuroligins -1, -2, -3, and -4 expressed in a heterologous system. We purified recombinant soluble neurexin-Fc (the leader sequence and neurexin LNS domain fused to the human Ig constant region) either with no insert at S4, nrx(−S4)-Fc, or with the 30 aa sequence inserted into S4 in the LNS domain, nrx(+S4)-Fc. We transfected COS cells with N-terminally HA-tagged neuroligin-1, -2, -3, or -4 and coincubated live with 50 ng/μl soluble neurexin-Fc protein and with anti-HA antibody to determine the amount of neuroligin at the surface. Unlike previous biochemical assays, we discovered that addition of the S4 insert does not abolish the binding of neurexin to neuroligin (Fig. 3A–H). Both soluble nrx(−S4)-Fc and soluble nrx(+S4)-Fc bound to COS cells transfected with each of the four neuroligin proteins but not to untransfected COS cells. To determine relative binding affinity, we quantified the ratio of the average intensity of bound neurexin-Fc to the average intensity of surface HA staining per transfected cell (Fig. 3L). Whereas the addition of the S4 insert had little effect on binding to neuroligin-3, it significantly decreased neurexin binding to neuroligin-1 and neuroligin-4 by 25 and 43%, respectively (∗∗p < 0.001). Addition of the S4 insert also caused a smaller but significant decrease in neurexin binding to neuroligin-2 (∗p < 0.05). We performed three controls for specificity of binding (Fig. 3I–K). First, nontransfected COS cells exhibited no binding (nontransfected cells are shown specifically in Fig. 3I but were also present in all experiments). Second, previous studies have shown that the binding of neurexin to neuroligin is Ca2+ dependent (Ichtchenko et al., 1995). Likewise, we found that soluble neurexin-Fc was unable to bind to neuroligins in Ca2+-free buffer containing 1 mm EGTA (the average intensity ratio of bound neurexin-Fc per HA-neuroligin-expressing COS cell was <4% of the intensity ratio in the presence of calcium). Third, deletion of the LNS domain of neurexin-Fc also abolished binding [the average intensity ratio of bound neurexin-dLNS-Fc per HA-neuroligin-expressing COS cell was <3% of the signal obtained for bound neurexin(−S4)-Fc].
Figure 3.

Addition of the splice insert at S4 modulates neurexin-binding affinity to neuroligins. A–H, Both insert-negative (A, C, E, G) and insert-positive (B, D, F, H) soluble neurexin-Fc bound to HA-tagged neuroligin-1 (A, B), neuroligin-2 (C, D), neuroligin-3 (E, F), and neuroligin-4 (G, H) expressed in COS cells. HA staining was performed live to reveal only surface neuroligin. I–K, Controls for specificity of binding include lack of neurexin-Fc binding to nontransfected cells visible in the phase contrast images (I, ∗), lack of neurexin-Fc binding to cells expressing HA-neuroligin under conditions of nominally zero calcium plus EGTA (J), and lack of binding of neurexin-dLNS-Fc bearing a deletion in the LNS domain (K). L, Quantification of the average intensity of bound neurexin-Fc normalized to average intensity of surface HA-neuroligin (NLG) showed a significant reduction of insert-positive neurexin binding to neuroligins-1 and -4 compared with insert-negative binding (∗∗p < 0.001; n = 40 cells each). Addition of the splice insert at S4 also slightly reduced neurexin binding to neuroligin-2 (∗p < 0.05; n = 40 cells each), although binding to neuroligin-3 was unaffected. Scale bar, 20 μm.
This finding that neurexin(+S4) is able to bind to all four neuroligins was surprising considering previous reports to the contrary. Indeed, while this report was under review, Boucard et al. (2005) also reported that neurexin(+S4) is able to bind neuroligin-1. The cell binding assay used here, and a similar assay by Boucard et al. (2005), may be more sensitive than previous assays, which relied on the ability of neurexin to pull down neuroligins from brain or cell extract (Ichtchenko et al., 1995, 1996) or on the cell adhesion activity of neurexins and neuroligins (Nguyen and Sudhof, 1997; Dean et al., 2003). The binding data reported here (Fig. 3) correlates well with the result that addition of the S4 insert fails to abolish clustering of neuroligins or gephyrin or PSD-95 in the coculture assay (Figs. 1, 2).
Structure-function analysis of neurexin LNS domain
We determined previously that the LNS domain was the only domain of neurexin-1β that was both necessary and sufficient to induce GABAergic and glutamatergic postsynaptic protein clustering (Graf et al., 2004). We further mapped out the regions of the LNS domain that were necessary for such clustering. The crystal structure of the neurexin-1β LNS domain shows it to be composed of two seven-stranded β sheets forming a jelly roll fold (Rudenko et al., 1999). The loops at either end of the β sandwich represent likely binding interfaces that may determine biological function. To determine which loops are necessary for synaptogenic activity, we created alanine substitutions of three to five residues in length in most of the loops between β strands in the nrx(−S4)-CFP construct (supplemental Fig. 1M, available at www.jneurosci.org as supplemental material). We created two separate mutations in the β2β3 loop: β2β3mid substituting residues 103–106 (NDRP) with alanines and β2β3end substituting residues 107–109 (STR) with alanines. We mutated residues 119–122 (TVQK) in the β3β4 loop, residues 132–135 (SSGL) in the β4β5 loop, residues 154–157 (VGTD) in the β6β7 loop, residues 167–170 (IIND) in the β7β8 loop, residues 190–193 (DSWP) in the β9β10 loop, residues 202–205 (RQLT) in the β10β11 loop, residues 218–222 (KEQGQ) in the β11β12 loop, and residues 244–246 (END) in the α1β13 loop. To ensure that the mutations did not affect protein processing and disrupt the ability of each neurexin protein to reach the cell surface, we assayed for surface localization. We added a myc tag to the extracellular N terminus of each neurexin construct, transfected them into COS cells, and stained live with anti-myc antibody. In this assay, myc staining showed only surface protein, whereas CFP expression showed total protein. Of these mutated constructs, only mutations in the β7β8 and β9β10 loops disrupted the ability of neurexin to reach the cell surface, as determined by the absence of myc staining (supplemental Fig. 1H,I, available at www.jneurosci.org as supplemental material). All of the other mutated proteins reached the cell surface with levels similar to the unmutated forms nrx(−S4)-CFP and nrx(+S4)-CFP (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and were analyzed in the coculture assay.
We ascertained the synaptogenic activity of the mutated proteins compared with unmutated nrx(−S4)-CFP by determining the percentage of transfected COS cells with extensive dendritic contact that induced clustering of gephyrin and PSD-95 in the coculture assay (Fig. 4A–D). Mutations in loops β3β4, β4β5, β11β12, and α1β13 had no effect on induction of gephyrin or PSD-95 clusters, demonstrating synaptogenic activity similar to unmutated neurexin. Conversely, the β2β3end, β6β7, and β10β11 mutations completely abolished gephyrin and PSD-95 clustering. No exogenously induced nonsynaptic clusters of gephyrin or PSD-95 were observed at the dendritic contacts of COS cells transfected with these mutated neurexin constructs, suggesting that residues in these loops are necessary for neurexin synaptogenic activity.
Figure 4.

Definition of regions of the LNS domain essential for neurexin synaptogenic activity. Mutations in individual loops of the neurexin(−S4)-CFP, each corresponding to alanine substitution of three to five residues, were tested for synaptogenic activity in the coculture assay. A–C, Representative examples of gephyrin and PSD-95 cluster induction by mutated neurexin proteins show that some alanine substitutions such as the β4β5 mutation (A) had no effect on neurexin synaptogenic activity and clustered both gephyrin and PSD-95 strongly, whereas the β2β3mid mutation (B) caused a reduction in PSD-95 clustering but induced strong gephyrin clustering. In contrast, fibroblasts transfected with neurexin containing the β6β7 mutation (C) were unable to induce gephyrin or PSD-95 clusters. Arrowheads indicate synapsin-negative induced clusters, whereas arrows indicate endogenous clusters that are synapsin positive. D, The effect of each mutation on synaptogenic activity was determined by quantifying the percentage of transfected fibroblasts overlaying dendrites that induced gephyrin or PSD-95 clustering. For gephyrin: +++, 80–100%; ++, 40–79%; +, 1–39%; −, 0%. For PSD-95: +++, 60–80%; ++, 25–59%; +, 1–24%; −, 0%. E, F, Model of the neurexin LNS domain shows the mutated regions that abolished induction of both gephyrin and PSD-95 clusters (red), reduced PSD-95 but not gephyrin clustering (green), or had no effect on neurexin synaptogenic activity (blue). Two views of the 3D structural model are shown. Scale bar, 10 μm.
Whereas the β2β3mid mutation did not abolish clustering of either gephyrin or PSD-95, and the percentage of transfected COS cells showing induced gephyrin clustering was similar to unmutated neurexin, fewer COS cells induced PSD-95 clustering. In addition, the level of PSD-95 clustering associated with COS cells transfected with the neurexin construct containing the β2β3mid mutation appeared less pronounced compared with induction by unmutated neurexin (Fig. 1A), whereas gephyrin clustering remained strong (Fig. 4B). Total integrated fluorescence intensity of synapsin-negative PSD-95 clusters associated with COS cells transfected with neurexin containing the β2β3mid mutation was significantly decreased compared with unmutated neurexin (48 ± 8 vs 110 ± 17, respectively; p < 0.01; n = 20). In contrast, the total fluorescence intensity of nonsynaptic gephyrin clusters induced by neurexin containing the β2β3mid mutation versus unmutated neurexin was similar (252 ± 39 vs 259 ± 34, respectively; p > 0.1; n = 20). Interestingly, the four mutations that either abolished or altered induction of gephyrin and PSD-95 clusters were all located on the same face of the LNS domain, suggesting that this may be the binding interface of neurexin-1β with neuroligins (Fig. 4E,F, red and green regions). Alternatively, the three mutations located on other faces of the protein had no effect on clustering activity (blue regions). The SSGL sequence in the β4β5 loop, although located at one edge of the apparent binding face, was also not essential for synaptogenic activity.
To fully characterize the binding interface of the neurexin LNS domain, we made single alanine substitutions of the charged or polar residues in the loops determined necessary for gephyrin and PSD-95 clustering in the previous assay. The following point mutations were made: S107A, T108A, and R109A in the β2β3 loop (residues that were in the original β2β3end mutation); T156A, D157A, and D158A in the β6β7 loop; and R202A, Q203A, and T205A in the β10β11 loop. In the previous assay, we did not mutate the small β8β9 loop, but because it is near the other loops found to be necessary for synaptogenic activity, we mutated the single charged residue in that loop, N184A. As mentioned above, the binding of neurexin to neuroligin is Ca2+ dependent (Ichtchenko et al., 1995) (Fig. 3J). The Ca2+-binding residues have been mapped out previously in the homologous laminin LNS domain (Hohenester et al., 1999; Tisi et al., 2000). By comparing the structure and amino acid sequence of the laminin and neurexin-1β LNS domains, we determined the putative Ca2+-binding amino acids in neurexin-1β to be D137 and N208. Thus single alanine mutations were made of D137 and N208 to determine whether they may play a role in postsynaptic protein clustering.
As in the previous assay, each mutated construct was analyzed in a fibroblast-neuron coculture assay by evaluating the percentage of transfected fibroblasts that induced gephyrin and PSD-95 clusters. Whereas neurexin that contained the D157A mutation clustered gephyrin and PSD-95 at levels comparable with the unmutated form, all other mutations either decreased postsynaptic protein clustering compared with unmutated neurexin or completely abolished the synaptogenic activity of the LNS domain altogether (compare Figs. 5 and 1A). Mutations S107A, T108A, T156A, N184A, and Q203A had no effect on gephyrin clustering but showed a moderate decrease for induction of PSD-95 clusters, both in terms of percentage of transfected cells that caused clustering as well as degree of clustering when induction did occur. Mutations D158A, R202A, and T205A disrupted synaptogenic activity more severely, moderately to greatly decreasing but not abolishing both gephyrin and PSD-95 cluster induction. Furthermore, whereas the longer β2β3end, β6β7, and β10β11 mutations all abolished clustering, the only point mutation made within these loops to completely eliminate the ability of neurexin to cluster gephyrin and PSD-95 was R109A. In addition, mutation of each putative Ca2+-binding residue, D137A in the β4β5 loop and N208A in the β10β11 loop, abolished all synaptogenic activity (Fig. 5E), supporting previous reports that suggested the activity of the neurexin-neuroligin link is Ca2+ dependent.
Figure 5.
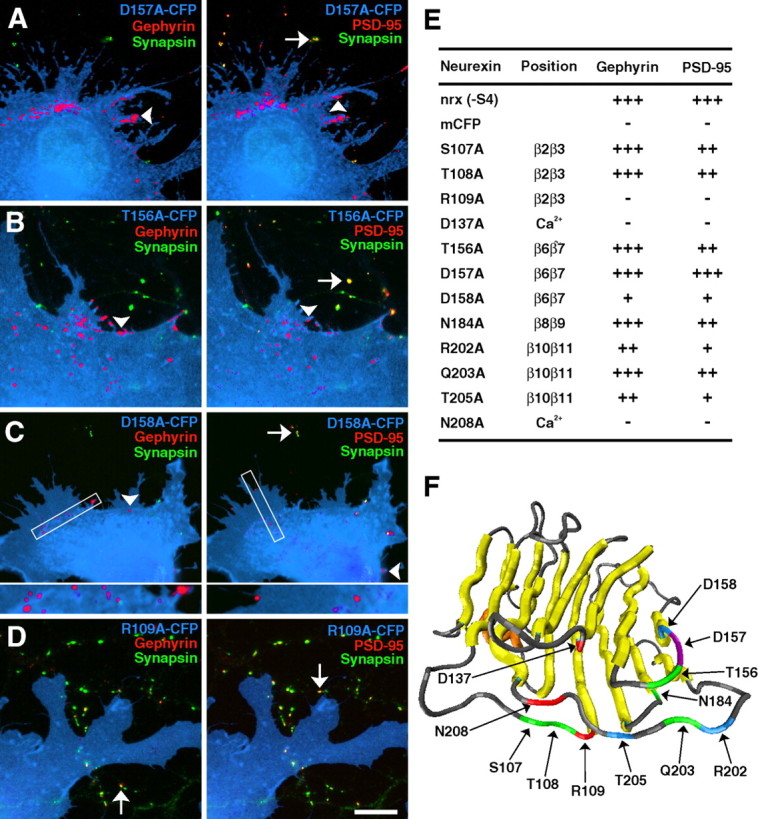
Definition of single residues essential for neurexin synaptogenic activity. A–D, Although the D157A mutation (A) showed strong induction of gephyrin and PSD-95 clusters, other mutations such as T156A (B) caused moderate reduction of PSD-95 clustering only. Other mutations such as D158A (C) caused more severe reductions in both gephyrin and PSD-95 clustering, whereas the R109A (D) mutation completely abolished gephyrin and PSD-95 clustering. Arrowheads indicate synapsin-negative induced clusters, whereas arrows indicate endogenous clusters that are synapsin positive. E, The effect of each mutation on synaptogenic activity was determined by quantifying the percentage of transfected fibroblasts overlaying dendrites that induced gephyrin or PSD-95 clustering. For gephyrin: +++, 80–100%; ++, 40–79%; +, 1–39%; −, 0%. For PSD-95: +++, 60–80%; ++, 25–59%; +, 1–24%; −, 0%. Position refers to the location of the mutation or its function as a predicted Ca2+-binding residue. F, The model shows the location of residues that abolished induction of both gephyrin and PSD-95 clustering (red), reduced both gephyrin and PSD-95 clustering (blue), reduced PSD-95 but not gephyrin clustering (green), or had no effect on neurexin synaptogenic activity (purple). Scale bar, 10 μm.
Interestingly, none of the mutations decreased gephyrin clustering while leaving PSD-95 unaffected. Instead, it appeared that most mutations decreased the overall synaptogenic activity of neurexin, but gephyrin clustering was more resistant to moderate decreases in activity than was induction of PSD-95 clusters. Only mutants that severely decreased PSD-95 clustering also decreased gephyrin clustering.
Effect of LNS domain mutations on recruitment of and binding to neuroligins
We further assessed the relationship between the ability of neurexins to induce gephyrin and PSD-95 clustering in the coculture assay and recruitment of and affinity of binding to neuroligins (Fig. 6) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). For this purpose, we chose a subset of the mutant neurexins representing a range of postsynaptic-inducing activities and assayed relative binding affinity to neuroligins-1 and -2 as representative of the major neuroligins at glutamatergic and GABAergic synapses, respectively (Song et al., 1999; Graf et al., 2004; Varoqueaux et al., 2004). For each of the mutant neurexins chosen, plus wild-type (WT) neurexin-1β positive control and the LNS deletion negative control, we quantified the ratio of the average intensity of bound mutant neurexin-Fc to the average intensity of surface HA-neuroligin-1 or HA-neuroligin-2 per transfected COS cell. As expected, neurexin mutations that abolished postsynaptic-inducing activity in the neuron coculture assay (D137A and N208A) also abolished binding to neuroligins-1 and -2 (p < 0.001 compared with wild type; p > 0.1 compared with dLNS). Neurexin mutations that reduced but did not abolish postsynaptic-inducing activity (β2β3mid, S107A, T108A, R202A, and T205A) also reduced but did not abolish binding to neuroligins (p < 0.005 compared with WT). The severity of these effects also correlated roughly (i.e., R202A and T205A showed the greatest reductions in postsynaptic inducing activity and the greatest reductions in neuroligin binding). We show additionally that R202A and T205A reduced recruitment of neuroligin-1/3/4, and -2 in the coculture assay by ≥75%, whereas T108A reduced recruitment of neuroligins by 35–57% (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Notably, all five of the partially active mutants analyzed reduced binding to neuroligin-1 to a greater extent than they affected binding to neuroligin-2 (p < 0.001). Together with the finding that these mutations more severely disrupted induction of clustering of PSD-95 compared with gephyrin (Figs. 4, 5), these results support the idea that binding to neuroligin-1 selectively promotes glutamatergic postsynaptic differentiation, whereas binding to neuroligin-2 promotes GABAergic postsynaptic differentiation.
Figure 6.

Neurexin mutations that reduce synaptogenic activity reduce binding affinity to neuroligins. A–H, Compared with wild-type soluble neurexin-Fc (nrx −S4) binding to HA-neuroligins (NLG) expressed in COS cells (A, E), neurexin mutations that reduced synaptogenic activity reduced binding to HA-neuroligins (B, C, F, G), and mutations that abolished synaptogenic activity abolished binding to HA-neuroligins (D, H). The mutations reduced binding to neuroligin-1 (B, C) to a greater extent than to neuroligin-2 (F, G). HA staining was performed live to reveal only surface neuroligin. I, Quantification of the average intensity of bound neurexin-Fc normalized to average intensity of surface HA-neuroligin and expressed as a percentage of binding of wild-type neurexin-Fc to HA-neuroligin-1 or -2. All mutations significantly reduced binding of neurexin to neuroligin-1 and -2 (p < 0.005; n ≥ 40 cells each), except that the β2β3mid mutation did not affect binding to neuroligin-2 (p > 0.1). Scale bar, 20 μm.
Discussion
We show here that insertion of the 30 aa alternatively spliced sequence at S4 of neurexin-1β does not abolish binding to neuroligins but alters binding in a more complex way. Furthermore, the presence of the S4 insert reduces glutamatergic but not GABAergic synaptogenic activity of neurexin-1β. Additional structure-function analysis reveals the glutamatergic synaptogenic activity and binding to neuroligin-1 to be more sensitive to mutations than the GABAergic synaptogenic activity and binding to neuroligin-2. We identify loops β2β3, β6β7, and β10β11 as the key interface of the neurexin-1β LNS domain mediating synaptogenic activity and neuroligin binding.
Physiological role of alternative splicing in the neurexin-1β LNS domain
The data reported here suggest that splice variations of neurexin may play an instructive role in glutamatergic and GABAergic postsynaptic differentiation. Whereas the S4 insert has no effect on the ability of neurexin to induce gephyrin clusters in a fibroblast-neuron coculture assay, induction of PSD-95 clusters is specifically reduced (Fig. 1). In parallel, whereas the S4 insert has no effect on the ability of neurexin to induce neuroligin-2 clusters in coculture assay, induction of neuroligin-1, -3, and -4 clusters is specifically reduced (Fig. 2). Furthermore, we show here that the S4 insert modulates but does not abolish the ability of neurexin-1β to bind to neuroligins. Whereas the S4 insert caused a small decrease in the ability of neurexin to bind to neuroligin-2, we observed a larger reduction of neurexin binding to neuroligins-1 and -4 (Fig. 3). We suggest that these differences in binding affinity are biologically significant in light of previous studies showing specificity of neuroligin isoform localization. Whereas neuroligin-1 is localized at glutamatergic synapses, neuroligin-2 is localized at GABAergic synapses (Song et al., 1999; Graf et al., 2004; Varoqueaux et al., 2004). In addition, whereas aggregation of all four neuroligin isoforms can coaggregate PSD-95, coaggregation of gephyrin is specific to neuroligin-2. Together with these previous findings, our current results suggest that addition of the S4 insert may alter neurexin function by specifically decreasing binding affinity to neuroligins associated with glutamate synapses and thus reducing glutamatergic synaptogenic activity. In contrast, the S4 insert has much less effect on neuroligin-2 binding or recruitment, allowing GABAergic synaptogenic activity to remain strong.
The apparent differential effects of the S4 insert on glutamatergic versus GABAergic postsynaptic differentiation suggest that neurexin splice site variations may be one mechanism by which neurons control synaptic development. It is intriguing to speculate that GABAergic neurons may be more likely to express S4 insert-positive neurexins, whereas glutamatergic neurons may express neurexins negative for the insert at S4, although more extensive in situ hybridization and single-cell reverse transcription-PCR studies are needed to determine whether this does indeed occur in vivo. Previous in situ studies have reported differential expression of neurexin splice forms, showing high levels of S4 insert-positive neurexins-1 and -2 in the striatum, substantia nigra, and cerebellar nuclei and high levels of insert-negative neurexins-1 and -2 in the hippocampus and cerebral cortex (Ichtchenko et al., 1995). Interestingly, this study also reported high levels of insert-negative neurexin-3 expression in the glutamatergic pyramidal neurons of the hippocampus but no detectable insert-positive mRNA. Furthermore, studies of neurexin expression in chick sympathetic neurons suggests that expression of neurexin splice variants may change during development (Patzke and Ernsberger, 2000). Additional work must be done to specifically identify how splice variations correlate with glutamatergic versus GABAergic neurons throughout brain development.
The results reported here of differential binding of neurexin-1β to neuroligins-1, -2, -3, and -4 also highlight the importance of considering a physiological role for each of the neuroligins. Neuroligins-1, -2, and -3 are widely expressed in brain from postnatal day 0 (P0) to adult, and neuroligin-4 has an even broader tissue distribution (Brose, 1999; Song et al., 1999; Bolliger et al., 2001; Varoqueaux et al., 2004). Binding affinity of neurexin-1β to the glutamatergic synaptic neuroligins was much greater for neuroligin-1 than neuroligins-3 or -4. However, viability of the neuroligin-1 knock-out mice (Song et al., 1999) suggests that other neuroligins can functionally substitute in the absence of neuroligin-1. Furthermore, linkage of mutations in human neuroligins-3 and -4 with autism and mental retardation indicate a specific role for these neuroligins in human cognition (Jamain et al., 2003; Laumonnier et al., 2004). Perhaps the insensitivity of neuroligin-3 to splice site 4 variations may be physiologically relevant. Finally, the relatively lower affinity of neurexin-1β for neuroligin-2 compared with neuroligin-1 is surprising given its more potent synaptogenic activity for GABAergic than glutamatergic postsynaptic proteins, suggesting that perhaps neuroligin-2 may be more abundant than the other neuroligins in most neurons.
Neurexin-1β structure-function analysis
We have shown previously that the neurexin LNS domain is both necessary and sufficient for induction of postsynaptic protein clusters (Graf et al., 2004). We report here the loops and individual residues in the LNS domain that are necessary for synaptogenic activity. Alanine substitution of multiple amino acids in the β2β3, β6β7, and β10β11 loops completely abolished neurexin synaptogenic activity, delineating the presumed binding interface for interaction with neuroligins. As direct confirmation, single residue substitutions in β2β3 and β10β11 reduced binding affinity for neuroligins (Fig. 6). These loops of the LNS domain at the rim of the β sandwich opposite the N and C termini may form a generally conserved binding interface. The homologous surface of the LNS domain of sex hormone-binding globulin was directly identified as the steroid binding site by crystallographic analysis (Grishkovskaya et al., 2000), and indirect studies support the idea that this surface is key to the function of agrin and laminins as well as neurexin (Rudenko et al., 2001). It is remarkable that the 30 residue S4 insert in the β10β11 loop did not completely destroy the neuroligin binding site; presumably, these residues fold away from the interface, thus only partially modifying the binding surface. The identification here of the β2β3 loop of neurexin as essential for synaptogenic activity is also interesting in light of the finding that alternative splicing at the z site in the equivalent loop of the third agrin LNS domain regulates its synaptogenic activity (Ferns et al., 1993; Burgess et al., 1999; Rudenko et al., 1999).
Of the 12 single residue mutations tested, only three completely abolished neurexin-1β synaptogenic activity. We suggest that two of these, D137A and N208A, abolish synaptogenic activity by disrupting the ability of neurexin to bind Ca2+. Previous studies (Ichtchenko et al., 1995), as well as our own observations (Fig. 3J), show that neurexin-neuroligin binding is Ca2+ dependent. Half-maximal binding of neuroligin-1 to immobilized neurexin-1β was reported at 2 μm free Ca2+ (Nguyen and Sudhof, 1997), suggesting that the Ca2+-binding sites are normally saturated in vivo. Because these two mutated residues are in an equivalent position to the identified Ca2+-binding aspartates in the similar laminin LNS domain (Hohenester et al., 1999; Tisi et al., 2000), and asparagine is also capable of coordinating a calcium ion, it is likely that D137A and N208A are responsible for Ca2+ binding in neurexin. The other essential single residue, R109A, is close to the calcium-binding residues in the 3D structure (Fig. 5F) and is presumably important for binding specific neuroligin residues. Identification of the individual residues that contribute to synaptogenic activity of neurexin-1β as performed here may be useful for mapping of specific interactions at the neurexin-neuroligin interface. Such analysis awaits identification of the residues of the neuroligin acetylcholinesterase domain important for neurexin binding.
It is interesting that no manipulation of the neurexin-1β LNS domain resulted in reduced gephyrin clustering or binding to neuroligin-2 while leaving induction of PSD-95 clusters or binding to neuroligin-1 unaffected. Whereas the R202A and T205A point mutations decreased gephyrin clustering and binding to neuroligin-2, they severely decreased PSD-95 clustering and binding to neuroligin-1 to an even greater degree. Several other point mutations as well as the larger β2β3mid mutation and the addition of the S4 insert reduced PSD-95 clustering while leaving gephyrin clustering relatively unaffected. Like the S4 insert, the β2β3mid mutation decreased binding of neurexin to neuroligin-1 but not neuroligin-2. These results reveal that glutamatergic synaptogenic activity and neurexin binding to neuroligins associated with glutamate synapses is very sensitive to neurexin modification. Conversely, all of the active neurexin mutants generated here, like wild-type neurexin-1β, had a more potent inducing activity for GABAergic than glutamatergic postsynaptic proteins. This is consistent with a recent report that reduction of endogenous neuroligins by RNAi resulted in morphological and functional defects that were more severe for GABAergic than glutamatergic synapses (Chih et al., 2005). It is possible that the neurexin-neuroligin system may be more important for GABAergic synaptogenesis or for controlling the balance of glutamate and GABA synapses (Graf et al., 2004; Prange et al., 2004) than for glutamatergic synaptogenesis per se. However, additional work is required to determine whether other neurexin isoforms and splice variants function in a complementary manner to promote glutamatergic differentiation more predominantly than GABAergic differentiation. The recent findings that α-neurexins bind neuroligins, and that neurexin binding is regulated by alternative splicing of neuroligin-1 (Boucard et al., 2005), raise further the potential for different physiological roles for different neurexin and neuroligin variants.
Footnotes
This work was supported by National Institutes of Health Grants MH70860 and NS34448, Canadian Institutes of Health Research and Michael Smith Foundation for Health Research establishment grants (A.M.C.), and a National Science Foundation predoctoral fellowship (E.R.G.). We thank Huaiyang Wu for excellent preparation of neuron cultures and Chenghua Wu and Matt Sibley for assistance with cloning and quantitation.
References
- Biederer T (2005). Progress from the postsynaptic side: signaling in synaptic differentiation. Sci STKE 2005:pe9. [DOI] [PubMed] [Google Scholar]
- Bolliger MF, Frei K, Winterhalter KH, Gloor SM (2001). Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem J 356:581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Sudhof TC (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to α- and β-neuerexins. Neuron 48:229–236. [DOI] [PubMed] [Google Scholar]
- Brose N (1999). Synaptic cell adhesion proteins and synaptogenesis in the mammalian central nervous system. Naturwissenschaften 86:516–524. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR (1999). Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron 23:33–44. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324–1328. [DOI] [PubMed] [Google Scholar]
- Craig AM, Graf ER, Linhoff MW (2006). How to build a central synapse: clues from cell culture. Trends Neurosci 29:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P (2003). Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci 6:708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z (1993). The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron 11:491–502. [DOI] [PubMed] [Google Scholar]
- Garner CC, Zhai RG, Gundelfinger ED, Ziv NE (2002). Molecular mechanisms of CNS synaptogenesis. Trends Neurosci 25:243–251. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G (1998). Rat hippocampal neurons in low-density culture. In: Culturing nerve cells (Banker G, Goslin K, eds) Ed 2 pp. 339–370. Cambridge, MA: MIT.
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishkovskaya I, Avvakumov GV, Sklenar G, Dales D, Hammond GL, Muller YA (2000). Crystal structure of human sex hormone-binding globulin: steroid transport by a laminin G-like domain. EMBO J 19:504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenester E, Tisi D, Talts JF, Timpl R (1999). The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol Cell 4:783–792. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14:33–38. 27–38. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC (1995). Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81:435–443. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Nguyen T, Sudhof TC (1996). Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem 271:2676–2682. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34:27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthelemy C, Moraine C, Briault S (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 74:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Sheng M (2003). Some assembly required: the development of neuronal synapses. Nat Rev Mol Cell Biol 4:833–841. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sudhof TC (1997). Binding properties of neuroligin 1 and neurexin 1beta reveal function as heterophilic cell adhesion molecules. J Biol Chem 272:26032–26039. [DOI] [PubMed] [Google Scholar]
- Patzke H, Ernsberger U (2000). Expression of neurexin Ialpha splice variants in sympathetic neurons: selective changes during differentiation and in response to neurotrophins. Mol Cell Neurosci 15:561–572. [DOI] [PubMed] [Google Scholar]
- Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A (2004). A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci USA 101:13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko G, Nguyen T, Chelliah Y, Sudhof TC, Deisenhofer J (1999). The structure of the ligand-binding domain of neurexin Ibeta: regulation of LNS domain function by alternative splicing. Cell 99:93–101. [DOI] [PubMed] [Google Scholar]
- Rudenko G, Hohenester E, Muller YA (2001). LG/LNS domains: multiple functions–one business end? Trends Biochem Sci 26:363–368. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101:657–669. [DOI] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Sudhof TC, Brose N (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA 96:1100–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama Y, Kawabata I, Sobue K, Okabe S (2005). Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods 2:677–684. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Sudhof TC (2002). Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics 79:849–859. [DOI] [PubMed] [Google Scholar]
- Tisi D, Talts JF, Timpl R, Hohenester E (2000). Structure of the C-terminal laminin G-like domain pair of the laminin alpha2 chain harbouring binding sites for alpha-dystroglycan and heparin. EMBO J 19:1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich B, Ushkaryov YA, Sudhof TC (1995). Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron 14:497–507. [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC (1992). Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science 257:50–56. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol 83:449–456. [DOI] [PubMed] [Google Scholar]