

Abstract
Background:
The Bosley-Salih-Alorainy syndrome (BSAS) variant of the congenital human HOXA1 syndrome results from autosomal recessive truncating HOXA1 mutations. We describe the currently recognized spectrum of ocular motility, inner ear malformations, cerebrovascular anomalies, and cognitive function.
Methods:
We examined nine affected individuals from five consanguineous Saudi Arabian families, all of whom harbored the same I75-I76insG homozygous mutation in the HOXA1 gene. Patients underwent complete neurologic, neuro-ophthalmologic, orthoptic, and neuropsychological examinations. Six individuals had CT, and six had MRI of the head.
Results:
All nine individuals had bilateral Duane retraction syndrome (DRS) type 3, but extent of abduction and adduction varied between eyes and individuals. Eight patients were deaf with the common cavity deformity of the inner ear, while one patient had normal hearing and skull base development. Six had delayed motor milestones, and two had cognitive and behavioral abnormalities meeting Diagnostic and Statistical Manual of Mental Disorders-IV criteria for autism spectrum disorder. MRI of the orbits, extraocular muscles, brainstem, and supratentorial brain appeared normal. All six appropriately studied patients had cerebrovascular malformations ranging from unilateral internal carotid artery hypoplasia to bilateral agenesis.
Conclusions:
This report extends the Bosley-Salih-Alorainy syndrome phenotype and documents the clinical variability resulting from identical HOXA1 mutations within an isolated ethnic population. Similarities between this syndrome and thalidomide embryopathy suggest that the teratogenic effects of early thalidomide exposure in humans may be due to interaction with the HOX cascade.
We recently reported a new Mendelian syndrome associated with truncating mutations in HOXA1,1 a homeodomain transcription factor critical for the proper development of hindbrain rhombomeres in mice.2,3 Homozygous 175-176insG guanine base-pair insertions were found in several families from Saudi Arabia, while a homozygous 84C>G nonsense mutation resulted in the substitution of a stop codon for a tyrosine residue (Y28X) in a Turkish individual. These two mutations cause a phenotype, referred to as the Bosley-Salih-Alorainy syndrome (BSAS; OMIM #601536), characterized by bilateral Duane retraction syndrome (DRS) type 3, deafness, malformations of the cerebral vasculature, and autism in some patients.
We also identified a third mutation in exon 1 of HOXA1, a homozygous 76C>T change resulting in the substitution of a stop codon for an arginine residue (R26X), in Native American children with the previously reported Athabascan brainstem dysgenesis syndrome (ABDS; OMIM #601536).1,4 These children share horizontal gaze restriction, deafness, and internal carotid artery (ICA) abnormalities with BSAS but also have central hypoventilation, facial and bulbar weakness, conotruncal heart defects, and mental retardation.
We now provide a more detailed clinical report of nine Saudi Arabian patients who harbor the same HOXA1 mutation in order to elucidate the phenotypic variability of BSAS and its relationship to ABDS. An abbreviated phenotypic description of eight of these patients was provided in the original BSAS report.1 The BSAS phenotype has implications for human brainstem development, for the etiology and variability of DRS, and for certain other congenital ocular motility syndromes and embryopathies.
METHODS
Patients
Nine affected individuals from five families were examined in neuro-ophthalmology clinics of the King Khaled Eye Specialist Hospital and the King Faisal Specialist Hospital and Research Centre and in the Pediatric Neurology Clinic of the King Khalid University Hospital in Riyadh, Saudi Arabia. The genetic features of eight of the individuals (Patients 1 through 8) were reported previously,1 while Patient 9 was the child of an unrelated consanguineous family. She was genotyped more recently, using the same primer sequences and PCR conditions detailed previously, and was found to have the same homozygous HOXA1 guanine insertion (I75-I76insG) founder mutation as other reported Saudi individuals. All families signed informed consent at the appropriate institution.
Clinical evaluation
All patients had complete neurologic, neuro-ophthalmologic, and orthoptic examinations, including videotaping of eye movements in eight individuals. Ocular motility and alignment was evaluated in all positions of gaze. Fusional status was evaluated using a variety of binocular vision tests including the Worth 4 Dot test, Titmus stereo-acuity test, Bagolini striated lenses, and ability to overcome base-out prism with convergence. Best-corrected visual acuity was assessed with methods appropriate for age and cognitive ability. All patients had dilated funduscopic examinations.
Neuropsychological evaluation
Eight members of four Saudi families were seen by a neuropsychologist (M.N.), seven of whom were evaluated with the Vineland Adaptive Behavior (VAB) scale. VAB was administered by an experienced native neuropsychological technician trained in its use, and scoring for age equivalency was done independently by the neuropsychologist. All children other than the youngest (Patient 9) had repeated neurodevelopmental evaluations over several years (M.A.S.), and Diagnostic and Statistical Manual of Mental Disorders (DSM)-4 criteria for autism were applied separately by a neuropsychologist and a pediatrician. Patient 3 was also evaluated with the Childhood Autism Rating Scale (CARS).
Neuroimaging
Six affected individuals had MR images of the brain or neck. Standard brain MR pulse sequences were acquired in five patients using a 1.5 Tesla GE Signa scanner (GE Medical Systems, Waukesha, WI) or 1.5 Tesla Siemens Magneton Vision (Siemens Medical Systems, Germany) including sagittal T1-weighted spin echo (350 to 500/10 to 15/1 [TR/TE/NEX]), coronal fluid-attenuated inversion recovery (8,800/130, inversion time, 2,200 msec), and axial dual echo (3,500/119 and 17). MR images were obtained in one patient on a 3.0 Tesla Siemens Magneton Allegra scanner (Siemens Medical Systems) including sagittal T1-weighted (624/7), axial fluid-attenuated inversion recovery (8,000/80, inversion time 2400 msec), and axial MP-RAGE (2,500/4.4, 1 mm thickness) through the brain, and axial 3D FT constructive interference in steady-state of the inner ear and cerebellopontine angle (11/5.6, 1 mm thickness).
MR angiogram (MRA) of the head was obtained in five patients using time-of-flight (TOF) sequence and of the neck in four of them using TOF or phase contrast sequence. One patient had CT of the brain (7 mm slice thickness), and five had CT of the temporal bone (1 mm slice thickness). Volume data from CT were used for reconstruction of coronal petrous bone images.
RESULTS
Clinical and neuropsychological observations
Patient numbers for Patients 1 through 8 correspond to Supplementary Table 1 Online of Tischfield et al.,1 while Patient 9 was proven more recently to have the same Saudi BSAS mutation. Table 1 includes basic demographics and certain general characteristics of these nine affected individuals (three male and six female, aged 3 to 20 years), while table E-1 (on the Neurology Web site at www.neurology.org) summarizes the developmental and cognitive characteristics of these patients, including the VAB scale.
Table 1.
General information
| Patient | Family | Age, y | Sex | Deaf | Motor milestones | Autistic spectrum disorder |
Other |
|---|---|---|---|---|---|---|---|
| 1 | KF | 17 | F | Yes | WNL | No | Flattened ear helix bilaterally |
| 2 | KF | 5 | M | Yes | Delayed | No | Polydactyly of left foot; brachydactyly of 4th and 5th toes; duplex ureteral system with urethral stricture |
| 3 | KF | 20 | M | Yes | Walking by 4 y | Yes | Smaller right side of face; flattened ear helix bilaterally; frequent grimacing |
| 4 | LF | 11 | F | Yes | Delayed | No | Recurrent choking until age 1½ years |
| 5 | LF | 4 | F | Yes | Not sitting or crawling by 4 y |
Yes | Chronic constipation; sleep disturbance; low amplitude horizontal pendular nystagmus |
| 6 | LG | 18 | F | Yes | Walking by 3 y |
No | Low amplitude vertical pendular nystagmus |
| 7 | LG | 4 | F | Yes | Walking by 3 y; still unsteady at 4 y |
No | Chronic constipation; low positioned ears with anteriorly angulated right auricle; frequent grimacing |
| 8 | NO | 4 | M | No | WNL | No | Right talipes equinovarus repaired surgically |
| 9 | KK | 3½ | F | Yes | WNL | No | Frequent grimacing |
Age = age at last examination; WNL = within normal limits.
Eight patients were deaf and mute while one had normal hearing and speech development. Eight had a variety of behavioral (sleep disturbance, grimacing) and somatic (affecting face, ear, gastrointestinal tract, genitourinary system, and feet) abnormalities. Two deaf patients with speech abnormalities (Patients 1 and 9) and the one patient with normal hearing (Patient 8) had grossly normal neurodevelopmental histories and examinations except for ocular motility. Six patients had developmental motor delay by report (two on examination when young), typically not walking until the age of 3 years. No patient had scoliosis, inappropriately small stature, or cervical spine malformation, but as a group they had a somewhat large cranial circumference. Three families reported a total of seven miscarriages.
Patients 3 and 5 met DSM-4 criteria for autistic spectrum disorder by examination, family report, and VAB scores. Patient 3, who was institutionalized, scored 42 on CARS examination, falling in the moderate/severe autistic range. Both children exhibited profound linguistic delay with virtually nonexistent nonverbal communication skills, impaired social functioning, lack of eye contact, stereotypic hand movements, tactile avoidance, and obsessional features, such as compulsively arranging pictures and objects in the environment and persistent preoccupation with bright or shiny parts of objects. They had no reciprocal play, and their interests were narrow. Self-care skills were uniformly poor. Families of both autistic individuals reported sleep disturbance, although the older individual had improved with age. Patient 7 also had autistic features with stereotyped movements, excessive shyness, and complete lack of language; however, she had nonverbal communication and socialized in a fashion typical of a deaf child her age.
Ocular motility
All patients had a moderately severe or complete limitation of adduction and abduction bilaterally with full vertical gaze, as detailed in table 2 and illustrated in figure 1. Six patients (Patients 1, 2, 4, 6, 8, and 9) had globe retraction and palpebral fissure narrowing on adduction bilaterally diagnostic of DRS type 3 (figure 1, A through F). Patient 7 had retraction documented only unilaterally; however, horizontal motility restriction was present bilaterally, and her older sister had unequivocal globe retraction bilaterally. Globe retraction and fissure narrowing was not observed in either eye of Patients 3 and 5 (figure 1, G through I). These patients had severe restriction of horizontal eye movement, and autism interfered with their ability to cooperate with the examination. Both patients had at least one BSAS-affected sibling with obvious DRS type 3 bilaterally. Bilateral globe retraction was readily apparent upon convergence in the three patients with strong convergence (Patients 4, 8, and 9).
Table 2.
Ocular motility
| Patient | Abduction | Adduction | Globe retraction | Alignment | Convergence | Vertical anomalies |
|---|---|---|---|---|---|---|
| 1 | 30% OU | 70% OU | Moderate OU | Moderate ET at distance; near NA |
NA | None |
| 2 | 0% OU | 40% OU | Mild OU | Moderate ET at distance and near |
Poor | Upshoot OS on left and right gaze when fixing with OD |
| 3 | 0% OU | 30% OU | None observed | Ortho at distance; small XT at near |
Absent (poor coop) |
None |
| 4 | 0% OU | 60% OD, 70% OS |
Marked OU | Small ET at distance and near |
Good* | Upshoot on adduction OU; upshoot OD on abduction when fixing with OS |
| 5 | 0% OU | 0% OU | None observed | Ortho | Poor | NA |
| 6† | 0% OU | 60% OU | Moderate OU | Moderate ET at distance; small ET at near |
NA | Upshoot in abduction and adduction of nonfixing eye |
| 7 | 0% OU | 10% OD, 0% OS |
Moderate OD | Ortho | Poor | None |
| 8 | 40% OD, 0% OS |
0% OD, 40% OS |
Moderate OU | Ortho (with left face turn) |
Good* | None |
| 9 | 0% OU | 45% OD, 75% OS |
Mild OU | Ortho | Moderate* | None |
Abduction and adduction expressed in % of normal excursion.
Globe retraction present OU during convergence.
Patient 6 = preoperative observations
OU = both eyes; ET = esotropia; NA = not available; OS = left eye; OD = right eye; XT = exotropia; ortho = orthophoria; coop = cooperation.
Figure 1. Variability of ocular alignment and motility.
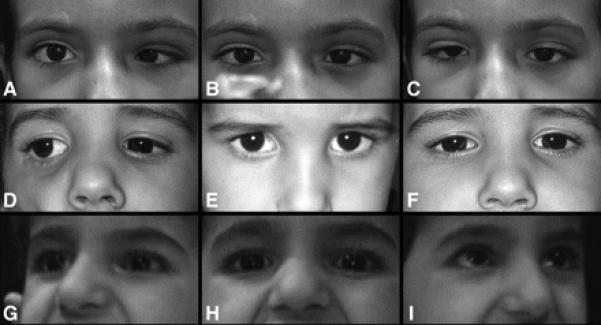
Images A, D, and G are right gaze, while images B, E, and H are in primary position, and images C, F, and I are left gaze. Top row (A, B, and C) are of Patient 4 who had a primary position esotropia with no anomalous head position when fixing with the right eye. This patient was unable to abduct either eye beyond the midline but demonstrated modest adduction bilaterally with globe retraction and fissure narrowing of the adducting eye. Middle row (D, E, and F) are of Patient 8 who was orthotropic in primary position but adopted a small face turn left to maintain comfortable binocular fixation. He had no eye movements to the left but modest symmetric movement of both eyes into right gaze. Bottom row (G, H, and I) are of Patient 5, who was orthotropic in primary position and had severely limited eye movements bilaterally with only minimal adduction the right eye. No abnormal head posture was present in primary position but she used a face turn to view objects to the right or left.
Five patients (Patients 3, 5, 7 through 9) were orthotropic in primary position at distance (figure 1, E and H), while four (Patients 1, 2, 4, and 6) had an esotropia in primary position (figure 1B). Patient 6 had an accommodative element to her esotropia, and she was the only patient who underwent strabismus surgery to correct the horizontal deviation. Two patients had low amplitude nystagmus that was horizontal pendular in one (Patient 5) and vertical pendular in another (Patient 6). Three patients (Patients 2, 4, and 6) developed a vertical deviation on attempted horizontal gaze.
Afferent functioning
Seven patients had normal visual acuity, ocular fundi, pupillary function, and visual fields. Patient 2 had 20/80 acuity bilaterally, possibly because of age and ability to cooperate. Three patients (Patients 6, 7, and 8) demonstrated fusion. Patient 6 had a monofixation syndrome with a tiny residual manifest deviation following early strabismus surgery, while Patient 7 had no manifest deviation in primary position, and Patient 8 required a small left face turn to maintain binocular alignment (figure 1E).
Neuroimaging
Table 3 details results of neuroimaging studies. No imaged patient had gross congenital brain anomalies or evidence of ischemia. Orbits and extraocular muscles appeared normal in all studies (figure E-1, A and B). Brainstem and 4th ventricle were normally developed (figure E-1, C and D), as were supratentorial structures. Patient 8 had a 3 Tesla scan with thin MR sections through the caudal pons. These images were degraded by a pulsation artifact anterior to the pons, but the exiting abducens cranial nerve (CN) could not be identified bilaterally (figure E-1, E through G) despite routine identification of these nerves on control images. Scout views of five of the six patients undergoing head CT allowed gross evaluation of the cervical spine, all of which appeared normal.
Table 3.
Neuroimaging
| Patient | Examinations available | Inner ear | Cerebral vasculature | Other |
|---|---|---|---|---|
| 1 | CT temporal bones; MRI brain; MRA brain and neck |
Right CCD with IAC stenosis; absent left inner ear structures and IAC |
Low right CCA bifurcation; hypoplastic left ICA with larger left PCom; duplication of proximal half of left vertebral artery |
|
| 2 | NA | |||
| 3 | CT temporal bones | Left CCD with IAC stenosis; absent right inner ear structures and IAC |
NA | Small right carotid canal |
| 4 | CT temporal bones; MRI brain; MRA brain and neck; US abdomen |
Bilateral CCD with bilateral IAC stenosis |
Absent left ICA; hypoplastic intracavernous right ICA; low right CCA bifurcation; large basilar system |
Absent left carotid canal; normal C-spine |
| 5 | CT temporal bones; MRI brain; MRA brain and neck; US abdomen |
Bilateral CCD with bilateral absent IAC |
Bilateral absent ICAs; large basilar system; duplication of intracranial left vertebral artery |
Absent carotid canals bilaterally; normal C-spine |
| 6 | CT temporal bones; MRI brain; MRA brain and neck |
Bilateral CCD with bilateral IAC stenosis |
Absent left ICA; low right CCA bifurcation; hypoplastic intracavernous right ICA; large basilar system |
Absent left carotid canal; normal C-spine |
| 7 | CT brain; MRI brain; MRA brain; US abdomen |
Bilateral CCD with bilateral absent IAC |
Absent left ICA; low right CCA bifurcation; large basilar system |
Normal C-spine |
| 8 | MRI brain | Normal inner ear bilaterally | Hypoplastic left ICA; large left PCom |
CN 6 not conclusively visible bilaterally |
| 9 | NA |
Columns 3 and 4 modified from Tischfield et al.,1 Supplementary Table 1 Online.
MRA = magnetic resonance angiography; CCD = common cavity deformity; IAC = internal auditory canal; CCA = common carotid artery; ICA = internal carotid artery; PCom = posterior communicating artery; NA = not available; C-spine = cervical spine; US = ultrasound.
The skull base was normal only in Patient 8, who had normal inner ear anatomy (figure 2A) and normal hearing, while the eight patients with sensorineural deafness had malformations involving the inner ear and petrous bone. Four patients (Patients 4 through 7) had the common cavity deformity (CCD) replacing cochlea, semicircular canals, and vestibule bilaterally (figure 2, B through D) and two (Patients 1 and 3) had CCD on one side and complete absence of inner ear structures on the other side (figure 2E). Carotid canals were absent bilaterally in Patient 5 and unilaterally in Patients 4 and 6 (figure 2B). Internal auditory canal (IAC) was absent or severely narrowed in the six patients with inner ear malformation (figure 2E). The combination of profound inner ear maldevelopment with absent carotid canal sometimes yielded the appearance of a small petrous bone with patulous Meckel's caves (figure 2F).
Figure 2. Variability of skull base neuroimaging.
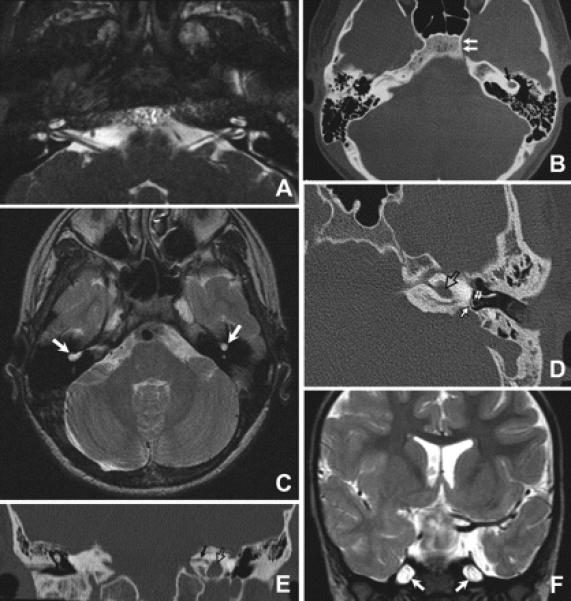
(A) High resolution axial constructive interference in steady-state MR image of the inner ear and low brainstem of Patient 8 demonstrating normal inner ear structures and normal lower cranial nerves. (B, C) Axial CT (B) and T2-weighted MR images (C) at the level of the inner ear of Patient 6 demonstrating bilateral common cavity deformity (black arrow in B and white arrows in C). The medial part of the left carotid canal is absent (double arrows in B) while the right one is present. (D) Axial CT of the petrous bone of Patient 3 showing common cavity deformity of the left inner ear (open arrow) with normal stapes (double arrows) and lack of development of the oval window opposite to the footplate of stapes (solid arrow). (E) Reconstructed coronal CT image of the petrous bone of Patient 3 showing the common cavity deformity of the left inner ear (open arrow) and severe stenosis of the left internal auditory canal (black arrow). Inner ear structures on the right side are absent. (F) Coronal T2-weighted MR image of Patient 7 showing patulous Meckle's caves (arrows).
All six patients with MR angiography or MR imaging had abnormalities of the internal carotid arteries. The left internal carotid artery (ICA) was hypoplastic in Patients 1 and 8 (figure 3A) and absent in Patients 4, 6, and 7 (figure 3B) where the left common carotid artery terminated into the external carotid artery. Four patients (Patients 1, 6, 7, and 8) with hypoplastic or absent left ICAs had a right common carotid artery bifurcation quite low in the neck, at about the C7 level (figure 3C). All six patients with ICA anomalies had an enlarged posterior communicating artery on the side of an absent or hypoplastic ICA (figure 3, B, D, and E). The ICAs were absent bilaterally in Patient 5, who had blood supply to the brain only through an enlarged vertebrobasilar system (figure 3, D and E). The intracranial portion of the left vertebral artery was duplicated in Patient 5 (figure 3F), while Patient 1 had duplication of the proximal half of the left vertebral artery.
Figure 3. Variability of MR angiography.
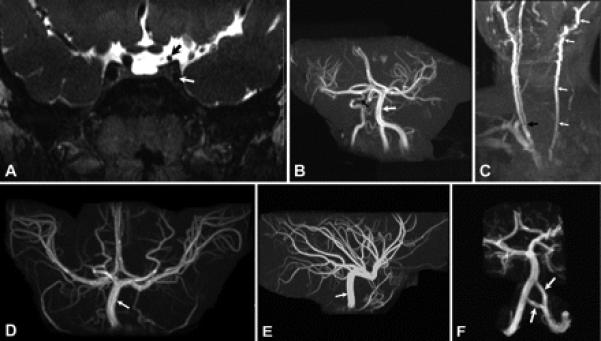
(A) Coronal T2-weighted MR image of Patient 8 demonstrating hypoplasia of the cavernous (white arrow) and supraclinoid (black arrow) portions of the left internal carotid artery. The cavernous and supraclinoid portions of the right internal carotid artery have normal caliber. (B) MR angiogram of the brain of Patient 6 showing absence of the left internal carotid artery and hypoplasia of the cavernous portion of the right internal carotid artery (black arrow). Vertebral and basilar arteries (white arrow) have larger caliber than the right internal carotid artery. (C) MR angiogram of the neck of Patient 6 demonstrating absence of the left internal carotid artery with the common carotid artery ending as a continuation of external carotid artery (white arrows). The right common carotid artery has a bifurcation (black arrow) at about the level of the C7 vertebra, while normal bifurcation of common carotid artery is about the level of C4. (D, E) Anterior (D) and lateral (E) projections of the MR angiogram of the brain of Patient 5 demonstrating absence of both internal carotid arteries and enlargement of the basilar (arrow) and posterior communicating arteries, which supply both the anterior and posterior cerebral circulations. (F) MR angiogram of the brain of Patient 5 showing duplication of the intracranial portion of the left vertebral artery (arrows).
DISCUSSION
This comprehensive phenotypic description of patients with genetically defined BSAS expands the original brief report1 and complements the description of ABDS that was published prior to identification of the underlying HOXA1 mutation.4 BSAS is characterized by variable DRS type 3 bilaterally, deafness with malformation of the inner ear, and abnormal development of cerebral vasculature. When patients are young or uncooperative or when individuals have atypical features such as severely restricted horizontal ocular motility or normal hearing, these HOXA1 syndromes may be difficult to distinguish phenotypically from autosomal recessive horizontal gaze palsy and progressive scoliosis (HGPPS; OMIM 607313). Table E-2 compares BSAS with ABDS and HGPPS,5,6 emphasizing genetic, clinical, and neuroimaging features important to the diagnosis.
The BSAS phenotype is variable among these nine patients. Horizontal ocular motility, globe retraction, and convergence were different between eyes and individuals. One patient had normal hearing with normal inner ear anatomy, while eight were deaf with a severe abnormality of cochlear and semicircular canal development. All studied patients had abnormalities of cerebral vasculature, but each had a unique constellation of abnormalities. Patients had a spectrum of somatic defects, most commonly involving external ears or extremities. Delayed motor development could be explained by the lack of a vestibular system rather than by a generalized brain disturbance in most patients. However, two were unequivocally autistic and a third had a number of autistic features, implying an abnormality extending beyond the brainstem. Some of the variability between patients could be interpreted as a severity gradient, from relatively mild involvement (e.g., Patient 8 with modest ocular motility, normal hearing, only left ICA hypoplasia, and normal cognition) to more severe disease (e.g., Patient 5 with no observed horizontal eye movement, deafness and CCD bilaterally, absent ICA bilaterally, and autism).
These patients with BSAS are distinct among patients with inherited DRS7-12 because they have bilateral DRS with limitations of both adduction and abduction. DRS is now understood as a neurogenic problem in which an abnormal abducens nerve results in a denervated lateral rectus muscle that is aberrantly innervated by the inferior division of the oculomotor nerve that also innervates the ipsilateral medial rectus muscle.13 This innervation pattern is believed to precipitate co-contraction of lateral and medial recti, causing the globe retraction and fissure narrowing on adduction and convergence that define DRS.14,15 Abducens nucleus interneurons are spared in unilateral DRS type 1,13,16 where the contralateral eye moves normally, but their status is uncertain in bilateral DRS type 3.
Given that the abducens cranial nerve is absent in the Hoxa1-−/− mouse model17 and probably also in one patient reported here, it seems likely that both BSAS and ABDS patients have partial or complete absence of the abducens nucleus and nerve bilaterally. Differences in residual adduction and the presence of mild abduction in two patients imply variable depletion of abducens motoneurons and interneurons. Similarly, variable dysinnervation of the lateral rectus by branches of the oculomotor nerve may explain differences in globe retraction and fissure narrowing on both voluntary adduction and convergence.
Surprisingly, brain neuroimaging is relatively normal in BSAS. The brainstem has a normal contour, implying that loss of tissue volume is minimal compared to Hoxa1 mutations in the mouse, which cause loss of rhombomere 5 and a 40% reduction in the size of the ventral pons.18 Extraocular muscles appear normal at the resolution of available images, suggesting that the lateral rectus muscle received sufficient innervation for myofiber survival. The supratentorial brain also has a normal appearance despite autism in two patients with BSAS and mental retardation in all patients with ABDS. Only patients with ABDS severely affected by central hypoventilation were reported to have diffuse cortical atrophy, possibly secondary to recurrent hypoxia.4
Deafness is arguably the most important disability in patients with BSAS. The eight deaf patients with BSAS all had the common cavity deformity and underdeveloped petrous bones. Hoxa1-−/− mice also have variable developmental abnormalities in the inner,17,19 middle,2,17 and external ear2 even though Hoxa1 expression has not been reported in the otic vesicle. Inductive signals from the adjacent hindbrain neuroectoderm and notochord are necessary for otic vesicle patterning,20,21 and hindbrain defects associated with Hoxa1/HOXA1 dysfunction may lead to inner ear malformations. Current patients with BSAS were enrolled primarily because of visual abnormalities, and the prevalence of HOXA1 abnormalities in deafness clinics is not yet known.
All six patients with BSAS studied appropriately had cerebrovascular malformations, and the majority of patients with ABDS had conotruncal heart defects.4 Carotid arteries and cardiac out-flow tract arise from symmetrically paired dorsal aortae, aortic arches, and the aortic sac, which appear in the third and fourth weeks of gestation.22 Hoxa1 is expressed in lateral plate and paraxial mesoderm as vasculogenesis commences in the mouse23 and could regulate aspects of angioblast migration or remodeling of primordial aortic arch vessels,24 a process dependent upon neural crest cells and lateral plate mesoderm.25,26 Vascular defects in these patients with BSAS were clinically silent but probably place patients at increased risk of cerebrovascular compromise.
Two patients with BSAS had autism spectrum disorder and another had autistic features. These observations are surprising because the CNS expression pattern of Hoxa1 in vertebrates has a rostral limit at the boundary of rhombomere 3 and 4 prior to neurogenesis27 and at the midbrain-forebrain boundary following neurogenesis.28 Loss of HOXA1 function might cause cortical and cerebellar abnormalities by disturbing development of serotonergic neurons in the brainstem1 that may modulate cerebral cortex columnar organization and cerebellar Purkinje cell arborization.29,30 The HOXA1 syndrome implies that correct hindbrain development may be necessary for normal cognition and behavior, and studies of patients with BSAS and ABDS and mutant mice are necessary to determine how HOXA1/Hoxa1 function regulates cognitive and behavioral maturation. Patients with BSAS had relatively large skulls, as reported previously in patients with a HOXA1 polymorphism.31
The constellation of congenital defects in BSAS is similar to abnormalities described in thalidomide exposure between 20 and 24 days post-fertilization.32,33 These thalidomide children sometimes manifest DRS, facial nerve palsy, deafness, external ear anomalies, other somatic abnormalities, autism, and mental retardation, suggesting that thalidomide may disrupt early rhombomere development,34 possibly by a direct effect on HOXA1 or downstream pathways.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants in this study.
Supported in part by NIH-RO1-EY015298 (E.C.E.).
GLOSSARY
- ABDS
Athabascan brainstem dysgenesis syndrome
- BSAS
Bosley-Salih-Alorainy syndrome
- CARS
Childhood Autism Rating Scale
- CCA
common carotid artery
- CCD
common cavity deformity
- CN
cranial nerve
- C-spine
cervical spine
- DRS
Duane retraction syndrome
- DSM-4
Diagnostic and Statistical Manual of Mental Disorders
- ET
esotropia
- IAC
internal auditory canal
- ICA
internal carotid artery
- MRA
magnetic resonance angiography
- NA
not available
- OD
right eye
- ortho
orthophoria
- OS
left eye
- OU
both eyes
- PCom
posterior communicating artery
- TOF
time-of-flight
- US
ultrasound
- VAB
Vineland Adaptive Behavior
- WNL
within normal limits
- XT
exotropia
Footnotes
Supplemental data at www.neurology.org
Disclosure: The authors report no conflicts of interest.
REFERENCES
- 1.Tischfield MA, Bosley TM, Salih MA, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37:1035–1037. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 2.Chisaka O, Musci TS, Capecchi MR. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature. 1992;355:516–520. doi: 10.1038/355516a0. [DOI] [PubMed] [Google Scholar]
- 3.Lufkin T, Dierich A, LeMeur M, Mark M, Chambon P. Disruption of the Hox-1.6 homeobox gene results in defects in a region corresponding to its rostral domain of expression. Cell. 1991;66:1105–1119. doi: 10.1016/0092-8674(91)90034-v. [DOI] [PubMed] [Google Scholar]
- 4.Holve S, Friedman B, Hoyme HE, et al. Athabascan brainstem dysgenesis syndrome. Am J Med Genet A. 2003;120:169–173. doi: 10.1002/ajmg.a.20087. [DOI] [PubMed] [Google Scholar]
- 5.Jen JC, Chan WM, Bosley TM, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509–1513. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosley TM, Salih MA, Jen JC, et al. Neurologic features of horizontal gaze palsy and progressive scoliosis with mutations in ROBO3. Neurology. 2005;64:1196–1203. doi: 10.1212/01.WNL.0000156349.01765.2B. [DOI] [PubMed] [Google Scholar]
- 7.Evans JC, Frayling TM, Ellard S, Gutowski NJ. Confirmation of linkage of Duane's syndrome and refinement of the disease locus to an 8.8-cM interval on chromosome 2q31. Hum Genet. 2000;106:636–638. doi: 10.1007/s004390000311. [DOI] [PubMed] [Google Scholar]
- 8.Demer J, Clark RA, Lim K-H, Engle EC. Magnetic resonance imaging evidence for widespread orbital dysinnervation in dominant Duane's retraction syndrome linked to the DURS2 locus. IOVS. 2007;48:194–202. doi: 10.1167/iovs.06-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung M, Stout JT, Borchert MS. Clinical diversity of hereditary Duane's retraction syndrome. Ophthalmology. 2000;107:500–503. doi: 10.1016/s0161-6420(99)00090-1. [DOI] [PubMed] [Google Scholar]
- 10.Appukuttan B, Gillanders E, Juo SH, et al. Localization of a gene for Duane retraction syndrome to chromosome 2q31. Am J Hum Genet. 1999;65:1639–1646. doi: 10.1086/302656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engle EC, Andrews C, Law K, Demer JL. Two pedigrees segregating Duane's retraction syndrome as a dominant trait map to the DURS2 genetic locus. Invest Ophthalmol Vis Sci. 2007;48:189–193. doi: 10.1167/iovs.06-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Baradie R, Yamada K, Hilaire C, et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet. 2002;71:1195–1199. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller NR, Kiel SM, Green WR, Clark AW. Unilateral Duane's retraction syndrome (Type 1) Arch Ophthalmol. 1982;100:1468–1472. doi: 10.1001/archopht.1982.01030040446016. [DOI] [PubMed] [Google Scholar]
- 14.Scott AB, Wong GY. Duane's syndrome. An electro-myographic study. Arch Ophthalmol. 1972;87:140–147. doi: 10.1001/archopht.1972.01000020142005. [DOI] [PubMed] [Google Scholar]
- 15.Papst W, Esslen E. Symptomatology and therapy in ocular motility disturbances. Am J Ophthalmol. 1964;58:275–291. doi: 10.1016/0002-9394(64)91577-6. [DOI] [PubMed] [Google Scholar]
- 16.Hotchkiss MG, Miller NR, Clark AW, Green WR. Bilateral Duane's retraction syndrome. A clinical-pathologic case report. Arch Ophthalmol. 1980;98:870–874. doi: 10.1001/archopht.1980.01020030864013. [DOI] [PubMed] [Google Scholar]
- 17.Mark M, Lufkin T, Vonesch JL, et al. Two rhombomeres are altered in Hoxa-1 mutant mice. Development. 1993;119:319–338. doi: 10.1242/dev.119.2.319. [DOI] [PubMed] [Google Scholar]
- 18.del Toro ED, Borday V, Davenne M, Neun R, Rijli FM, Champagnat J. Generation of a novel functional neuronal circuit in Hoxa1 mutant mice. J Neurosci. 2001;21:5637–5642. doi: 10.1523/JNEUROSCI.21-15-05637.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavalas A, Studer M, Lumsden A, Rijli FM, Krumlauf R, Chambon P. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development. 1998;125:1123–1136. doi: 10.1242/dev.125.6.1123. [DOI] [PubMed] [Google Scholar]
- 20.Bok J, Bronner-Fraser M, Wu DK. Role of the hind-brain in dorsoventral but not anteroposterior axial specification of the inner ear. Development. 2005;132:2115–2124. doi: 10.1242/dev.01796. [DOI] [PubMed] [Google Scholar]
- 21.Barald KF, Kelley MW. From placode to polarization: new tunes in inner ear development. Development. 2004;131:4119–4130. doi: 10.1242/dev.01339. [DOI] [PubMed] [Google Scholar]
- 22.Larsen WJ. Human embryology. 3rd ed. Churchill Livingstone; 2001. [Google Scholar]
- 23.Murphy P, Hill RE. Expression of the mouse labial-like homeobox-containing genes, Hox 2.9 and Hox 1.6, during segmentation of the hindbrain. Development. 1991;111:61–74. doi: 10.1242/dev.111.1.61. [DOI] [PubMed] [Google Scholar]
- 24.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 25.Etchevers HC, Vincent C, Le Douarin NM, Couly GF. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 2001;128:1059–1068. doi: 10.1242/dev.128.7.1059. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Grimmer JF, Van De Water TR, Lufkin T. Hmx2 and Hmx3 homeobox genes direct development of the murine inner ear and hypothalamus and can be functionally replaced by Drosophila Hmx. Dev Cell. 2004;7:439–453. doi: 10.1016/j.devcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter E, Goddard J, Chisaka O, Manley N, Capecchi M. Loss of Hox-A1 (Hox-1.6) function results in the reorganization of the murine hindbrain. Development. 1993;118:1063–1075. doi: 10.1242/dev.118.4.1063. [DOI] [PubMed] [Google Scholar]
- 28.McClintock JM, Jozefowicz C, Assimacopoulos S, Grove EA, Louvi A, Prince VE. Conserved expression of Hoxa1 in neurons at the ventral forebrain/midbrain boundary of vertebrates. Dev Genes Evol. 2003;213:399–406. doi: 10.1007/s00427-003-0335-7. [DOI] [PubMed] [Google Scholar]
- 29.Janusonis S, Gluncic V, Rakic P. Early serotonergic projections to Cajal-Retzius cells: relevance for cortical development. J Neurosci. 2004;24:1652–1659. doi: 10.1523/JNEUROSCI.4651-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kondoh M, Shiga T, Okado N. Regulation of dendrite formation of Purkinje cells by serotonin through serotonin1A and serotonin2A receptors in culture. Neurosci Res. 2004;48:101–109. doi: 10.1016/j.neures.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Conciatori M, Stodgell CJ, Hyman SL, et al. Association between the HOXA1 A218G polymorphism and increased head circumference in patients with autism. Biol Psychiatry. 2004;55:413–419. doi: 10.1016/j.biopsych.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Miller MT. Thalidomide embryopathy: a model for the study of congenital incomitant horizontal strabismus. Trans Am Ophthalmol Soc. 1991;89:623–674. [PMC free article] [PubMed] [Google Scholar]
- 33.Stromland K, Miller MT. Thalidomide embryopathy: revisited 27 years later. Acta Ophthalmol (Copenh) 1993;71:238–245. doi: 10.1111/j.1755-3768.1993.tb04997.x. [DOI] [PubMed] [Google Scholar]
- 34.Miller MT, Stromland K. The Möbius sequence: a re-look. JAAPOS. 1999;3:199–208. doi: 10.1016/s1091-8531(99)70003-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.