

Abstract
Objective
CCR5 and its ligands (CCL3, CCL4 and CCL5) may play a role in inflammatory cell recruitment into the joint. However, recently it has been reported that CCR5 on T cells and neutrophils acts as a decoy receptor for CCL3 and CCL5 to assist in resolution of inflammation. To determine whether CCR5 functions as a pro-inflammatory or anti-inflammatory mediator in arthritis, we examined the role of CCR5 in proteoglycan (PG)-induced arthritis (PGIA).
Methods
PGIA was induced by immunization of BALB/c wild type (WT) and CCR5-deficient (CCR5−/−) mice with human PG in adjuvant. The onset and severity were monitored overtime. Met-RANTES was used to block CCR5 in vivo. Arthritis was transferred to SCID mice with spleen cells from arthritis WT and CCR5−/− mice. Cytokines and chemokines were measured by ELISA.
Result
Treatment with the CCR5 inhibitor, Met-RANTES, and CCR5−/− mice developed exacerbated arthritis late in the course of disease. The increase in arthritis severity in CCR5−/− correlated with elevated serum levels of CCL5. However, exacerbated arthritis was not intrinsic to the CCR5−/− lymphoid cells as arthritis transferred into SCID recipients was similar in WT and CCR5−/− mice. CCR5 expression in the SCID was sufficient to clear CCL5 as serum levels of CCL5 were the same in SCID recipients receiving WT or CCR5−/− cells.
Conclusion
These data demonstrate that CCR5 is a key player in controlling the resolution of inflammation in experimental arthritis.
Keywords: Autoimmunity, Inflammation, Rheumatoid Arthritis, Chemokine and Rodent
Rheumatoid arthritis (RA) is a chronic, progressive autoimmune disease characterized by joint infiltration by several populations of leukocytes including T cells, B cells, macrophages and neutrophils (1). These infiltrating cells, in collaboration with resident cells, mediate cartilage destruction and bone erosion through a combination of cytokines, chemokines and matrix metalloproteases, (2)(3). In synovial fluid of RA patients, all four groups of chemokine families, CXC, CC, C and CX3C, and their receptors have been identified (3). The inhibition of infiltrating cells into the joint therefore represents an important step for therapeutic intervention.
Chemokine (C-C motif) receptor 5 (CCR5), is a G-protein coupled chemokine receptor composed of seven transmembrane helices that is expressed by activated T cells, monocytes, dendritic cells, tissue macrophages, and neutrophils (4, 5). CCR5 is the natural receptor for the ligands CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) all of which contribute to chemotactic activity in the synovial fluid of RA patients (6–8). The accumulation of CCR5+ T cells in the synovium of patients with RA suggests an important role in disease pathology (8). However, the chemotactic responses to RA synovial fluid were not dependent solely on a functional CCR5 receptor (9). The CCR5+ T cells express other chemokine receptors such as CCR1 which are also capable of responding to CCL3 and CCL5 (9). An allelic form of the CCR5 gene contains a 32-bp deletion (Δ32CCR5) which results in a nonfunctional CCR5 receptor. Several studies have addressed whether a loss of function mutation could have a protective effect against RA. Two of these studies demonstrated a statistical significant negative correlation between Δ32CCR5 and RA (10, 11) whereas three other studies were not significant (12–14). More recently, a meta-analysis of these 5 published studies demonstrated a significant negative association of Δ32CCR5 with RA (15) suggesting that - at least in the population of European ancestry - it may be protective.
Treatment with a modified form of RANTES known as Met-RANTES, mildly inhibits arthritis in collagen-induced arthritis (CIA) and adjuvant induced arthritis (AIA) (15–17); however, these studies did not exclude the possibility that Met-RANTES interaction with CCR1 mediates inhibition. Use of a non-peptide antagonist of CCR5 in CIA revealed intact cellular immune responses but impaired T cell migration (16). Contrary to these findings, CIA in CCR5-deficient mice is similar to wild type mice (17).
A novel role for CCR5 in the clearance of inflammation was recently elucidated. Early clues suggesting an anti-inflammatory role of CCR5 showed that the CCR5 antagonist Met-RANTES affected the uptake of apoptotic cells in a model of glomerulonephritis (18). Similarly, a deficiency in CCR5 or blocking CCR5 enhanced pathogenic inflammatory responses in hepatic liver disease, pancreatitis and glomerulonephritis (18–20). An important mechanistic explanation for these phenomena revealed that CCR5 may act as a novel decoy receptor on late apoptotic neutrophils and T cells. In this way, CCR5 can function to remove excess CCL3, CCL4, and CCL5 from tissue in the presence of pro-resolution lipid mediators (5).
In this study we use an established model of RA, proteoglycan-induced arthritis (PGIA), to examine the function of CCR5 in disease. We found that CCR5 function is important in the clearance of CCL5 thereby promoting the resolution of inflammation.
MATERIAL AND METHODS
Mice
CCR5-deficient mice (CCR5−/−) on the BALB/c background were originally generated by Dr. Rodrigo Bravo (21) and were subsequently backcrossed to BALB/c for 10 generations. CCR5−/− mice were generously supplied by Dr. Don Moser (The Scripps Research Institute, La Jolla, CA). Mice were genotyped using primers specific for CCR5. Wild type (WT) BALB/cJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were maintained in the Rush University Medical Center facility. All animal experiments were approved by the institutional Animal Care and Use Committee at Rush University Medical Center (Chicago, IL).
Induction and Assessment of Arthritis
Cartilage obtained from human joint replacement surgery was obtained via the Orthopedic Tissue and Implant Repository of Rush University Medical Center, with the approval of the Institutional Review Board. PG (aggrecan) was isolated as previously described (22). Age matched, female BALB/c mice, 12–14 wks of age, were used in all experiments. Mice were immunized i.p. with 150 μg human PG (measured as protein) in dimethyl-dioctadecyl ammonium bromide (DDA) (Sigma Aldrich, St. Louis, MO) as described (23). Booster immunizations were given at three and six weeks with 100 μg PG in DDA. Mice were monitored for arthritis twice weekly and scored in a blinded manner. Paw swelling was scored based on an established scoring system on a scale from one to four as follows: 0, normal; 1, mild erythema and swelling of several digits; 2, moderate erythema and swelling; 3, more diffuse erythema and swelling; and 4, severe erythema and swelling of complete paw with ankylosis. Incidence of arthritis denotes the percentage of mice that develop PGIA. Scores range from 0 to 16, based on individual paw scores of 0 to 4. Hind ankle joints of immunized mice were isolated. Joints were fixed in formalin, decalcified in 5% formic acid, embedded in paraffin and stained with hematoxylin and eosin. Cellular infiltration was measured on a scale of 0–4 by a blinded observer.
Transfer of arthritis to SCID mice
Spleen cells (2.5×107 spleen cells/mouse) from immunized WT or CCR5−/− mice and 100μg PG was injected i.p. into SCID mice. Mice were monitored by a blinded observer for onset and severity of disease as described above. SCID mice were treated with Met-RANTES (100μg) in PBS or PBS alone every third day for 9 days starting on day 6 or day 9 depending on the experiment. Met-RANTES was generously provided by Dr. Amanda Proudfoot, Geneva Research Centre, Merck Serono SA, Geneva Switzerland.
Assessment of T cell activation and autoantibody production
CD4+ T-cells were isolated from the spleens of PG-immunized mice by negative selection using CD4 isolation kits and AutoMACS automated separation (Miltenyi Biotech, Auburn, CA). CD4+ T cells (2.5×105 cells/well) were stimulated in the presence or absence of PG (10 μg/ml) and naïve irradiated spleen cells (2.5×105 cells/well) (2500 rad) in 96-well plates (Fisher Scientific, Fair Lawn, NJ) in serum-free medium HL-1 media (Fisher Scientific) containing 100μg/ml penicillin, 100μg/ml streptomycin, and 2mM L-glutamine (complete media). Cells were cultured in quadruplicate. Cultures were incubated at 37°C in 5% CO2 for 5 days and pulsed with [3H] thymidine (0.5 μCi/well) for the last 18h. Cells were harvested (Tomtec, Orange, CT) and incorporated [3H]-thymidine assessed by scintillation counting (EC&G Wallac, Galesburg, MD).
Serum was obtained from immunized and assessed for antibodies to mouse PG (mPG). EIA tissue culture “half area” plates (Costar Corning, NY) were coated with 0.75 μg of native mouse PG in carbonate buffer. Sera were serially diluted in PBS containing 0.5% Tween 20. Samples were incubated with the immobilized mPG and mouse PG-specific autoantibodies detected using peroxidase-conjugated rabbit IgG against mouse IgG1 and IgG2a (Zymed Laboratories, San Francisco, CA). Secondary antibodies were detected with the substrate o-phenylenediamine. Relative concentration was determined using a standard curve of known concentrations of unlabeled murine IgG1 and IgG2a. Data represent the mean ± SEM of IgG1 and IgG2a autoantibodies from 7–10 mice.
Assessment of cytokines/chemokines
Spleens were harvested from PG-immunized mice. CD4+ T cells were purified using CD4 isolation beads (Miltenyi Biotech) and cultured with naïve irradiated spleen cells (2500 rad) in the absence or presence of PG (10 μg/ml) in complete media. Supernatants were harvested on day 4 and examined for cytokines by enzyme-linked immunosorbent assay (ELISA) using the OPT EIA mouse IFN-γ or IL-4 kit (BD PharMingen, San Diego, CA) and a mouse IL-17 ELISA kit (R&D Systems, Minneapolis, MN). Synovial fluid was obtained from ankle joints of arthritic mice and samples pooled from 3 mice and serum samples from individual mice assessed for CCL3, CCL4, and CCL5 by ELISA (R&D Systems).
Statistical analysis
The Mann-Whitney U test was used to compare nonparametric data for statistical significance. P values less than 0.05 were considered significant.
RESULTS
CCR5 ligands expressed in synovial fluid of arthritic mice
We first examined the expression of CCR5 ligands CCL3, CCL4, and CCL5 and CCR5 in arthritic mice (Figure 1). Synovial fluid was obtained from ankle joints of arthritic hind paws, pooled and assayed for CCL3, CCL4, and CCL5 by ELISA. CCL3, CCL4, and CCL5 were expressed in the synovial fluid of arthritic mice (Figure 1A). Arthritic ankle joints were collagen digested and cells stained with antibodies to CD4, IL-6G/C (GR-1), and CCR5 (Figure 1B). Synovial tissue contained CD4 low and CD4 high expressing cells and CCR5 was expressed predominantly on the CD4 low cells. Neutrophils are the predominant cell population in the synovial tissue. Neutrophils, identified by the GR-1 antibody, detected a population of GR-1 lo and GR-1 hi cells with CCR5 expressed on the GR-1 lo cells. The presence of CCL3, CCL4, and CCL5 and CCR5 expressing CD4+ T cells and neutrophils suggest a functional role for these chemokines/chemokine receptors in PGIA.
Figure 1.
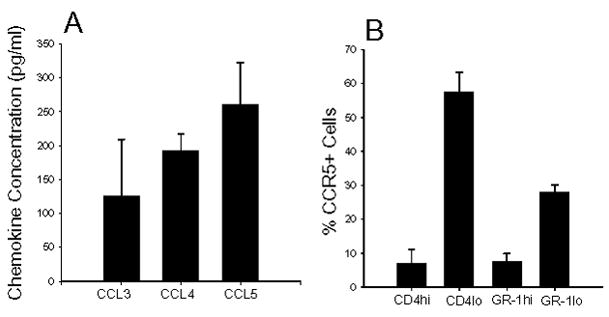
Expression of CCR5 ligands in synovial fluid of arthritic mice. (A) Synovial fluid was obtained from inflamed joints of arthritic WT mice during peak of inflammation and examined by ELISA for CCL3, 4, 5 (MIP1α, MIP1β and RANTES respectively). Results shown are average ± SEM. (B) Synovial tissue cells were obtained from joints of arthritic mice and CD4+ T cells and GR-1+ neutrophils were stained for CCR5 expression by flow cytometry. Results are average ± SD.
Transient inhibition of arthritis by CCR5 blockade
In order to determine if CCR5 plays a role in PGIA, we used a peptide antagonist, Met-RANTES, to transiently block the engagement of CCR5 and CCR1 with its natural ligands. To facilitate the effective use of the peptide antagonist, we used the adoptive transfer model of PGIA. In this system, transfer of spleen cells from arthritic mice and PG i.p. into naïve SCID mice produces rapid and reproducible arthritis within a few weeks. Recipient mice were treated starting at day 6 after cell transfer with 100μg of Met-RANTES in PBS or PBS alone every third day for nine days. Our results showed a brief, significant delay in the onset and severity of disease in mice treated with Met-RANTES in comparison to PBS treated mice (Figure 2A and B). These data suggest CCR5/CCR1 contributes to the inflammatory process in PGIA. Interestingly, as arthritis progressed there appeared to be enhanced swelling and erythema in the Met-RANTES treated mice. To assess the possibility that blocking CCR5/CCR1 might have a negative affect on inflammation we repeated the adoptive transfer experiment. In this experiment, spleen cells from arthritic mice were transferred into SCID recipients but treatment with Met-RANTES was delayed until day 12 (Figure 2C and D). At day 12, 100μg of Met RANTES was administered every three days for nine days. The early suppression of arthritis was blunted, probably because of the delay in Met-RANTES treatment. However, when development of arthritis is monitored over several weeks, disease severity was significantly elevated in the Met-RANTES treated mice. The normal waxing and waning of inflammation observed in PBS treated mice does not occur in the Met-RANTES treated animals. These data demonstrate that CCR5/CCR1 have an important regulatory function in PGIA.
Figure 2.
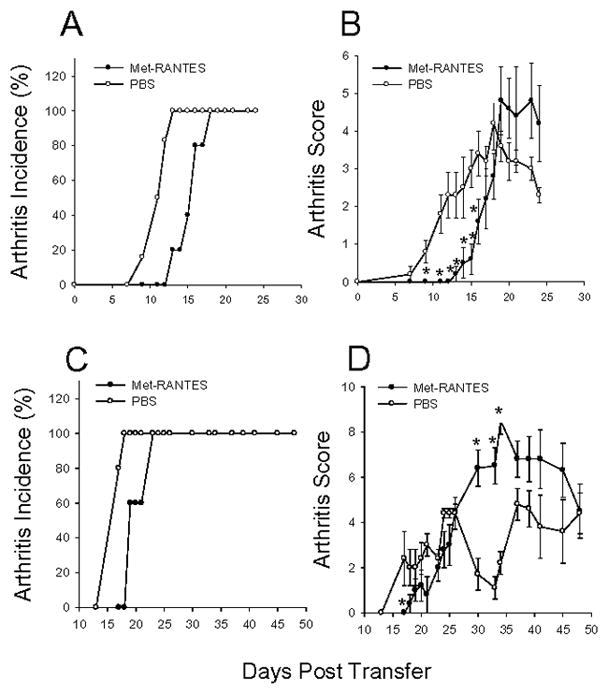
Suppression of early arthritis and exacerbation of late arthritis in mice treated with CCR5/CCR1 antagonist Met-RANTES. (A–D) Spleen cells (2.5×107) from arthritic WT mice were transferred to SCID mice along with 100 μg hPG i.p. on day 0. (A and B) Mice were administered 100 μg Met-RANTES in PBS (n=5) or PBS (n=5) as control on day 6 after cell transfer and every third day for nine days. (C and D) Mice were administered 100 μg Met-RANTES in PBS (n=5) or PBS (n=5) as control on day 12 after cell transfer and every third day for nine days. Arthritis incidence (A and C) and arthritis score (B and D) were monitored daily by a blinded observer. Data represents daily averages ±SEM. Asterisks (*) denote significant differences (p≤0.05). Data are representative of three experiments.
CCR5 deficient mice exhibit sustained inflammation as disease in WT mice wanes
To begin to understand the role of CCR5 in regulating PGIA, we used mice deficient in CCR5 (CCR5−/−). We immunized groups of age-matched BALB/c WT and CCR5−/− mice with PG in adjuvant on day 0, 21, and 42 and monitored disease onset and severity over time (Figure 3A and B). CCR5−/− mice succumbed to disease early with similar onset and severity of arthritis as WT mice. However, after week 11 post immunization, the CCR5−/− mice began to develop more severe disease than WT mice and by week 16 disease was significantly more severe. These data support a role for CCR5 in the regulation of inflammation, rather than in recruitment of inflammatory cells late in disease. Histological assessment of ankle joints in arthritic WT and CCR5−/− mice exhibited leukocyte infiltration and synovial lining proliferation, with cartilage loss and bone erosion (Figure 3C and D). Although the clinical score was more severe in the CCR5−/− mice late in disease development compared to WT mice, the histological appearance was not significantly different. This may reflect the fact that the histological appearance was very severe in both CCR5−/− and WT mice.
Figure 3. Arthritis is exacerbated late in CCR5−/− mice.
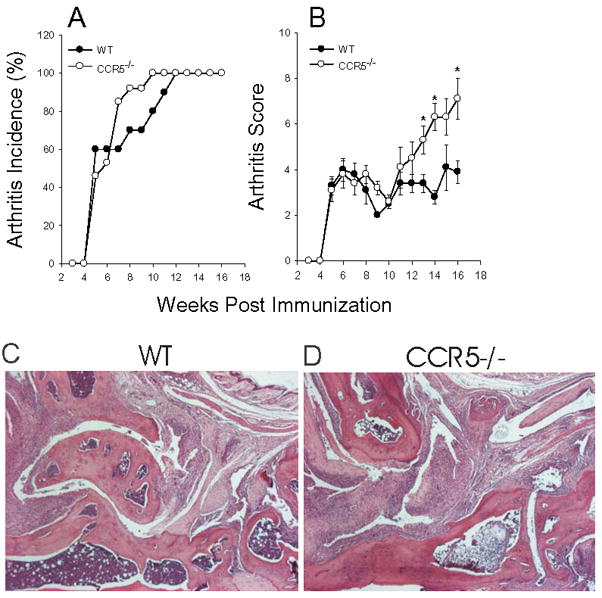
WT (n=10) and CCR5−/− (n=13) age matched female BALB/c mice were immunized i.p. with human PG in adjuvant three times at three week intervals and monitored for arthritis onset and severity by a blinded observer. (A) Arthritis incidence is expressed as the percentage of mice that developed arthritis. (B) Arthritis score is the sum of paw inflammation scores for each mouse divided by the number of arthritic mice. Results are shown as weekly mean scores ±SEM. Asterisks (*) denote significant differences (p≤0.05) compared to WT. (C) WT and (D) CCR5−/− ankle joint histology. Sections were stained with hematoxylin and eosin.
Cytokines are important in the development of PGIA, thus, it is possible that CCR5−/− mice may have an altered cytokine profile later in the course of disease when the CCR5−/− mice exhibit severe arthritis. To examine this possibility we harvested spleen cells from WT and CCR5−/− mice 16 weeks after the initial immunization. We determined whether T cell inflammatory cytokines IFN-γ, IL-17, and IL-4 were increased in CCR5−/− mice. CD4+ T cells were purified from spleen and re-stimulated in the presence of naïve antigen presenting cells (APCs) and PG and supernatants assessed for the production of cytokines by ELISA (Figure 4A, B, and C). In addition, the frequency of CD4+ T cell expressing cytokines was measured by intracellular staining for IFN-γ, IL-17, and IL-4. We found no difference between WT and CCR5−/− mice in the production or the frequency of CD4+ T cells expressing IFN-γ, IL-17, and IL-4 (Figure A and B) (data not shown). T cell activation was also measured by the ability of T cells to specifically response to PG in vitro culture. There was no difference between WT and CCR5−/− mice in their CD4+ T cells recall response to PG (Figure 4D).
Figure 4.

Inflammatory cytokine expression, autoantibody production, and T cell proliferation expression similar in WT and CCR5−/− mice. (A, B, and C) Purified T cells were obtained from immunized WT and CCR5−/− mice and stimulated in the presence of PG and irradiated naïve spleen cells. Culture supernatants were harvested at 4 days and assayed for IFN-γ, IL-17, and IL-4 by ELISA. (D) Purified T cells were stimulated as above in (A, B, and C). PG-specific T cell proliferation was measured by the incorporation of [3H]-thymidine. (E) Serum was obtained and anti-murine PG (mPG) antibody isotypes (IgG1 and IgG2a) were measured by ELISA. Results are the average ± SEM. (WT n=7–10, CCR5 n=10–11).
Autoantibodies specific for mouse PG are a hallmark of PGIA. To determine if the concentration of PG-specific autoantibodies is increased in CCR5−/− mice in comparison to WT, mice were bled after the 3rd immunization with PG. There was no difference in the levels of autoantibodies to PG in WT and CCR5−/− mice (Figure 4E). These data demonstrate that the increase in disease severity in CCR5−/− mice was not governed by altered T helper cytokine concentration, PG-specific T cell activation or autoantibody level.
CCL5 significantly elevated in naïve and PG immunized CCR5 deficient mice
CCR5 deficiency in mice and humans is associated with elevated concentrations of CCL5 in tissue (24–26). Due to the redundancy of the chemokine/chemokine receptor system, it is possible that elevated levels of CCL5 in CCR5 impaired people (or mice) could promote enhanced influx of leukocytes via CCR1, which is also expressed on T-cells (25, 26), (19). To determine if this occurs in CCR5−/− mice, we examined the serum concentrations of CCL3, CCL4 and CCL5 in naïve mice and one week after each PG immunization. CCL3 and CCL4 were undetectable in the serum of WT and CCR5−/− mice, however, CCL5 was significantly elevated in CCR5−/− mice compared to WT at each time point tested (Figure 5).
Figure 5.
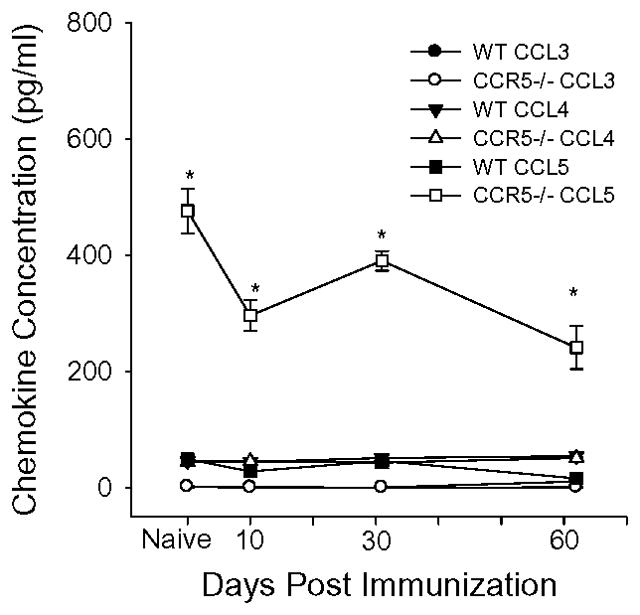
Elevated expression of CCL5 in CCR5−/− mice. Serum was taken from WT and CCR5−/− mice before immunization and 10 days after each immunization. Individual serum samples were tested for CCL3, CCL4, and CCL5 by ELISA. Results are the average ± SEM. (WT n=13, CCR5 n=13.). Asterisks (*) denote significant differences (p≤0.05) compared to WT.
Severe disease in CCR5−/− mice resolved upon SCID transfer
CCR5 has emerged as an important chemokine receptor on T cell and neutrophils in the clearance of inflammation by scavenging CCL3, CCL4, and CCL5 (5). In PGIA, CD4+ T cells and B cells are required for the development of disease although T cells are a minor cell population in the inflamed joint where neutrophils dominate. To determine if the severe disease observed late in the CCR5−/− mice was due to a defect in CD4+ T cells or a defect in CD4+ T cells and neutrophils, we used the SCID transfer model of PGIA. In this model, SCID non-lymphoid cells express CCR5 whereas the transferred spleen cells are CCR5 negative. We transferred spleen cells from WT or CCR5−/− mice into SCID mice and monitored disease onset and severity (Figure 6A and B). Disease developed in the SCID mice with similar onset and severity whether WT or CCR5−/− spleen cells were transferred. These data suggest that the presence of CCR5 on neutrophils or other non T or B cells in the SCID mouse was sufficient to allow for the resolution of inflammation similar to WT recipients. To assess the ability of the CCR5-sufficient cell in the SCID recipient to scavenge chemokines, we again examined the CCL5 concentrations in serum from the arthritic mice by ELISA (Figure 6C). In contrast to the elevated concentrations of CCL5 in arthritic CCR5−/− mice, serum concentration of CCL5 in the SCID recipients of WT or CCR5−/− spleen cells were similar. These data demonstrate that expression CCR5 on non-lymphoid cells was able to control the levels of circulating CCL5, supporting the association between elevated CCL5 and sustained inflammation.
Figure 6.
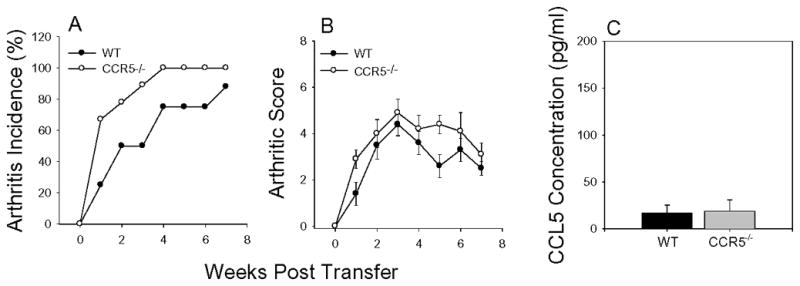
Disease transferred to SCID mice effectively. Spleen cells (4×107) from WT or CCR5−/− mice were injected i.p. into SCID recipients with PG and monitored for arthritis incidence (A) and severity (B) by a blinded observer. Results shown are average ± SEM. (WT n=8, CCR5 n=9). (C) SCID recipients of CCR5−/− spleen cells controlled serum CCL5 concentrations similar to WT cells. Individual serum samples from SCID mice repopulated with WT or CCR5−/− spleen cells at week 3 were examined by ELISA for CCL5 expression. Results shown are average ± SEM. (WT n=6, CCR5 n=6).
DISCUSSION
The present study was undertaken to determine the function of CCR5 in PGIA based on the findings that CCR5 ligands are expressed in synovial fluid of arthritic mice and CCR5 is expressed on CD4+ T cells and neutrophils in synovial tissue. In this study, we identify CCR5 as an important molecule in the resolution phase of inflammation. In mice deficient in CCR5, arthritis is similar to WT mice early in disease. These data suggest that the early infiltration of leukocytes into the joint may be mediated by other CXC, CC, C and CX3C family members such as CXCL5 which is known to precede the development of arthritis in adjuvant-induced arthritis (27). However, as arthritis begins to wane in WT mice, arthritis increases in CCR5−/− mice. Similarly, exacerbation in the late phase of arthritis was observed in Met-RANTES treated mice. However, treatment with Met-RANTES was also inhibitory in the early phase of disease. Since Met-RANTES is not specific for CCR5 but also inhibits CCR1, it is possible that the difference between Met-RANTES treatment and CCR5−/− mice is the inhibition of CCR1. It has been reported that another compound capable of inhibiting CCR1 also suppresses the development of CIA (28). In support of an immunoregulatory role for CCR5, several other models of inflammation report that a deficiency in CCR5 leads to an increase in inflammation. In models of T-cell-mediated hepatitis, influenza A virus, graft-versus-host disease, and Mycobacterium tuberculosis infection, CCR5−/− mice exhibit exacerbated disease (19, 26, 29–31).
A role for CCR5 in the resolution of inflammation has recently been reported (5). The mechanism for this down-regulation of inflammation involves the use of CCR5 as a decoy receptor on the surface of late apoptotic T-cells and neutrophils. Binding of local chemokines to CCR5 - without subsequent signaling - has the effect of reducing the concentration of chemokines and thereby assisting the resolution of inflammation. We show in CCR5−/− mice that CCL5 was detectable in the serum of both naïve and PG-immunized mice whereas in WT mice CCL5 was almost undetectable. The presence of elevated CCL5 in CCR5−/− mice correlates with the increase in arthritis exhibited in the late phase of disease. These data suggest that the inability to clear CCL5 in CCR5−/− mice contributes to the enhanced inflammation. To determine whether clearance of CCL5 correlates with reduction in arthritis, we used an adoptive transfer models. In this model, spleen cells from arthritic CCR5−/− were transferred into CCR5 positive SCID mice. Spleen cells from CCR5−/− were unable to transfer accelerated arthritis indicating that the enhanced disease was not intrinsic to the transferred cell population. Thus, a deficiency in CCR5 expression on cells of the spleen is not the cause of the enhanced arthritis observed in CCR5−/− mice but is rather due to another factor such as high levels of CCL5 in CCR5−/− mice. Moreover, the level of CCL5 in the serum of SCID mice was similar whether the cells were transferred from WT or CCR5−/− mice. These data suggest that the CCR5 positive myeloid, natural killer and dendritic cells in the SCID cleared CCL5 and clearance of CCL5 prevented the development of enhanced arthritis.
Regulatory T cells (Tregs) play an important role in the prevention of autoimmunity. Treg migration to sites of inflammation use several different chemokine receptors including CCR5 (32–34). A deficiency in Treg migration to the joints might account for the increase in arthritis in the CCR5−/− mice. If this were the case, we would predict that the transfer of lymphocytes from CCR5−/− mice would transfer Tregs that could not migrate to arthritic joints, however, we found no difference in the development of arthritis in SCID recipients whether they received WT or CCR5−/− cells. In addition, if Tregs were involved in exacerbated arthritis CCR5−/− mice we would have expected an increase in PG-specific T and B cell responses, however, these responses were similar in arthritic WT and CCR5−/− mice.
The ability to remove chemokines in a timely manner to dampen chemokine activity and stop leukocyte infiltration could be a useful therapy in RA for resolution of inflammation. Treatment with soluble CCR5 or with cells engineered to express non-signaling CCR5 might function as a chemokine-scavenging device for the treatment of RA or other chronic autoimmune inflammatory diseases.
In conclusion we have demonstrated that CCR5 is unnecessary for initiation of PGIA, but rather plays a role in the clearance of pro-inflammatory chemokines to resolve inflammation.
Acknowledgments
This work was supported by National Institutes of Health Grants AR045652 (T.T.G and A.F.).
References
- 1.Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 2.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–42. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 3.Koch AE. Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum. 2005;52(3):710–21. doi: 10.1002/art.20932. [DOI] [PubMed] [Google Scholar]
- 4.Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol Today. 1998;19(12):568–74. doi: 10.1016/s0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 5.Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE, Luster AD, et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7(11):1209–16. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, et al. Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. J Clin Invest. 1994;93(3):921–8. doi: 10.1172/JCI117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson E, Keystone EC, Schall TJ, Gillett N, Fish EN. Chemokine expression in rheumatoid arthritis (RA): evidence of RANTES and macrophage inflammatory protein (MIP)-1 beta production by synovial T cells. Clin Exp Immunol. 1995;101(3):398–407. doi: 10.1111/j.1365-2249.1995.tb03126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norii M, Yamamura M, Iwahashi M, Ueno A, Yamana J, Makino H. Selective recruitment of CXCR3+ and CCR5+ CCR4+ T cells into synovial tissue in patients with rheumatoid arthritis. Acta Med Okayama. 2006;60(3):149–57. doi: 10.18926/AMO/30745. [DOI] [PubMed] [Google Scholar]
- 9.Santiago B, Galindo M, Rivero M, Brehmer MT, Mateo I, Pablos JL. The chemoattraction of lymphocytes by rheumatoid arthritis - synovial fluid is not dependent on the chemokine receptor CCR5. Rheumatol Int. 2002;22(3):107–11. doi: 10.1007/s00296-002-0203-1. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Reino JJ, Pablos JL, Carreira PE, Santiago B, Serrano L, Vicario JL, et al. Association of rheumatoid arthritis with a functional chemokine receptor, CCR5. Arthritis Rheum. 1999;42(5):989–92. doi: 10.1002/1529-0131(199905)42:5<989::AID-ANR18>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 11.Pokorny V, McQueen F, Yeoman S, Merriman M, Merriman A, Harrison A, et al. Evidence for negative association of the chemokine receptor CCR5 d32 polymorphism with rheumatoid arthritis. Ann Rheum Dis. 2005;64(3):487–90. doi: 10.1136/ard.2004.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garred P, Madsen HO, Petersen J, Marquart H, Hansen TM, Freiesleben Sorensen S, et al. CC chemokine receptor 5 polymorphism in rheumatoid arthritis. J Rheumatol. 1998;25(8):1462–5. [PubMed] [Google Scholar]
- 13.Zapico I, Coto E, Rodriguez A, Alvarez C, Torre JC, Alvarez V. CCR5 (chemokine receptor-5) DNA-polymorphism influences the severity of rheumatoid arthritis. Genes Immun. 2000;1(4):288–9. doi: 10.1038/sj.gene.6363673. [DOI] [PubMed] [Google Scholar]
- 14.Cooke SP, Forrest G, Venables PJ, Hajeer A. The delta32 deletion of CCR5 receptor in rheumatoid arthritis. Arthritis Rheum. 1998;41(6):1135–6. doi: 10.1002/1529-0131(199806)41:6<1135::AID-ART24>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Prahalad S. Negative association between the chemokine receptor CCR5-Delta32 polymorphism and rheumatoid arthritis: a meta-analysis. Genes Immun. 2006;7(3):264–8. doi: 10.1038/sj.gene.6364298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, et al. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol. 2002;32(8):2124–32. doi: 10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 17.Quinones MP, Ahuja SK, Jimenez F, Schaefer J, Garavito E, Rao A, et al. Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. J Clin Invest. 2004;113(6):856–66. doi: 10.1172/JCI20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anders HJ, Frink M, Linde Y, Banas B, Wornle M, Cohen CD, et al. CC chemokine ligand 5/RANTES chemokine antagonists aggravate glomerulonephritis despite reduction of glomerular leukocyte infiltration. J Immunol. 2003;170(11):5658–66. doi: 10.4049/jimmunol.170.11.5658. [DOI] [PubMed] [Google Scholar]
- 19.Moreno C, Gustot T, Nicaise C, Quertinmont E, Nagy N, Parmentier M, et al. CCR5 deficiency exacerbates T-cell-mediated hepatitis in mice. Hepatology. 2005;42(4):854–62. doi: 10.1002/hep.20865. [DOI] [PubMed] [Google Scholar]
- 20.Moreno C, Nicaise C, Gustot T, Quertinmont E, Nagy N, Parmentier M, et al. Chemokine receptor CCR5 deficiency exacerbates cerulein-induced acute pancreatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;291(6):G1089–99. doi: 10.1152/ajpgi.00571.2005. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Kurihara T, Ryseck RP, Yang Y, Ryan C, Loy J, et al. Impaired macrophage function and enhanced T cell-dependent immune response in mice lacking CCR5, the mouse homologue of the major HIV-1 coreceptor. J Immunol. 1998;160(8):4018–25. [PubMed] [Google Scholar]
- 22.Finnegan A, Mikecz K, Tao P, Glant TT. Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J Immunol. 1999;163(10):5383–90. [PubMed] [Google Scholar]
- 23.Hanyecz A, Berlo SE, Szanto S, Broeren CP, Mikecz K, Glant TT. Achievement of a synergistic adjuvant effect on arthritis induction by activation of innate immunity and forcing the immune response toward the Th1 phenotype. Arthritis Rheum. 2004;50(5):1665–76. doi: 10.1002/art.20180. [DOI] [PubMed] [Google Scholar]
- 24.Locati M, Murphy PM. Chemokines and chemokine receptors: biology and clinical relevance in inflammation and AIDS. Annu Rev Med. 1999;50:425–40. doi: 10.1146/annurev.med.50.1.425. [DOI] [PubMed] [Google Scholar]
- 25.Carr DJ, Ash J, Lane TE, Kuziel WA. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J Gen Virol. 2006;87(Pt 3):489–99. doi: 10.1099/vir.0.81339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol. 2000;156(6):1951–9. doi: 10.1016/S0002-9440(10)65068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szekanecz Z, Halloran MM, Volin MV, Woods JM, Strieter RM, Kenneth Haines G, 3rd, et al. Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43(6):1266–77. doi: 10.1002/1529-0131(200006)43:6<1266::AID-ANR9>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 28.Amat M, Benjamim CF, Williams LM, Prats N, Terricabras E, Beleta J, et al. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br J Pharmacol. 2006;149(6):666–75. doi: 10.1038/sj.bjp.0706912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welniak LA, Wang Z, Sun K, Kuziel W, Anver MR, Blazar BR, et al. An absence of CCR5 on donor cells results in acceleration of acute graft-vs-host disease. Exp Hematol. 2004;32(3):318–24. doi: 10.1016/j.exphem.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Wysocki CA, Burkett SB, Panoskaltsis-Mortari A, Kirby SL, Luster AD, McKinnon K, et al. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pretransplant conditioning. J Immunol. 2004;173(2):845–54. doi: 10.4049/jimmunol.173.2.845. [DOI] [PubMed] [Google Scholar]
- 31.Algood HM, Flynn JL. CCR5-deficient mice control Mycobacterium tuberculosis infection despite increased pulmonary lymphocytic infiltration. J Immunol. 2004;173(5):3287–96. doi: 10.4049/jimmunol.173.5.3287. [DOI] [PubMed] [Google Scholar]
- 32.Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood. 2005;106(9):3300–7. doi: 10.1182/blood-2005-04-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med. 2006;203(11):2451–60. doi: 10.1084/jem.20060956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–55. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]