

Abstract
Systemic infection with Escherichia coli on postnatal day (P) 4 in rats results in significantly altered brain cytokine responses and behavioral changes in adulthood, but only in response to a subsequent immune challenge with lipopolysaccharide [LPS]. The basis for these changes may be long-term changes in glial cell function. We assessed glial and neural cell genesis in the hippocampus, parietal cortex (PAR), and pre-frontal cortex (PFC), in neonates just after the infection, as well as in adulthood in response to LPS. E. coli increased the number of newborn microglia within the hippocampus and PAR compared to controls. The total number of microglia was also significantly increased in E. coli-treated pups, with a concomitant decrease in total proliferation. On P33, there were large decreases in numbers of cells coexpressing BrdU and NeuN in all brain regions of E. coli rats compared to controls. In adulthood, basal neurogenesis within the dentate gyrus (DG) did not differ between groups; however, in response to LPS, there was a decrease in neurogenesis in early-infected rats, but an increase in controls to the same challenge. There were also significantly more microglia in the adult DG of early-infected rats, although microglial proliferation in response to LPS was increased in controls. Taken together, we have provided evidence that systemic infection with E. coli early in life has significant, enduring consequences for brain development and subsequent adult function. These changes include marked alterations in glia, as well as influences on neurogenesis in brain regions important for cognition.
1. Introduction
Microglia are the primary immunocompetent cells of the brain, with demonstrated roles in both protection and pathology. For instance, they appear to be neuroprotective following stroke, by producing trophic factors that aid in cellular repair (Lalancette-Hebert et al., 2007). In contrast, chronic or exaggerated microglial activation is associated with multiple neuroinflammatory diseases, including Parkinson's, Alzheimer's, and Huntington's disease (Perry, 2004). Activated microglia produce many factors, including superoxide, nitric oxide, and cytokines, that may lead to neuronal damage or interfere with neuronal function directly (Block et al., 2007). Microglial activation is also associated with multiple neurodevelopmental disorders with known or suspected immune etiologies, including autism, schizophrenia, and cerebral palsy, although the direction of causality in the majority of these disorders is unknown (Bilbo and Schwarz, in press; Meyer et al., 2005; Vargas et al., 2005).
We have reported that systemic infection with Escherichia coli on postnatal day (P) 4 in rats is associated with marked hippocampal-dependent memory impairments in adulthood. However, these impairments are only observed if a second immune challenge (bacterial lipopolysaccharide [LPS]) is administered in close proximity to learning in adulthood (Bilbo et al., 2005a; Bilbo et al., 2005b). The impairment is linked to exaggerated pro-inflammatory cytokine production to the LPS challenge, and decreased learning-induced brain-derived-neurotrophic factor (BDNF) expression within the hippocampus, a growth factor critical for memory consolidation (Bilbo et al., 2008; Bilbo et al., 2007). Importantly, this low dose of LPS does not impair memory in control rats. Thus, infection during the neonatal period appears to act as a “vulnerability” factor, by amplifying the adult rat's central cytokine response to the LPS challenge, which then impairs cognition.
We have hypothesized that the basis for this vulnerability may be long-term changes in glial cell function. Microglia are the primary cytokine producers within the brain, and are an excellent candidate for long-term changes, because they are long-lived and can become and remain activated chronically (Town et al., 2005). There is increasing support for the concept of “glial priming”, in which cells can become sensitized by an insult, challenge, or injury, such that subsequent responses to a challenge are exaggerated (Perry et al., 2003). Notably, the first week of life in rats is a time of extensive brain growth, when microglial proliferation, migration, and density peak (Bayer et al., 1993; Wu et al., 1992). Thus, the early postnatal stage may be particularly vulnerable to insult, with subsequent long-term changes in microglial cell function.
In support of this hypothesis, we have reported increases in glial cell “activation” markers in our infection model (e.g., CD11b [complement 3 receptor], and major histocompatibility complex [MHC] II), both acutely in response to the E. coli, as well as in adulthood following the LPS challenge (Bilbo et al., 2005a, 2007; in press). However, the functional consequences of observed “activation” is unknown. For instance, an increase in overall activation markers could indicate an increase in the reactivity of the same number of cells, or conversely an increase in the number of cells, without a change in function. Distinguishing between these two possibilities is critical for establishing causality, as well as eventual treatment options. Moreover, the neuronal responses to both the early E. coli and the adult LPS are completely unknown. Neurons constitutively inhibit microglial activity via interactions with factors such as the CD200 receptor expressed exclusively on microglia (Barclay et al., 2002; Lyons et al., 2007). Thus, any decrease in neurogenesis or survival by early-life infection could have enduring effects on glial cell reactivity via reduced inhibitory tone. Finally, an emerging literature implicates inflammation as an important factor in regulating neurogenesis within the adult brain (Whitney et al., 2009), but the influence of early-life inflammation on later-life responses is virtually unknown. Thus, the goal of these experiments was to 1) assess glial and neural cell proliferation and survival following infection with E. coli on day 4, and 2) assess the glial and neuronal cell responses to the subsequent LPS challenge in adulthood.
2. Materials and Methods
2.1. Animals
Adult male and female Sprague–Dawley rats (70 days) were obtained from Harlan (Indianapolis, IN) and housed in same sex pairs in polypropylene cages with food and water freely available. The colony was maintained at 22 °C on a 12:12-h light:dark cycle (lights on at 0600 MST). Following acclimation to experimental conditions, males and females were paired into breeders. Sentinel animals were housed in the colony room and screened periodically for the presence of common rodent diseases; all screens were negative. All experiments were conducted with protocols approved by the University of Colorado Animal Care and Use Committee.
2.2. Experimental procedures
2.2.1. Experiment 1
The effects of neonatal E. coli infection on precursor cell proliferation and neuro/gliogenesis.
2.2.1.1. Neonatal manipulations
Female breeders were visually examined daily for confirmation of pregnancy, and male breeders were removed from cages prior to the birth of pups (postnatal day [P] 0). Litters were culled on P4 to a maximum of 10 pups/litter, retaining two female and as many male pups as possible. All litters were born within 1 week of each other, and all studies were limited to males. Escherichia coli culture (ATCC 15746; American Type Culture Collection, Manassas, VA) vial contents were hydrated and grown overnight in 30 ml of brain-heart infusion (BHI; Difco Labs, Detroit, MI) at 37°C. Cultures were aliquoted into 1 ml stock vials supplemented with 10% glycerol and frozen at −20°C. One day before injections, a stock culture was thawed and incubated overnight in 40 ml of BHI at 37°C. The number of bacteria in cultures was read using a microplate reader (Bio-Tek Instruments Inc., Winooski, VT) and quantified by extrapolating from previously determined growth curves. Cultures were centrifuged for 15 min at 2880 xg, the supernatants were discarded, and the bacteria were re-suspended in the dose-appropriate volume of sterile Dulbecco's PBS (Invitrogen Corp., Carlsbad, CA). Male pups were injected subcutaneously using a 30G needle on P4 with either 0.1 × 106 colony forming units (CFU) of live bacterial E. coli/g suspended in 0.1 ml PBS, or 0.1 ml PBS. All pups were removed from the mother at the same time and placed into a clean cage with bedding, weighed and injected individually, and returned to the mother as a group. Elapsed time away from the mother was less than 5 min. All pups from a single litter received the same treatment due to concerns over possible cross-contamination from E. coli. All injections were given between 1530 and 1600 h. To control for possible litter effects, a maximum of two pups/litter were assigned to a single experimental group.
2.2.1.2. BrdU injections and tissue harvest
5-Bromo-2′-deoxyuridine (BrdU, Sigma-Aldrich, St. Louis, MO) was dissolved (10 mg/ml) in sterile 0.01 M PBS. Pups were injected subcutaneously using a 30G needle with BrdU (50 mg/kg ip, volume 5 ml/kg) 3 times: once immediately following the E. coli or PBS vehicle injection on P4, and twice (once at 1000 and once at 1600 h) on P5. Rats were killed on either P6 (for analysis of precursor cell proliferation and microgliogenesis) or P33 (for analysis of neuro/gliogenesis and proliferating cell survival). Rats were deeply anesthetized with sodium pentobarbital and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.01 M PBS. Brains were postfixed for 4 hours in 4% paraformaldehyde in 0.01 M PBS and cryoprotected in 30% sucrose for at least 3 days, then quickly frozen in −30°C isopentane. Brains were sliced through the PFC and hippocampus in a 1:6 series at 40 m in a −20°C cryostat using the atlas of Paxinos and Watson (1998) as a guide. Sections were stored at 4°C in cryoprotectant until immunohistochemistry was performed.
2.2.1.3. Immunohistochemistry
The ionized calcium-binding adaptor molecule (Iba)-1 protein was used to identify microglia, because it is specific to microglia and its expression is constitutive, and binds cell bodies and processes in every state of activation (Imai et al., 1996). Double immunofluorescence was performed for BrdU (to label proliferating cells) and Iba1 (to label microglia) on tissue from P6 and P33 rats. Triple immunofluorescence for BrdU, Neuronal Nucleus (NeuN, to label neurons), and GFAP, to label astrocytes, was performed on tissue from P33 rats. Fluorescence double- or triple-labeling of BrdU and Iba1, or BrdU, GFAP, and NeuN was performed (Figure 1). Free-floating sections were washed 3 times in 0.01 M PBS first and between each subsequent step except as noted. Sections were incubated in 10% normal goat serum (NGS) and 0.3% Triton-X in PBS to block and permeabilize, respectively. DNA was denatured by incubation in 2N HCl at 37°C for 30 min followed by a 0.1 M boric acid wash. Sections were incubated overnight at RT in a combination of primary antibodies: rat monoclonal anti-BrdU (1:100, Accurate Chemical, Westbury, VT) and either rabbit anti-Iba1 (1:1,000, Wako Pure Chemical Industries, Richmond, VA), or rabbit anti-GFAP (1:1,000, DakoCytomation, Carpinteria, CA) and mouse monoclonal anti-NeuN (1:200, Chemicon, Billerica, MA). Sections were then incubated for 2 hr in a combination of fluorescent secondary antibodies: goat anti-rat Alexa 594 and goat anti-rabbit Alexa 488, or goat anti-rat Alexa 594, goat anti-rabbit Alexa 488, and goat anti-mouse Alexa 350 (each 1:200, Molecular Probes/Invitrogen, Carlsbad, CA). Sections were mounted on slides and coverslipped with Vectashield (Vector Laboratories, Burlingame, CA).
Figure 1.
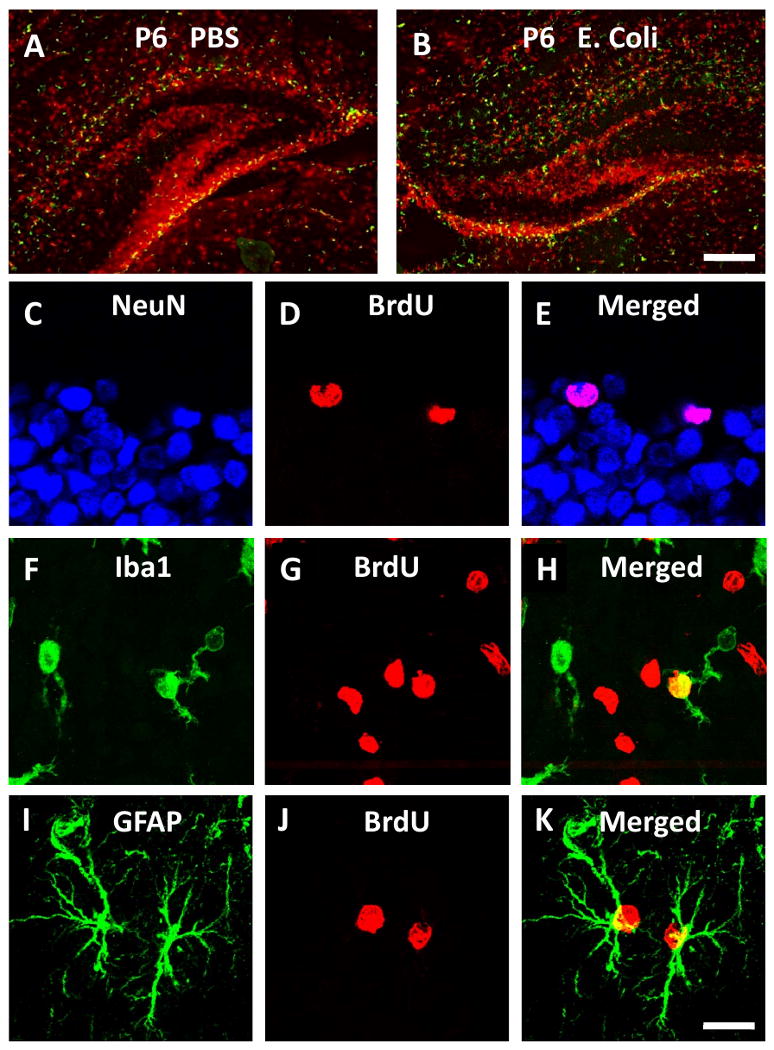
Photomicrographs showing confocal images of BrdU labeling of different cell types. BrdU and Iba1 staining in the P6 hippocampus, where significantly more Iba1 (green) is apparent in E. coli (B) compared to PBS (A); Scale bar = 100 μm. BrdU colocalized with NeuN in the dentate gyrus (C-E), Iba1 in the cortex (F-H) and GFAP in CA3 (I-K) indicating proliferation of neurons, microglia, and astrocytes, respectively; Scale bar = 20 μm.
2.2.1.4. Cell counts
Counts of BrdU labeled cells (proliferating cells, red; Fig 1), Iba1 labeled cells (microglia, green), and BrdU + Iba1 labeled cells (proliferating microglia, yellow) were obtained from tissue from P6 and P33 rats. Microglia were classified as either active (having a large, rounded cell body and thick processes) or inactive (having a small cell body and many fine processes). Counts of BrdU labeled cells (cells that had proliferated on P4-P6, red), BrdU + GFAP labeled cells (astrocytes that had proliferated on P4-P6, yellow), and BrdU + NeuN labeled cells (neurons that had proliferated on P4-P6, fuschia) were obtained from tissue from P33 rats. Counts were obtained from hippocampal subfields CA1, CA3, and dentate gyrus (DG) as well as from pre-frontal cortex (PFC) and parietal cortex (PAR) of tissue from P6 rats. We have reported significant changes in IL-1 expression within the HP and PAR, but not in PFC, of neonatally-infected rats injected with LPS as adults (Bilbo et al., 2005a), and so analyzed those same regions here—i.e., with the prediction that neonatal group differences would be observed in HP and PAR, but less so in PFC. Eight images of each subregion were acquired with an Olympus BX61 Fluorescence microscope (Olympus America, Center Valley, PA) at 40× using Microsuite software (Olympus America). Cells counts and area measures were obtained using Image J software (http://rsb.info.nih.gov/ij/index.html). Cell counts are expressed within each subregion as cells/mm2.
2.2.2. Experiment 2
The effects of an adult immune challenge with LPS in rats treated neonatally with E. coli on precursor cell proliferation, and neuro/gliogenesis.
2.2.2.1. Neonatal manipulations
Neonatal rats were treated with E. coli or PBS as in Experiment 1. To control for possible litter effects, a maximum of two pups/litter were assigned to a single experimental group. Pups were weaned on P21 into sibling pairs and remained undisturbed until adulthood.
2.2.2.2. Adult manipulations
Adult rats (∼70 d) from each neonatal group were injected intraperitoneally (i.p.) with 0.1 ml of 25 g/kg LPS (from E. coli, Serotype 0111:B4, Lot 072K4096, Sigma-Aldrich) or an equal volume of sterile saline. Immediately after the LPS or saline injections, rats were injected i.p. with 100 mg/kg BrdU (10 mg/ml in sterile PBS, injection volume 1 ml/100 g). Rats were killed either 2 hr (for proliferation) or 28 days (for neuro/gliogenesis and proliferating cell survival) after LPS and BrdU injections. Rats were perfused and tissue was harvested and sectioned as in Experiment 1.
2.2.2.3. Immunohistochemistry
For the 2 hr time point, immunohistochemistry was performed for determination of total BrdU (proliferating cells), total Iba1, and BrdU + Iba1 colocalization (proliferating microglia). For the 28 day time point, immunohistochemistry was performed for determination of total BrdU (cells that had proliferated soon after LPS and BrdU injections), total Iba1, total NeuN, and colocalization of BrdU with Iba1 (surviving microglia that had proliferated after LPS and BrdU injections) or NeuN (neurogenesis). For assessment of total BrdU, Iba1, and NeuN labeled cells as well as microglial cell volumes, single immunohistochemistry with 3-3′-diaminobenzidine (DAB) as a chromogen was performed with each antibody (Figure 2). All sections were processed within the same run. Free-floating sections were washed 3 times in 0.01 M PBS first and between each subsequent step except as noted. Sections were incubated in 0.3 % hydrogen peroxide then blocked and permealized as in Experiment 1. For BrdU, DNA was denatured as above. Sections were incubated overnight at RT in either mouse monoclonal anti-BrdU (1:100, Roche, Indianapolis, IN), mouse monoclonal anti-NeuN (1:500, Chemicon), or rabbit anti-Iba1 (1:1,000, Wako) in PBS with 10% NGS and 0.3% Triton-X. Sections were then incubated in biotinylated goat anti-mouse or goat anti-rabbit IgG (1:200, Jackson ImmunoResearch, West Grove, PA) for 2 hr, followed by incubation in avidin biotin complex (ABC kit, Vector Laboratories) for 2 hr. Following three washes in 0.1 M PB, immunoreactivity was visualized with DAB (DAB substrate kit, Vector Laboratories) with nickel ammonium sulphate (Sigma-Aldrich). Sections were mounted on slides, dehydrated and delipidated, and coverslipped with Permount (Sigma-Aldrich). Fluorescence double- labeling of either BrdU and Iba1 or BrdU and NeuN was performed as in Experiment 1.
Figure 2.
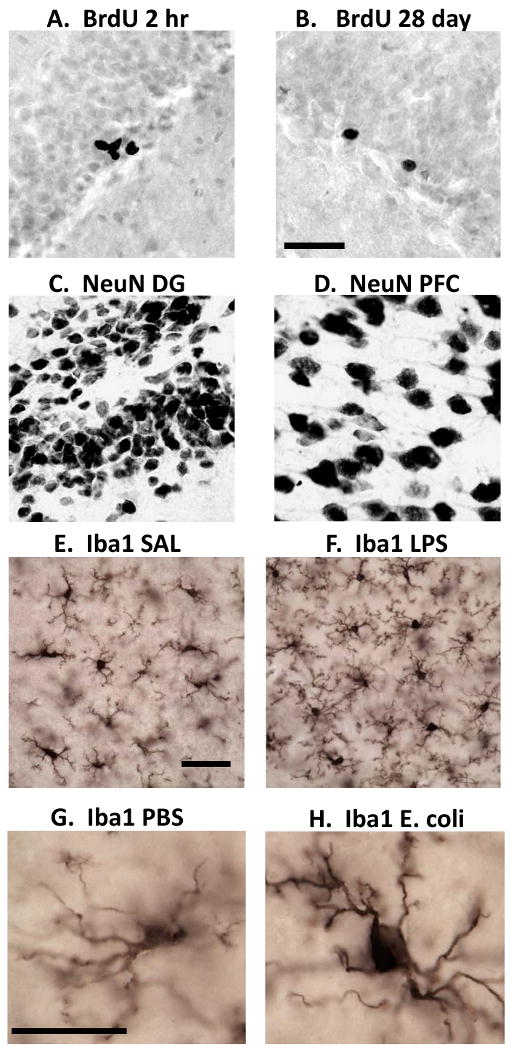
Photomicrographs showing immunohistochemistry for proliferating cells (BrdU), neurons (NeuN), and microglia (Iba1). BrdU labeled cells in the DG 2 hr (A) and 28 days (B) after BrdU injection. NeuN labeled cells in the DG (C) and in the PFC (D); Scale bar for A-D = 50 μm. Iba1 labeled cells in the adult hippocampus of a rat injected with saline (E) versus LPS (F); Scale bar for E-F = 25 μm. Iba1 labeled cell in the adult CA1 of a rat treated neonatally with PBS (G) versus E. coli (H); Scale bar for G-H = 10 μm.
2.2.2.4. Cell counts
Unbiased stereology (Gundersen & Jensen, 1987) was used to estimate total numbers of BrdU, Iba1, and NeuN labeled cells. Cell numbers were estimated using the Cavalieri principal and the optical dissector method, with a 2 mm guard zone. The sampling fractions were 100% for BrdU, 1% for NeuN, and 0.5% for Iba1. Microglial volumes were estimated using five rays of independent isotropic probes within the “Nucleator” function of StereoInvestigator software (MBF Labs, Williston, VT). Stereology for BrdU and NeuN was performed using an Olympus BX61 microscope at 100× (oil) and NewCAST software (VisioPharm, Hørsholm, Denmark). Stereology for Iba1 was performed using a Nikon Eclipse 80i microscope at 100× (oil) and StereoInvestigator software.
Colocalization of BrdU and Iba1, or BrdU and NeuN labeling was assessed in double-labeled sections using a Leica TCS SP2 AOBS confocal microscope at 63× using an oil-immersion objective. At least 50 BrdU positive cells per rat were sampled for identification of colocalization of BrdU with either Iba1 or NeuN. A ratio was computed and then multiplied by total BrdU numbers within each subregion to determine the number of BrdU labeled neurons, microglia, or undifferentiated/unknown cell types (no co-expression with BrdU).
2.3. Statistics
Experiment 1. Data were analyzed with one-way ANOVA with neonatal treatment (PBS, E. coli) as the between-groups variable. Experiment 2. BrdU, colocalization, and NeuN data were analyzed with two-way ANOVA with neonatal treatment (PBS, E. coli) and adult treatment (saline, LPS) as between-groups variables. Iba1 data (counts and microglial volumes) data were analyzed with three-way ANOVA with time (2 hr, 28 days), neonatal treatment (PBS, E. coli) and adult treatment (saline, LPS) as between-groups variables. When a significant interaction was obtained, Fisher's LSD was used for post-hoc analysis. For all analyses was set at 0.05. All statistics were performed using StatView for Windows (SAS Institute, Inc., Cary, NC).
3. Results
3.1. Experiment 1
The effects of neonatal E. coli infection on precursor cell proliferation and neuro/gliogenesis.
3.1.1. Proliferation on P4-5
In the hippocampus, E. coli infection on P4 increased microglial proliferation and decreased proliferation of non-microglial cells in the CA subfields observed on P6 (Figure 3). In CA1 (Figure 3A), ANOVA revealed that E. coli treatment produced a near-significant increase in total microglia, F(1,17)= 3.54, p= .07 and produced significant increases in active microglia, F(1,17)= 6.64, p < .01 and proliferating microglia, F(1,17) = 8.78, p < .01 while producing a decrease in proliferating non-microglial cells (BrdU-only), F(1,17) = 5.85, p < .05. In CA3 (Figure 3B), E. coli treatment also produced increases in total microglia F(1,17) = 9.73, p < .01, active microglia, F(1,17) = 6.64, p < .01, and proliferating microglia F(1,17) = 6.56, p < .05, as well as decreases in proliferating non-microglial cells, F(1,17) = 4.88, p < .05. There were no significant effects of neonatal E. coli on microglia or proliferation of microglial or non-microglial cells in the dentate gyrus (Figure 3C), although trends were in a similar direction. In the cortex, the effects of E. coli infection on P4 on proliferation were more robust in PAR than in PFC, as predicted (Figure 4). In the PFC (Figure 4A), there was a trend for decreased proliferation of non-microglial cells, F(1,15) = 3.80, p = .07. In PAR, E. coli infection produced significant increases in total microglia, F(1,15) = 4.89, p < .05 and proliferating microglia, F(1,15) = 7.21, p < .05 and decreases in proliferating non-microglial cells, F(1,15) = 7.27, p < .05. There was no effect of neonatal E. coli treatment on active microglia in the cortex (data not shown).
Figure 3.
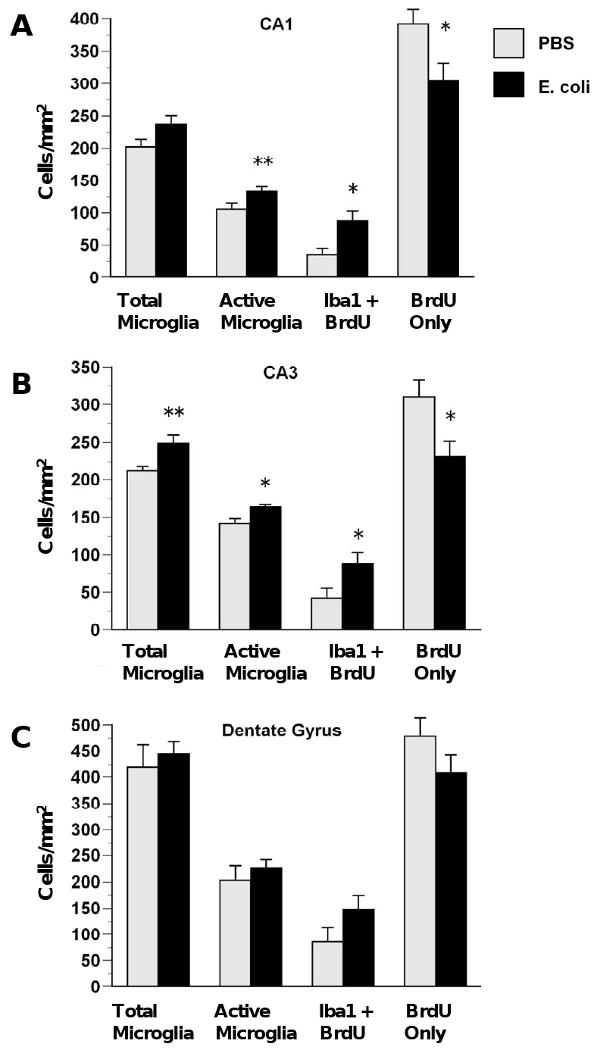
E. coli infection on P4 increases microgliogenesis and decreases proliferation of non-microglial cells in the hippocampus. Rats received injections of either E. coli or PBS vehicle on P4 and BrdU on P4 and P5 and were killed on P6. E. coli infection produced increases in total microglia, activated microglia, and proliferating microglia (BrdU + Iba1) and decreases in proliferating non-microglial cells (BrdU only) in CA1 (A) and CA2 (B) but not DG (C). Values are means ± SEMs of 9 to 11 rats/group. * Significantly different from PBS, p < .05. ** Significantly different from PBS, p < .01.
Figure 4.
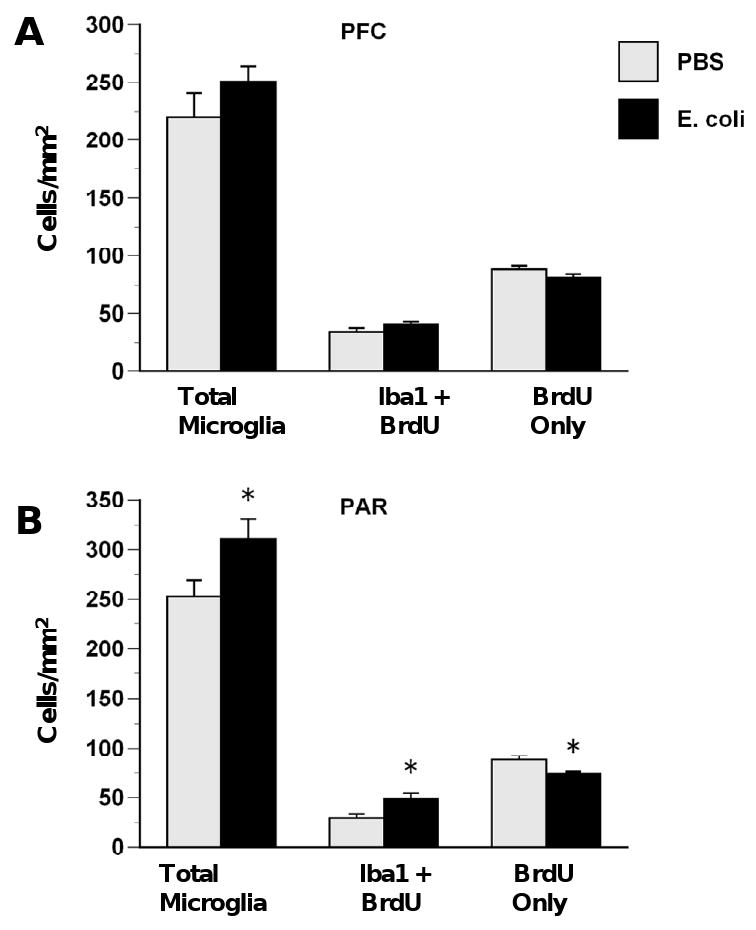
E. coli infection on P4 increases microgliogenesis and decreases proliferation of non-microglial cells in the cortex. Rats received injections of either E. coli or PBS vehicle on P4 and BrdU on P4 and P5 and were killed on P6. No group differences were observed in PFC (A). E. coli infection produced increases in total microglia and proliferating microglia (BrdU + Iba1) and decreases in proliferating non-microglial cells (BrdU only) in PAR (B). Values are means ± SEMs of 9 to 11 rats/group. * Significantly different from PBS, p < .05.
3.1.2. Survival and phenotypes of cells on P33 that proliferated during P4-6
Throughout the hippocampus, neonatal E. coli reduced both neurogenesis and proliferation or survival of undifferentiated/unknown cell types, with no enduring effects on gliogenesis, 28 days after E. coli injections (Figure 5). In CA1 (Figure 5A), neonatal E. coli infection produced significant decreases in neurogenesis (NeuN + BrdU), F(1,16) = 9.06, p < .01 and BrdU labeled undifferentiated/unknown cells (BrdU-only), F(1,16) = 11.19, p < .01. In CA3 (Figure 5B), neonatal E. coli also produced significant decreases in neurogenesis, F(1,16) = 5.89, p < .05 and BrdU-only cells, F(1,16) = 23.72, p < .001. In the DG (Figure 5C), neonatal E. coli also produced significant decreases in neurogenesis, F(1,16) = 4.87, p < .05 and BrdU-only cells, F(1,16) = 12.85, p < .01. There were no significant effects of neonatal E. coli on microglial or astrocytic proliferation in the hippocampus.
Figure 5.
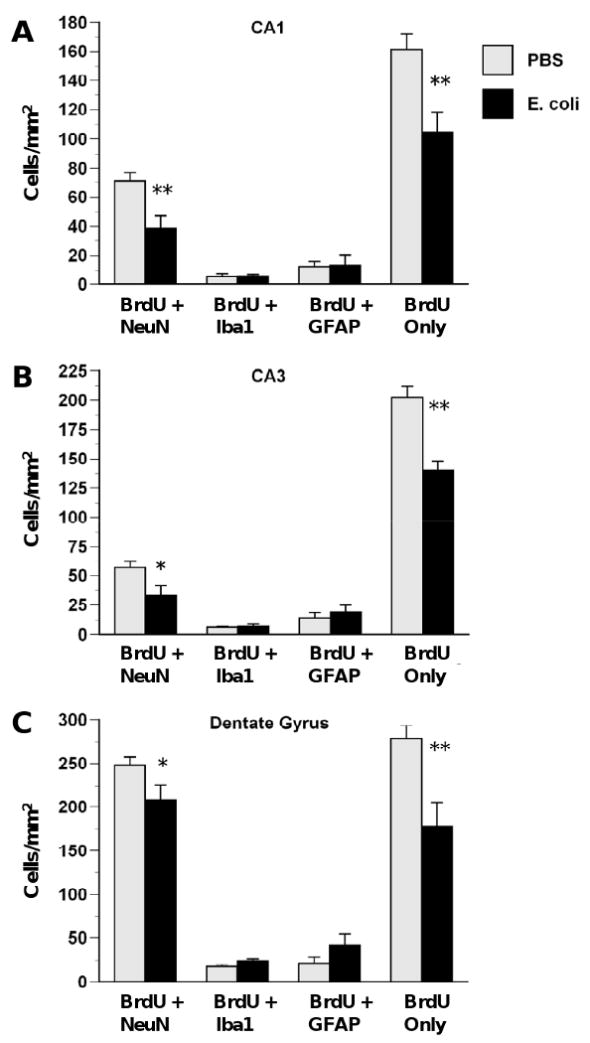
E. coli infection on P4 decreases the survival of undifferentiated cells in CA1 (A), CA3 (B) and DG (C). Rats received injections of either E. coli or PBS vehicle on P4 and BrdU on P4 and P5 and were killed on P33. E. coli infection produced decreases in neurogenesis (BrdU + NeuN) and proliferation of undifferentiated cells (BrdU only) but produced no changes in proliferating microglia (BrdU + Iba1) or astrocytes (BrdU + GFAP) at this time point. Values are means ± SEMs of 7 to 8 rats/group. * Significantly different from PBS, p < .05. ** Significantly different from PBS, p < .01.
In the cortex, the effect of neonatal E. coli 28 days after E. coli injection (Figure 6) was primarily a reduction of proliferation or survival of undifferentiated/unknown cell types in the PFC. In the PFC, neonatal E. coli produced significant decreases in BrdU-only cells (Figure 6A), F(1,16) = 5.23, p < .05, and neonatal E. coli produced a trend for decreased neurogenesis, F(1,16) = 3.70, p = .07. In PAR (Figure 6B), neonatal E. coli produced a trend for increased BrdU-only cells, F(1,16) = 3.71, p = .07. There were no effects of neonatal E. coli on proliferation of astrocytes (BrdU + GFAP) in either cortical subregion. No Iba1/BrdU double-labeled cells were observed in either cortical region.
Figure 6.
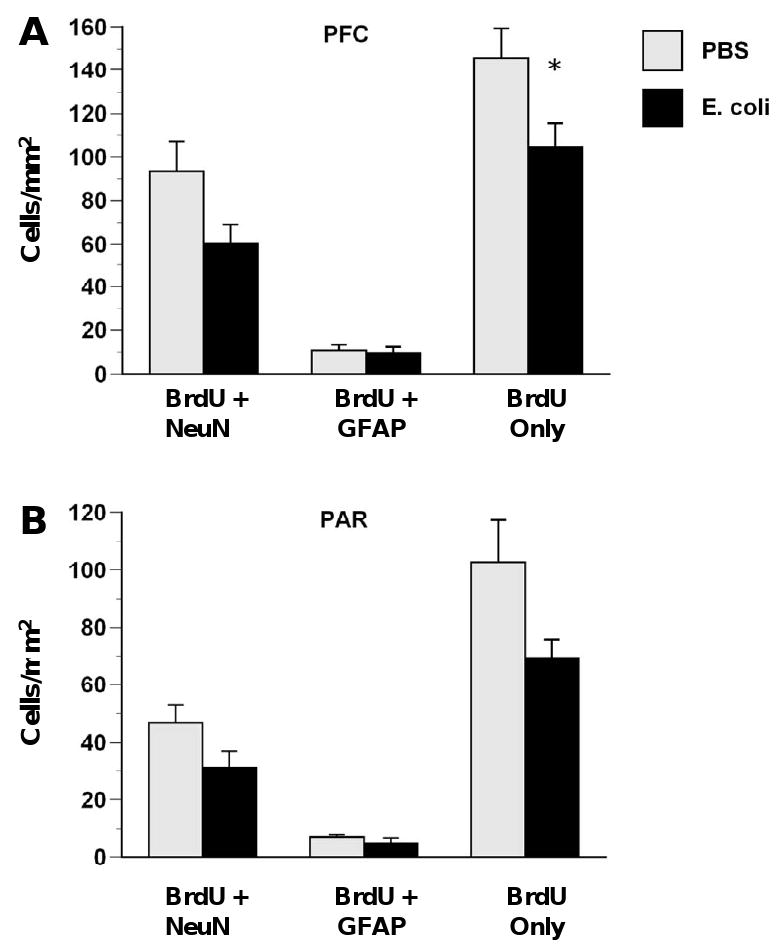
E. coli infection on P4 decreases the survival of undifferentiated cells in the cortex. Rats received injections of either E. coli or PBS vehicle on P4 and BrdU on P4 and P5 and were killed on P33. E. coli infection produced decreases in proliferation of undifferentiated cells (BrdU only) in the PFC (A) but no significant differences in PAR (B). Values are means ± SEMs of 7 to 8 rats/group. * Significantly different from PBS, p < .05
3.2. Experiment 2
The effects of an adult immune challenge with LPS in rats treated neonatally with E. coli on precursor cell proliferation, and neuro/gliogenesis.
3.2.1. Cell proliferation and neuro/microgliogenesis
The effects of adult LPS in rats that had been treated neonatally with E. coli were restricted to neuro/microgliogenesis in the DG. At 2 hr after LPS and BrdU there were no effects of neonatal or adult treatment on proliferation of undifferentiated cells (BrdU-only) or on microgliogenesis (BrdU + Iba1) in any brain region (data not shown). At 28 days after LPS and BrdU there were no effects of neonatal or adult treatment on proliferation of undifferentiated cells (BrdU-only) or on microgliogenesis (BrdU + Iba1) in cortex, or in the CA regions (not shown). However, there were significant neonatal treatment-dependent effects of adult LPS on DG neurogenesis, F(1,26) = 10.85, p < .01, and microgliogenesis, F(1,26) = 7.55, p < .05, at 28 days after LPS and BrdU (Figure 7). Post-hoc tests indicated that adult LPS produced increases in both neurogenesis and microgliogenesis in rats that had been treated neonatally with PBS, and a decrease in neurogenesis in rats that had been treated neonatally with E. coli, all p < .05.
Figure 7.
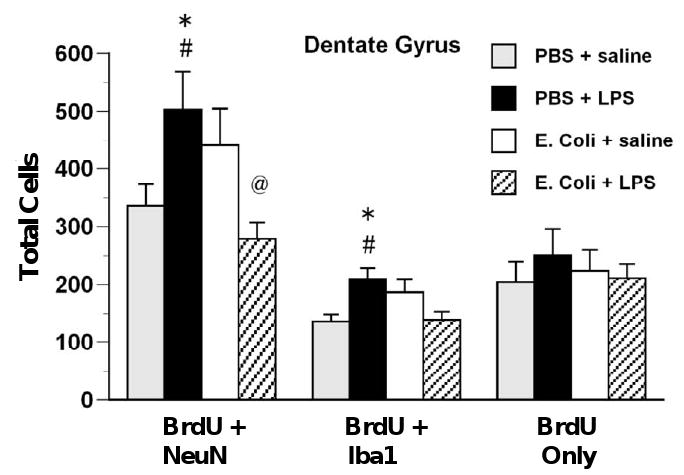
Immune challenge during adulthood decreases DG neurogenesis in rats treated neonatally with E. coli but increases neurogenesis and microgliogenesis in control rats. Rats received injections of either E. coli or PBS vehicle on P4 and BrdU on P4 and P5 and of either LPS or saline during adulthood. BrdU was injected immediately after LPS or saline and rats were killed 28 days later. In the DG, LPS injection during adulthood increased neurogenesis (BrdU + NeuN) in rats treated neonatally with PBS but decreased it in rats treated neonatally with E. coli. LPS during adulthood increased microgliogenesis (BrdU + Iba1) in rats treated neonatally with PBS. Values are means ± SEMs of 7 to 8 rats/group. * Significantly different from PBS + saline, p < .05. # Significantly different from E. coli + LPS, p < .05. @ Significantly different from E. coli + saline, p < .05
3.2.2. Stereological estimates of microglial cell volumes and numbers
Neonatal E. coli infection produced increased microglial volumes in CA1 and increased microglial numbers in DG of adult rats. Three-way ANOVA revealed a significant main effect of neonatal treatment on microglial cell volumes in CA1, F(1,55) = 14.68, p < .001 (Figure 8). Post-hoc tests indicated that rats that had been treated neonatally with E. coli and during adulthood with saline had significantly greater microglial cell volumes than rats treated neonatally with PBS and during adulthood with saline at both 2 hr and 28 days, p < .05. In addition, rats treated neonatally with E. coli and during adulthood with LPS had greater cell volumes than rats treated neonatally with PBS and during adulthood with LPS at 28 days, all p < .05. There were no significant effects of neonatal or adult treatment on microglial cell volumes in PFC, CA3 or DG (not shown). There were significant main effects of time on microglial cell volume in all subregions: CA1, F(1,55) = 89.76, p < .001; CA3, F(1,55) = 89.76, p < .001; DG, F(1,55) = 74.33, p < .001; and PFC, F(1,54) = 177.42, p < .001. In every region, microglial cell volumes were greater at 28 days than at 2 hr. The cause of this increase is unknown, but may be an effect of BrdU treatment itself.
Figure 8.
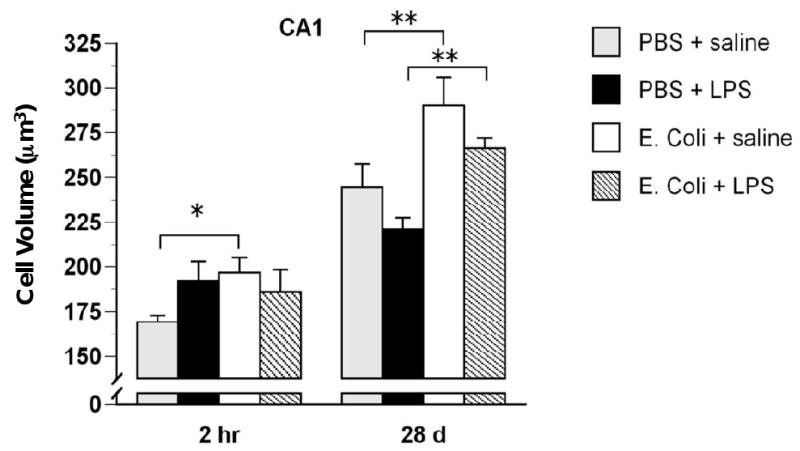
Microglial cell volumes are increased in the CA1 of rats treated neonatally with E. coli. Rats received injections of either E. coli or PBS vehicle on P4 and of either LPS or saline during adulthood and were killed 28 days later. Microglial cell volumes were increased in rats neonatally treated with E. coli. Values are means ± SEMs of 7 to 8 rats/group. * p < .05. ** p < .01.
For microglial cell counts (Table 1), three-way ANOVA revealed that in the DG there was a significant main effect of neonatal treatment F(1,55) = 5.38, p < .05; rats that had been treated neonatally with E. coli had more microglia in DG. LPS treatment also produced increases in microglia in the PFC, as revealed by a main effect of adult treatment, F(1,53) = 4.03, p < .05.
Table 1.
Stereological estimates of microglia and neuron numbers in CA1, CA3, dentate gyrus (DG) and the PFC. Rats received E. coli or PBS vehicle on P4 and LPS or saline vehicle during adulthood and were killed either 2 hr or 28 days after adult treatment.
| Neonatal PBS | Neonatal E. coli | |||
|---|---|---|---|---|
| Adult saline | Adult LPS | Adult saline | Adult LPS | |
| Microglia, 2 hr | ||||
| CA1 | 1.9 ± 0.1 | 1.8 ± 0.1 | 1.8 ± 0.1 | 1.8 ± 0.1 |
| CA3 | 2.3 ± 0.1 | 2.2 ± 0.1 | 2.4 ± 0.1 | 2.3 ± 0.1 |
| DG* | 2.8 ± 0.1 | 2.8 ± 0.1 | 2.9 ± 0.1 | 3.1 ± 0.1 |
| PFC # | 5.2 ± 0.4 | 6.3 ± 0.4 | 5.9 ± 0.4 | 6.3 ± 0.5 |
| Microglia, 28 days | ||||
| CA1 | 1.9 ± 0.1 | 1.9 ± 0.1 | 2.0 ± 0.1 | 1.9 ± 0.1 |
| CA3 | 2.7 ± 0.1 | 2.5 ± 0.1 | 2.6 ± 0.1 | 2.7 ± 0.2 |
| DG * | 2.8 ± 0.1 | 2.8 ± 0.1 | 2.9 ± 0.1 | 2.9 ± 0.1 |
| PFC # | 6.8 ± 0.4 | 7.2 ± 0.3 | 7.3 ± 0.1 | 7.4 ± 0.3 |
| Neurons, 28 days | ||||
| CA1 | 19.6 ± 1.0 | 16.2 ± 1.2 | 16.6 ± 1.7 | 19.5 ± 1.7 |
| CA3 | 14.6 ± 1.2 | 13.0 ± 1.7 | 13.6 ± 1.1 | 13.8 ± 1.3 |
| DG | 50.6 ± 5.1 | 40.1 ± 3.5 | 44.5 ± 4.9 | 48.2 ± 4.3 |
| PFC *# | 26.9 ± 2.2 | 24.0 ± 1.7 | 23.2 ± 1.4 | 18.4 ± 1.2 |
Values are means × 10-4 ± SEMs of 7 or 8 rats per group.
Main effect of neonatal treatment, p < .05.
Main effect of adult treatment, p < .05.
3.2.3. Stereological estimates of neuronal numbers 28 days after LPS treatment
In the CA1, there was a significant neonatal treatment by adult treatment interaction F(1,27) = 4.45, p < .05, but post-hoc tests indicated no significant differences between treatment groups (Table 1). In the PFC, neonatal E. coli infection produced decreases in total numbers of neurons, as revealed by a main effect of neonatal treatment, F(1,27) = 7.43, p < .01. In addition, LPS treatment during adulthood also produced decreases in total numbers of neurons in the PFC, as revealed by a significant main effect of adult treatment, F(1,27) = 5.23, p < .05.
4. Discussion
We have hypothesized that the basis for early-life infection-induced vulnerability to altered cytokine expression and cognitive deficits in adulthood may be due to long-term changes in glial cell function and/or influences on subsequent neural development. E. coli infection on P4 markedly increased microglial proliferation in the CA regions of the hippocampus and PAR of newborn pups, compared to a PBS injection (Fig 3-4). The total number of microglia, and specifically microglia with an “active” morphology (amoeboid, with thick processes), were also increased as a consequence of infection. There was a concomitant decrease in non-microglial newborn cells (BrdU+ only) in the early-infected rats, in the same regions. In order to assess cell survival, and to characterize which cell types were decreased by the neonatal infection, we counted glial and neuronal markers 28 d post-BrdU (P33). There was a profound reduction of BrdU+ neurons in every region of the hippocampus, as well as a reduction in newborn uncharacterized cells (BrdU+ only) in the hippocampus and PFC, in the early-infected rats compared to controls (Fig 5-6). Notably, the marked increase in microglial cell number was gone by P33, and GFAP also did not differ by neonatal group in any region. Thus, systemic bacterial infection transiently increased microglial proliferation, but decreased overall proliferation, within the hippocampus and cortex of newborn pups, and this decrease was at least in part neuronal. The phenotype of the remaining uncharacterized cells remains unknown, but could be due to decreases in oligodendrocytes and/or NG2 cells, as there is a significant literature on the influence of perinatal infection on white matter damage in both humans and animals (Dammann et al., 2002; Pang et al., 2003).
In order to assess whether neonatal infection–induced changes in neuronal and glial cell genesis within the brain endure into adulthood, total cell numbers were assessed in adult animals injected with saline. Despite large decreases in hippocampal neurogenesis/survival in early-infected rats at P33, there were no group differences in overall neuronal numbers in this brain region in adulthood (Table 1). In contrast, there were decreased total numbers of neurons within the PFC of E. coli rats compared to controls, which interestingly was the only region we assessed that did not show differences earlier in life. Thus, the developing brain is clearly capable of normalizing acute infection-induced changes in cell number, likely via compensatory mechanisms, as well as manifesting long-term changes that were not previously apparent.
In order to assess the response to a subsequent inflammatory challenge in adulthood, rats treated neonatally with E. coli or PBS were injected with either saline or LPS along with BrdU, and brains were collected 2 h (proliferation) or 28 d (survival) later. Fig 7 illustrates that basal neurogenesis (adult saline-injected) within the hippocampus did not differ by neonatal group. This is in contrast to prenatal LPS injection of rat dams, which significantly reduces basal neurogenesis in the adult offspring hippocampus, independent of any subsequent challenge (Cui et al., 2009), as does neonatal viral infection with lymphocytic choriomeningitis (Sharma et al., 2002). However, in response to adult LPS, there was a significant decrease in DG neurogenesis (BrdU+ NeuN) in neonatally-infected rats, whereas control rats exhibited an increase in neurogenesis in the same region (Fig 7B). It appears the infection produced a latent or hidden change within the brain, which was only “unmasked” by a second immune challenge. An emerging literature implicates inflammation as an important factor in neurogenesis within the CNS (Das and Basu, 2008; Jakubs et al., 2008; Seguin et al., 2009); in general, mild inflammation appears to stimulate neurogenesis, whereas more severe or prolonged inflammation is inhibitory (Whitney et al., 2009). Thus, these data well reflect our previous behavioral findings, in which memory disruption in early-infected rats is only observed in the face of an adult immune challenge (Bilbo et al., 2006), and are consistent with the idea that inflammation in the adult brain is exaggerated and/or prolonged as a result of early-life infection. It appears that the neonatally-infected animals have responded to the low dose LPS as if it is a much higher dose, which would be predicted to suppress neurogenesis in both groups.
Along with neurogenesis, a primary goal of this study was to characterize long-term changes in microglial cell number, proliferation and survival in adulthood, changes which may underlie differences in neurogenesis. There were significantly more microglia overall within the DG as a consequence of early-life infection compared to controls (Table 1). This difference occurred only in the DG, a region that did not exhibit differences in proliferation early in life (P6). Similarly, there were no differences in CA1/CA3, despite large initial increases in proliferation in these regions in early-infected rats. In response to LPS, there were surprisingly more newly born microglia (BrdU+ Iba1) in the DG of control rats compared to early-infected rats. This increase co-occurs with the increase in neurogenesis in the DG of controls, and raises the intriguing possibility that microgliogenesis may contribute to neurogenesis in these animals. This possibility remains speculation at this point, but there is increasing evidence that the immune system, and microglia in particular, are important for normal, “homeostatic” neurogenesis within the healthy brain (Ziv et al., 2006). For instance, microglia promote neurogenesis in cultured neural progenitor cells (Walton et al., 2006), and voluntary exercise markedly increases both microglial and neural genesis within the brains of mice (Ehninger and Kempermann, 2003).
Microglial activation during immune activation has been associated with both enhanced and suppressed neurogenesis. For instance, chronic LPS-induced inflammation severely impairs neurogenesis, and preventing microglial activation with minocycline, a specific inhibitor, rescues the impairment (Ekdahl et al., 2003). In marked contrast, minocycline treatment following stroke significantly impairs neurogenesis (Kim et al., 2009). Several glial-derived cytokines and chemokines are pro-neurogenic; interferon- stimulates neuronal differentiation (Wong et al., 2004), and chemokines such as monocyte-chemoattractant-protein (MCP)-1 influence both leukocyte and neural progenitor cell migration, a key step in inflammation-induced neurogenesis (Widera et al., 2004). In contrast, IL-1 and IL-6 each promote proliferation but decrease the survival of newborn neurons (Vallieres et al., 2002; Wang et al., 2007). We assessed the survival of mature neurons only in this study, and so the initial proliferative responses to both the E. coli infection and adult LPS challenges will need to be characterized in future studies.
Although the mechanisms remain largely unknown, the “glial cell priming” hypothesis posits that these cells have the capacity to become chronically sensitized by an inflammatory event within the brain (Perry et al., 2003). We assessed whether glial priming may be a likely factor in the current study by measuring the volume of each counted microglial cell within our stereological analysis. The morphology of primed glial cells is similar to that of “activated” cells (e.g. amoeboid, phagocytic), but primed glial cells do not chronically produce cytokines and other pro-inflammatory mediators typical of cells in an activated state. There was a striking increase in cell volume within the CA1 region of adult rats infected as neonates (Figs 2 and 8), the same region in which a marked increase in newborn glia was observed at P6. These data are consistent with the hypothesis that an inflammatory environment early in life may prime the surviving cells long-term, such that they over-respond to a second challenge, which we have demonstrated at the mRNA level in previous studies (Bilbo et al., 2005a; Bilbo et al., 2007; Bilbo and Schwarz, in press). It is unclear why this increase was only observed in CA1, although a similar pattern was apparent in all hippocampal regions, as well as in the PFC (not shown). It is also unclear why cell volumes increased so markedly from 2 h to 28 d in every treatment group. BrdU may have activated microglia in all animals, a possibility that deserves further investigation. Importantly, however, the difference in volume in CA1 (i.e., larger in E. coli rats) remains consistent across time points. These data are of course a preliminary step -future experiments will be needed to assess functional markers (e.g., CD68/ED1, MHCII), as well as secreted factors (e.g., cytokine/chemokine production), and finally putative molecular candidates underlying “priming”, and these are currently underway.
In summary, we have provided evidence that systemic infection with E. coli early in life has significant, enduring consequences for brain development. These changes include marked alterations in glia, both in number and in morphology, as well as influences on neurogenesis and likely other cells (e.g., oligodendrocytes), in several brain regions important for cognition. Altogether, these data support the hypothesis that glia underlie, at least in part, long-term “perinatal programming” of later-life vulnerability to inflammatory challenge. Early-life infection appears to sensitize the “inflammatory homeostat”, with significant long-term implications for neurogenesis within the brain, cognitive function, and likely other outcomes.
Acknowledgments
The authors thank Andrea Eads, Jessica Bolton, and Brandon Marsh for technical assistance. Supported in part by a NARSAD Young Investigator Award to STB, and R01MH083698 to SDB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors maybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002;23:285–290. doi: 10.1016/s1471-4906(02)02223-8. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Bilbo SD, Barrientos RM, Eads AS, Northcutt A, Watkins LR, Rudy JW, Maier SF. Early-life infection leads to altered BDNF and IL-1beta mRNA expression in rat hippocampus following learning in adulthood. Brain Behav Immun. 2008;22:451–455. doi: 10.1016/j.bbi.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. 2005a;25:8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005b;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Newsum NJ, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Differential effects of neonatal handling on early life infection-induced alterations in cognition in adulthood. Brain Behav Immun. 2007;21:332–342. doi: 10.1016/j.bbi.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Rudy JW, Watkins LR, Maier SF. A behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res. 2006;169:39–47. doi: 10.1016/j.bbr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Frontiers in Behavioral Neuroscience. doi: 10.3389/neuro.08.014.2009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Cui K, Ashdown H, Luheshi GN, Boksa P. Effects of prenatal immune activation on hippocampal neurogenesis in the rat. Schizophr Res. 2009 doi: 10.1016/j.schres.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Dammann O, Kuban KC, Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev. 2002;8:46–50. doi: 10.1002/mrdd.10005. [DOI] [PubMed] [Google Scholar]
- Das S, Basu A. Inflammation: a new candidate in modulating adult neurogenesis. J Neurosci Res. 2008;86:1199–1208. doi: 10.1002/jnr.21585. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Kempermann G. Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex. 2003;13:845–851. doi: 10.1093/cercor/13.8.845. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Jakubs K, Bonde S, Iosif RE, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Inflammation regulates functional integration of neurons born in adult brain. J Neurosci. 2008;28:12477–12488. doi: 10.1523/JNEUROSCI.3240-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Kim MJ, Park JM, Lee SH, Kim YJ, Ryu S, Kim YH, Yoon BW. Reduced neurogenesis after suppressed inflammation by minocycline in transient cerebral ischemia in rat. J Neurol Sci. 2009;279:70–75. doi: 10.1016/j.jns.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Pang Y, Cai Z, Rhodes PG. Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide. Brain Res Dev Brain Res. 2003;140:205–214. doi: 10.1016/s0165-3806(02)00606-5. [DOI] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Seguin JA, Brennan J, Mangano E, Hayley S. Proinflammatory cytokines differentially influence adult hippocampal cell proliferation depending upon the route and chronicity of administration. Neuropsychiatr Dis Treat. 2009;5:5–14. [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Valadi N, Miller AH, Pearce BD. Neonatal viral infection decreases neuronal progenitors and impairs adult neurogenesis in the hippocampus. Neurobiol Dis. 2002;11:246–256. doi: 10.1006/nbdi.2002.0531. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallieres L, Campbell IL, Gage FH, Sawchenko PE. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 2002;22:486–492. doi: 10.1523/JNEUROSCI.22-02-00486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, Marshall GP, 2nd, Scheffler B, Steindler DA. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54:815–825. doi: 10.1002/glia.20419. [DOI] [PubMed] [Google Scholar]
- Wang X, Fu S, Wang Y, Yu P, Hu J, Gu W, Xu XM, Lu P. Interleukin-1beta mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol Cell Neurosci. 2007;36:343–354. doi: 10.1016/j.mcn.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widera D, Holtkamp W, Entschladen F, Niggemann B, Zanker K, Kaltschmidt B, Kaltschmidt C. MCP-1 induces migration of adult neural stem cells. Eur J Cell Biol. 2004;83:381–387. doi: 10.1078/0171-9335-00403. [DOI] [PubMed] [Google Scholar]
- Wong G, Goldshmit Y, Turnley AM. Interferon-gamma but not TNF alpha promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Exp Neurol. 2004;187:171–177. doi: 10.1016/j.expneurol.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Wu CH, Wen CY, Shieh JY, Ling EA. A quantitative and morphometric study of the transformation of amoeboid microglia into ramified microglia in the developing corpus callosum in rats. J Anat. 1992;181(Pt 3):423–430. [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]