

Abstract
Casiopeínas are a series of mixed chelate copper complexes that are being evaluated as anticancer agents. Their effects in the cell include oxidative damage and mitochondrial dysfunction, yet the molecular mechanisms leading to such effects remain unclear. We tested whether [Cu(4,7-dimethyl-phenanthroline)(glycinate)]NO3 (Casiopeína IIgly or Cas IIgly) could alter cellular glutathione (GSH) levels by redox cycling with GSH to generate ROS and cellular oxidative stress. Cas IIgly induced a dramatic drop in intracellular levels of GSH in human lung cancer H157 and A549 cells, and is able to use GSH as source of electrons to catalyze the Fenton reaction. In both cell lines, the toxicity of Cas IIgly (2.5–5 μM) was potentiated by the GSH synthesis inhibitor L-buthionine sulfoximine (BSO) and diminished by the catalytic antioxidant manganese(III) meso-tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin (MnTDE-1,3-IP5+), thus supporting an important role for oxidative stress. Cas IIgly also caused an over-production of reactive oxygen species (ROS) in the mitochondria and a depolarization of the mitochondrial membrane. Moreover, Cas IIgly produced mitochondrial DNA damage that resulted in an imbalance of the expression of the apoproteins of the mitochondrial respiratory chain, which also can contribute to increased ROS production. These results suggest that Cas IIgly initiates multiple possible sources of ROS overproduction leading to mitochondrial dysfunction and cell death.
Keywords: copper-phenanthroline, glutathione, catalytic antioxidant, mtDNA, H157, A549
1. Introduction
It is generally accepted that oxidative damage plays a role in carcinogenesis. On the other hand, growing data suggest that oxidative stress is an important mechanism of action for a number of anticancer drugs, in particular alkylating agents. There is mounting evidence that cancer cells exist in an elevated oxidative state, and therefore are more vulnerable to increased levels of reactive oxygen species (ROS) (Nicco et al., 2005; Leitner et al., 2007; Armstrong, 2006). The prooxidant effects of cisplatin, for instance, include inhibition of thioredoxin reductase and increased ROS production in the mitochondria (Simons et al., 2007; Schweyer et al., 2004; Huang et al., 2003; Müeller et al., 2003; Henkels et al., 1999). One possible cause of cisplatin-induced over-production of ROS is by damaging mitochondrial DNA (mtDNA), which translates into an imbalance of the expression in the mitochondrial respiratory chain apoproteins and, in turn, lead to an increased “leakage” of superoxide (O2•−) (Yang et al., 2006; Kachadourian et al., 2007).
Cancer cells are able to adapt to increased ROS levels by elevating their intracellular antioxidants such as glutathione (GSH) and heme oxygenase-1 (HO-1) (Allen and Balin, 2003; Wang et al., 2006; Kim et al., 2008; Wang et al., 2008). This adaptive response is controlled by Nrf2, a transcription factor that mediates the induction of genes that contain an antioxidant response element (ARE). Along with catalase and the thioredoxin-assisted peroxiredoxins, the glutathione/glutathione peroxidase (GSH/GPx) system plays a key role in controlling hydrogen peroxide (H2O2) levels. L-Buthionine sulfoximine (BSO) is an irreversible inhibitor of GSH synthesis that depletes intracellular levels of GSH. BSO has been studied in combination with ionizing radiation and is currently in phase II clinical trials in combination with melphalan (Batist et al., 1996; Bailey et al., 1997).
Copper is an essential trace metal in living systems. However, depending on its concentration and the nature of its ligands, it can be very toxic. Like iron, copper catalyzes the Fenton reaction, generating the highly reactive hydroxyl radical (HO•) (Valko et al., 2007). The copper-phenanthroline complex Casiopeína IIgly (Cas IIgly, Fig. 1) is currently under investigation as a potential new anti-cancer drug. Cas IIgly is 10- to 100-fold more active than cisplatin in culture cell models and has good therapeutic indexes in animal tumor models (Leal-García et al., 2007). Cisplatin is a DNA alkylating agent and Cas IIgly a DNA intercalating agent, yet both exert prooxidant effects in the cell and have been associated with mitochondrial dysfunction and caspase-dependent and -independent programmed cell death (apoptosis) (Marín-Hernández et al., 2003; Trejo-Solís et al., 2005; Rivero-Müller et al., 2007; Alemón-Medina et al., 2008).
Figure 1.
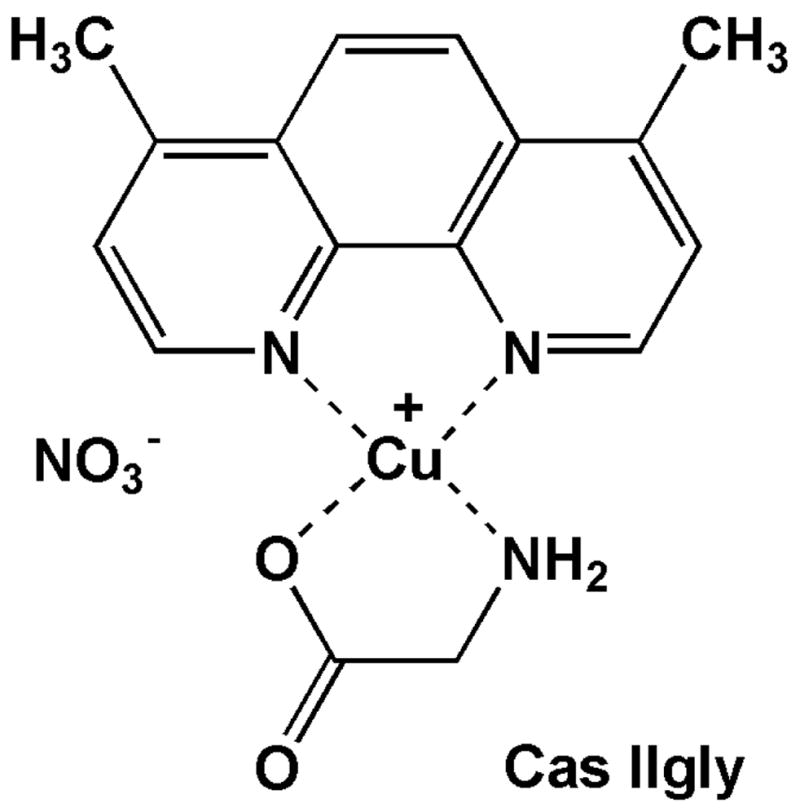
Structure of Casiopeína IIgly.
This study further explores the mechanisms of Cas IIgly-induced oxidative stress and mitochondrial dysfunction in two human lung cancer cell lines, i.e. H157 and A549. As shown in this study, these two cell lines showed a differential sensitivity to Cas IIgly treatment, reason why they were used for more in-dept studies. Cas IIgly induced a dramatic decrease in intracellular levels of GSH, most of which was oxidized to GSSG. We demonstrate that Cas IIgly catalyzes the Fenton reaction and can use GSH as source of electrons. Cas IIgly also caused mitochondrial dysfunction and the over-production of ROS, possibly due to mtDNA damage and an imbalance on the expression of the apoproteins of the mitochondrial respiratory chain.
2. Materials and methods
2.1. Chemicals and reagents
L-Buthionine sulfoximine (BSO), 5,5-dimethyl-1-pyrroline N-oxide (DMPO), L-glutathione, glutathione reductase from bakers yeast, hydrogen peroxide solution (H2O2, 30%), pyruvate (sodium salt), phosphoric acid, meta-phosphoric acid, sodium phosphate (monobasic), Triton X-100, EDTA, NADH, NADPH, K2HPO4, KH2PO4, HEPES, D-mannitol, DMSO and DMF were from Sigma-Aldrich (St. Louis, MO). Tris-HCl, NaCl, 2-mercaptoethanol, perchloric acid and methanol were from Fisher (Pittsburgh, PA). SDS was from Bio-Rad Laboratories (Hercules, CA). MitoSOX was from Molecular Probes (Eugene, OR). Phosphate-Buffered Saline (PBS) was from Cellgro (Herndon, VA). Protease inhibitor cocktail tablets supplemented with EDTA were from Roche Diagnostics (Indianapolis, IN). Manganese(III) meso-tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin (MnTDE-1,3-IP5+) was prepared as previously described in US patent #6,544,975B1 and was a kind gift from Aeolus Pharmaceuticals (Laguna Nigel, CA).
2.2. Synthesis of Cas IIgly
[Cu(4,7-dimethylphenanthroline)(glycinate)]NO3 (Casiopeína IIgly or Cas IIgly) was synthesized as previously described in US patent #5,576,326. Briefly, equimolar solution of copper(II) nitrate and the suitable substituted diimine were mixed together followed by addition of equimolar amount of the corresponding N–O donor (gly) previously deprotonated. Products were precipitated by partial evaporation of the solvent, recrystallized from water/ethanol at least two times, characterized by infrared spectroscopy (IR) and its purity was confirmed by elemental analysis. The ternary complexes exhibit IR absorptions typical of coordinated ligands: phen: 1625–1590 cm−1, 1524–1516 cm−1, 1430–1421 cm−1 (fused phenyl rings), 736–711 cm−1 and 896–811 cm−1 (δC–H out of plane); gly: 1634–1602 cm−1 (γas COO−), 648–597 cm−1 (δNH2), 3300–3240 cm−1 and 3449–3395 cm−1 (δNH2). The IR spectrum also showed an absorption band in 1384.7 cm−1 associated with the NO3− counter ion. Elemental analysis calculated for CuC16H16N4O5.2H2O (MW 443.90 g.mol−1): N (12.62), C (43.29), H (4.54); Found: N (12.96), C (43.00), H (4.42). The purity was estimated at 99%.
2.3. Cell lines and culture conditions
Human lung cancer A549 cells were purchased from ATCC (Manassas, VA) were grown in Ham’s F-12 medium with 2 mM L-glutamine (ATCC) supplemented with 10% fetal bovine serum (FBS) and 1% pen/strep (10,000 unit, Cellgro), as commonly used for this cell line, at 37°C and 5% CO2 air atmosphere. Human lung cancer H157 cells were kindly provided by Dr. Daniel Chan (University of Colorado Denver, Aurora, CO) and were grown in RPMI-1640 medium (ATCC) supplemented with 10% FBS. It is worth noting that growing A549 cells in RPMI-1640 medium did not significantly alter the response of these cells to Cas IIgly treatment (data not shown). Cells grown in 24-well plates were used for polymerase chain reactions (PCR), flow cytometry studies, the assessment of intracellular GSH and GSSG levels and cytotoxicity. Cells grown in T-150 flasks were used for mitochondrial isolation. Cas IIgly and BSO were added from aqueous stock solutions (2.5 mM and 10 mM, respectively).
2.4. Intracellular levels of GSH and GSSG
Intracellular GSH levels were determined by HPLC with electrochemical detection (HPLC-EC) (Bode and Rose, 1999). GSSG content was calculated as the difference between glutathione reductase (GR)-treated samples and GR-untreated samples. Cultured cells from 24-well plates were washed once with 1 ml of PBS, re-suspended in 0.5 ml of PBS and sonicated. Each sample was split into three samples (2 × 0.2 ml + 1 × 0.1 ml). The 0.2 ml samples were treated with 25 μl of GR reagent solution (11.25 U/ml GR, 0.675 mM NADPH, 25 mM Tris-HCl, 2.8 mM EDTA, pH 7.2) as described previously (Tietze, 1969), and other 0.2 ml sample received 25 μl of PBS. Next, 10% meta-phosphoric acid (25 μl) were added to both GR-treated and untreated samples (1% final concentration), the samples centrifuged at 20,000 g for 10 min, and the supernatants used for HPLC analysis. The HPLC column used was a Synergi 4u Hydro-RP 80A (150 × 4.6 mm) from Phenomenex (Torrance, CA) and the mobile phase was sodium phosphate buffer (125 mM sodium phosphate monobasic, pH adjusted to 3 with phosphoric acid) and 0.9% methanol. The flow rate was 0.5 ml.min−1. The retention time for GSH under these conditions was 7.5 min. The HPLC instrument was from ESA, Inc. (Chelmsford, MA), equipped with an autosampler (model 540) and a Coul array detector (model 5600A). The potential applied was + 0.75 V vs. H/Pd electrode, and the injection volume was 5 μl. The remaining 0.1 ml sample was used to measure protein content as described above.
2.5. Assessment of cytotoxicity
The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay is commonly used to measure cancer cell survival, yet it has revealed artifacts when measuring the cytotoxicity of prooxidant agents (Bernhard et al., 2003). Another simple method to evaluate drug-induced cytotoxicity is using membrane integrity as an index, which can be assessed by monitoring the release of cytosolic lactate dehydrogenase (LDH). LDH activity was measured in the culture medium and cell lysates (50 mM HEPES, Triton X-100 0.5%, pH 7) using a plate reader format as previously described (Day et al., 1995). Briefly, 5 μl of cell culture supernatant and lysates were incubated with 0.24 mM NADH in a Tris/NaCl pH 7.2 buffer in 96-well plates for 5 min at 25oC. The reaction was started by the addition of 9.8 mM pyruvate and the consumption of NADH was followed at 340 nm for 5 min at 30oC. Percent LDH release was calculated by the following equation: supernatant LDH/(supernatant LDH + lysate LDH) × 100.
2.6. EPR studies
EPR spectra were obtained in 1 mm ID pyrex capillaries supported in a 4 mm OD quartz tube. Spectra were run on a Bruker EMX-Plus spectrometer with a Bruker rectangular resonator at the University of Denver. Instrument parameters were 9.42 GHz microwave frequency, 100 gauss scan width, 1 gauss modulation amplitude at 100 kHz, and microwave power 20 mW. Since signals are time dependent, data acquisition was started at 2 min. after mixing, for all samples shown in figures and reported in comparisons. Five scans with a time/scan of 42 sec were averaged. Due to the low solubility of Cas IIgly in phosphate buffers, experiments were carried out in water.
2.7. Flow cytometry
MitoSOX is an analog of hydroethidine and has been used to detect mitochondrial superoxide by flow cytometry (Julian et al., 2005). The results obtained with this method remain qualitative rather than quantitative (Zhao et al., 2005), yet flow cytometry allows the study of specific groups of cells. The oxidation products of MitoSOX were detected using the FL2 channel. Rhodamine 123 (Rh123) is a dye commonly used to assess the depolarization of the mitochondrial membrane, and therefore mitochondrial dysfunction. Such effect can be detected by a decrease in fluorescence using the FL1 channel (Follstad et al., 2000). Briefly, control and drug-treated A549 cells (approximately 5 × 104) were exposed to 5 μM MitoSOX or 2 μM Rh123 for 20 min. The supernatant was removed and the cells scraped in 0.5 ml ice-cold PBS, centrifuged at 2000 g for 15 min, and re-suspended in 0.5 ml ice-cold PBS. Cells were analyzed within 30 min using a FACS Calibur flow cytometer (Becton Dickinson Biosciences, San Jose, CA). The total number of gated cell counted was 10,000.
2.8. Polymerase chain reaction of mitochondrial DNA
H157 and A549 cells were grown to near confluence and treated with 2.5 and 5 μM Cas IIgly, respectively. Following treatment, the media was removed and the cells were treated with 0.04% trypsin and collected by centrifugation at 500 g. After two washes with PBS to remove residual media, the cells were resuspended in 250 μl of PBS, and total DNA was extracted using the DNeasy blood & tissue kit from Qiagen (Valencia, CA). The resulting DNA recovery was measured by UV absorption (260 nm) using a ND-100 UV/vis spectrophotometer from Nanodrop Technologies (Wilmington, DE). Although the DNA extracted from cells grown in one well of a 24-well plate was sufficient for one PCR reaction, more reliable readings of DNA extracts were obtained by combining the cells from 2 wells (total number of approximately 100,000 cells, leading to DNA concentrations of ~ 40 ng/μl). As described previously, the L2 (5′-GCC CGT ATT TAC CCT ATA GC-3′) and H3 (5′-GTC TAG GGC TGT TAG AAG TC-3′) primers, obtained from Integrated DNA Technologies (Coralville, IA), were used to amplify a 5595 bp product of the human mitochondrial genome (Velsor et al., 2004). The amount of extracted DNA used for each PCR was 5 ng in a 25-μl reaction cocktail. Final concentrations were 300 nM of each primer, 2 mM MgSO4, 200 μM of each dNTP and 0.5 U of Platinum Taq Polymerase High Fidelity in the corresponding buffer (Invitrogen, Carlsbad, CA). The total number of cycles was 20. The purity of the amplification product was checked by electrophoresis using FlashGel DNA cassettes (1.2% agarose) from Lonza (Rockland, ME). The amount of DNA resulting from the reaction was measured by fluorescence using the Quant-Ti™ Picogreen dsDNA reagent from (Invitrogen) and a multi-well fluorescence plate reader (Cytofluor 4000 from PerSeptive Biosystems, Framingham, MA).
2.9. Mitochondria isolation
Isolation of mitochondria was achieved through differential centrifugation as previously described (Velsor et al., 2004). Briefly, cells were trypsinized (0.04% trypsin in Puck’s EDTA), pelleted by centrifugation (2,000 g for 10 min at 4°C), re-suspended in PBS and spun down (2,000 g for 10 min at 4°C) to yield a final cell pellet. The pellet was re-suspended in 550 μL of ice-cold hypotonic buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl, pH 7.5). The cell suspension was homogenized (Kontes glass homogenizer, Fisher-Scientific, Fair Lawn, NJ) and, immediately after homogenization, 400 μL of 2.5-X mannitol-sucrose buffer (525 mM mannitol, 175 mM sucrose, 12.5 mM Tris-HCl, 2.5 mM EDTA, pH 7.5) was added. Cellular debris was pelleted by centrifugation at 1,300 g for 10 min at 4°C. Centrifugation was repeated twice and mitochondria from the supernatant were isolated by centrifugation at 17,000 g for 15 min at 4°C. The mitochondrial pellet was washed with a mannitol-sucrose buffer and centrifuged again at 17,000 g for 15 min at 4°C to limit cytosolic contamination. Mitochondrial enrichment was determined by the relative activity of a cytosolic enzyme marker (LDH) and a mitochondrial enzyme marker (glutamate dehydrogenase, GDH) in the fractions as previously described (Velsor et al., 2004). The purity, according to LDH contamination, was 95%.
2.10. Immunoblotting of mitochondrial respiratory complexes I and II
Mitochondria pellets were lysed in a ground glass homogenizer with 50 μl of 50 mM HEPES, 0.5% Triton-X-100, pH 7.0 lysis solution. After homogenization, total volume was brought to 10 μl with protease inhibitor cocktail in H2O. Total protein concentration was measured at 595 nm on Spectra Max 340PC micro plate reader (Molecular Devices Corp., Sunnyvale, CA) using Coomassie Plus kit (Pierce, Rockford, IL). PAGEr® Gold Precast Polyacrylamide 4–20% Tris-Glycine (Cambrex Bio Science, Rockland, ME) were loaded with the mitochondrial fractions obtained as described above. Samples were run at 150 volts for 60 min and transferred to PVDF-plus membrane (Osmonics Inc., Westborough, MA) at 100 volts for one hour. Blocking, washing, and stripping solutions were prepared as suggested by manufacturer for optimal results with the ECL Plus Western Blotting Detection Reagents Kit (Amersham Biosciences, Buckinghamshire, UK). All wash steps were performed in triplicate for 10 min in Tris-buffered-saline-Tween (TBS-T). Membranes were blocked for one hour at room temperature in TBS-T and plus 10% horse serum. Complex I primary antibody (0.25 μg/ml of monoclonal 39 kDa antibody #A-21344, Molecular Probes, Eugene, OR) was applied for 2.5 h. Secondary antibody (peroxidase-conjugated AffiniPure goat anti-mouse IgG, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was diluted 1:40,000 in TBS-T and applied for 30 min. ECL Plus Western Blotting Detection Reagents were used to detect proteins. Following complex I detection membranes were submerged in stripping buffer (100 mM 2-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl pH 6.7) and incubated at 55°C for 45 min with occasional agitation. Membranes were re-probed for complex II (0.125 μg/ml of monoclonal 70 kDa antibody #A-11142, Molecular Probes). The secondary antibody (same as used above) was diluted 1:40,000 in TBS-T and applied for 30 min.
2.11. Immunoblotting of HO-1
Cells were grown in 24-well plates and, after treatment, washed with PBS. Cells were then sonicated in 200 μl of water. Cell debris was spun down and the supernatant volume reduced to approximately 30 μl by evaporation using a speed vacuum system. The resulting cytosolic proteins were run in a gradient gel and blotted as described above. The membrane was probed using heme hoxygenase 1 (HO-1) antibody (2.0 μg/ml of monoclonal 34.6 kDa antibody ab13248, Abcam, Cambridge, MA) applied over-night. The secondary antibody (same as used above) was diluted 1:40,000 in TBS-T and applied for 30 min. The membrane was re-probed using glyceraldehyde 3 phosphate dehydrogenase (GAPDH) antibody applied for 2.5 h (0.2 μg/ml of monoclonal 40.2 kDa antibody ab9484, Abcam, Cambridge, MA). The secondary antibody was the same as used above.
2.12. Statistical analysis
Data are presented as means ± standard error. For percentage of LDH relase and GSH levels measurements, results were reported from experimental groups consisting of three to four wells and experiments replicated once. Data were subsequently analyzed for significant differences using ANOVA analysis coupled with a Tukey’s range test where significance was preset at P < 0.05 (Prizm v.4, GraphPad, San Diego, CA).
3. Results
3.1. Cas IIgly-induced GSH depletion and cytotoxicity
Treatment of A549 cells with 5 μM Cas IIgly for 6 h induced a 75% loss of intracellular GSH compared to control (Fig. 2A). In H157 cells, the loss was even more dramatic, where 2.5 μM Cas IIgly induced an 85% loss (Fig. 2A). The loss of GSH was dose-dependent. Using membrane integrity as an index of cytotoxicity, Cas IIgly was also more toxic to H157 cells than to A549 cells, with IC50s of 3 μM and 6 μM, respectively (Fig. 2B and C). In both cell lines, the GSH synthesis inhibitor BSO (20 μM) further depleted GSH levels (not shown) and potentiated the Cas IIgly-induced toxicity (Fig. 2B and C). As shown with A549 cells, most of the lost GSH was accounted for as its oxidized disulfide form, GSSG (Fig. 3A). The mechanism for GSH depletion is likely through redox-cycling as shown in vitro, where Cas IIgly readily oxidizes GSH to GSSG (Fig. 3B).
Figure 2.

(A) Cas IIgly (6 h treatment) produced a dramatic loss (>70%) in intracellular glutathione (GSH) levels in A549 and H157 cells using 5 μM and 2.5 μM Cas IIgly, respectively, compared to control (Co). The 100% values for both cell lines were around 90 nmoles/mg of protein (±10% from one experiment to another). (B and C) Using membrane integrity (percentage of LDH release) as an index of cytotoxicity (24 h treatment), Cas IIgly was more cytotoxic in H157 cells compared to A549 cells (IC50s 3 μM and 6 μM, respectively). In both cell lines, the cytotoxicity was potentiated by the GSH synthesis inhibitor BSO (20 μM).
Figure 3.

(A) As shown in A549 cells, most of the intracellular GSH loss accounted as its oxidized disulfide form GSSG; following treatment of the lysate with glutathione reductase (GR), 63% of the GSH loss was recovered. Bars with different letters are statistically different from one another (n = 3, P < 0.05). (B) In a test tube using a Tris buffer (pH 7), Cas IIgly reacts with GSH, which 90% is recovered by adding NADPH and GR (samples were analyzed one hour after mixing compounds) (n = 3, P < 0.05). Oxygen likely acts as the acceptor of electrons to give O2•−.
3.2. Cas IIgly redox-cycles with H2O2 and GSH
In order to demonstrate the ability of Cas IIgly to catalyze the Fenton reaction, electronic paramagnetic resonance (EPR) experiments were carried out using 5,5-dimethyl-1-pyrroline N-oxide (DMPO, 100 mM) as spin trapping agent for the hydroxyl radical (HO•) (Zhu et al., 2002). The EPR spectrum of DMPO in the absence of H2O2 and Cas IIgly, showed no detectable EPR signals. The EPR spectrum for Cas IIgly is at lower field than the spectrum of DMPO/HO•, so at 0.1 mM the Cu(II) signal makes a negligible contribution to the 100 G scans that were used for the spin trapping experiments. In presence of H2O2 (5 mM), the 4-line EPR spectrum in Figure 4A is characteristic of DMPO/HO•, as expected if Cas IIgly catalyzes the Fenton reaction. Addition of 0.5 mM GSH caused the signal for DMPO/HO• to approximately double (Fig. 4B), thus demonstrating that GSH can be a source ofn electron for the Cas IIgly-catalyzed Fenton reaction, and explaining the conversion of GSH into GSSG observed in the test tube and in the cells.
Figure 4.
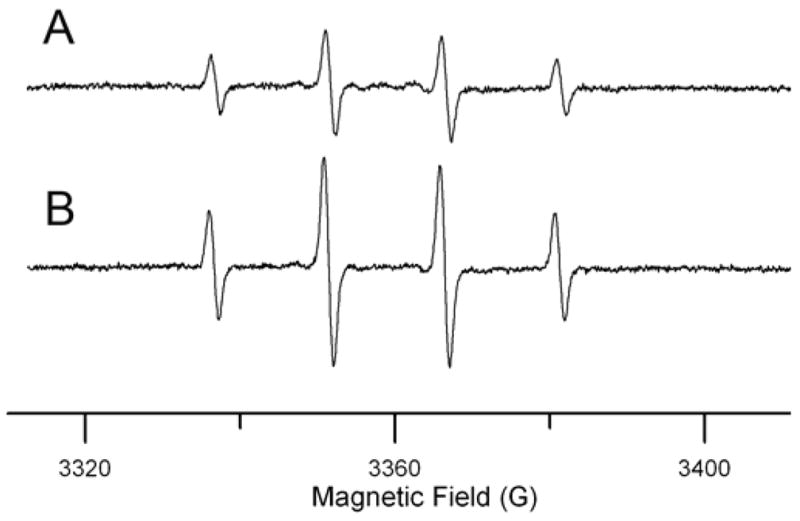
EPR spectra of DMPO/HO• produced by reaction of Cas IIgly with H2O2 in the absence (A) and the presence (B) of GSH (0.5 mM). Both reaction mixtures contained 100 mM DMPO, 5 mM H2O2, and 0.1 mM Cas IIgly.
3.3. MnTDE-1,3-IP5+ protects against Cas IIgly-induced toxicity
The catalytic antioxidant manganese(III) meso-tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin (MnTDE-1,3-IP5+, 5 μM), which has substantial superoxide dismutase and catalase-like activities, protected A549 cells from Cas IIgly-induced toxicity (7.5 μM, 24 h treatment) (Fig. 5A). This data suggest that some of Cas IIgly-induced effects are mediated by ROS or dependent upon superoxide (O2•−) or H2O2 to drive Fenton chemistry. In H157 cells, higher amounts of MnTDE-1,3-IP5+ (20 μM) were required for a protective effect toward Cas IIgly (5 μM) (Fig. 5B), thus supporting the observed increased sensitivity of H157 to Cas IIgly. In order to examine mechanisms associated with this oxidative stress, subsequent studies were carried out using 5 and 2.5 μM Cas IIgly in A549 and H157 cells, respectively, to avoid cell death.
Figure 5.

(A) The catalytic antioxidant MnTDE-1,3-IP5+ (MnP, 5 μM) protected A549 cells against Cas IIgly-induced injury (7.5 μM, 24 h treatment). (B) H157 cells required higher amounts of MnTDE-1,3-IP5+ (MnP, 20 μM) to protect against Cas IIgly-induced injury (5 μM). Membrane integrity (percentage of LDH release) was used as an index of cytotoxicity. Bars with different letters are statistically different from one another (n = 3, P < 0.05).
3.4. Delayed ROS production and mitochondrial dysfunction
MitoSOX fluorescence was used as a marker of mitochondrial ROS production. Treatment of A549 cells with 5 μM Cas IIgly led to a significant increase in ROS production at 12 h treatment, which was further increased at 24 h (Fig. 6A and B). The depolarization of the mitochondrial membrane reflects mitochondrial dysfunction, and was observed by a decrease in rhodamine 123 (Rh123) fluorescence. A549 cells showed a clear decrease of fluorescence 16 h after treatment with 5 μM Cas IIgly (Fig. 6C and D). Similar effects were obtained in H157 with 2.5 μM Cas IIgly (16 h after treatment) (Fig. 7A and B). This data provides evidence for Cas IIgly-induced mitochondrial dysfunction associated with ROS production.
Figure 6.

(A and B) A549 cells treated with Cas IIgly (5 μM, 12 and 24 h) showed increased levels of ROS, as shown by flow cytometry using MitoSOX fluorescence (FL2 channel). (C and E) A549 cells also showed a loss of Rho123 fluorescence (FL1 channel) and therefore a depolarization of the mitochondrial membrane. Bars with different letters are statistically different from one another (n = 3, P < 0.05).
Figure 7.
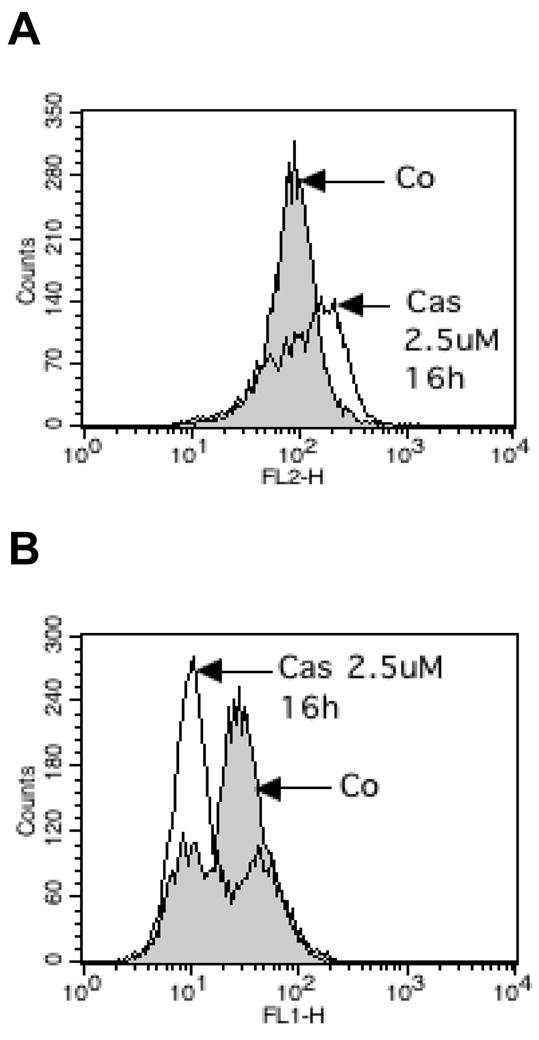
(A) H157 cells treated with 2.5 μM Cas IIgly (16 h) showed increased levels of ROS, as shown by flow cytometry using MitoSOX fluorescence (FL2 channel), and (B) a loss of Rho123 fluorescence (FL1 channel) and therefore a depolarization of the mitochondrial membrane.
3.5. Early mitochondrial DNA damage
Amplification efficiency assay using polymerase chain reaction (PCR) is commonly used to assess mtDNA damage (Milano and Day, 2000). Treatment of A549 cells for 6 h with 5 μM Cas IIgly decreased mtDNA amplification efficiency by 20% compared to untreated controls (Fig. 8A). This effect was dose-dependent, leading to approximately a 35% decrease using 10 μM Cas IIgly (not shown). Similar results were observed in H157 cells and using 2.5 μM Cas IIgly at 12 h after treatment (not shown). These results support an early mtDNA damage occurring before changes in membrane integrity were apparent.
Figure 8.
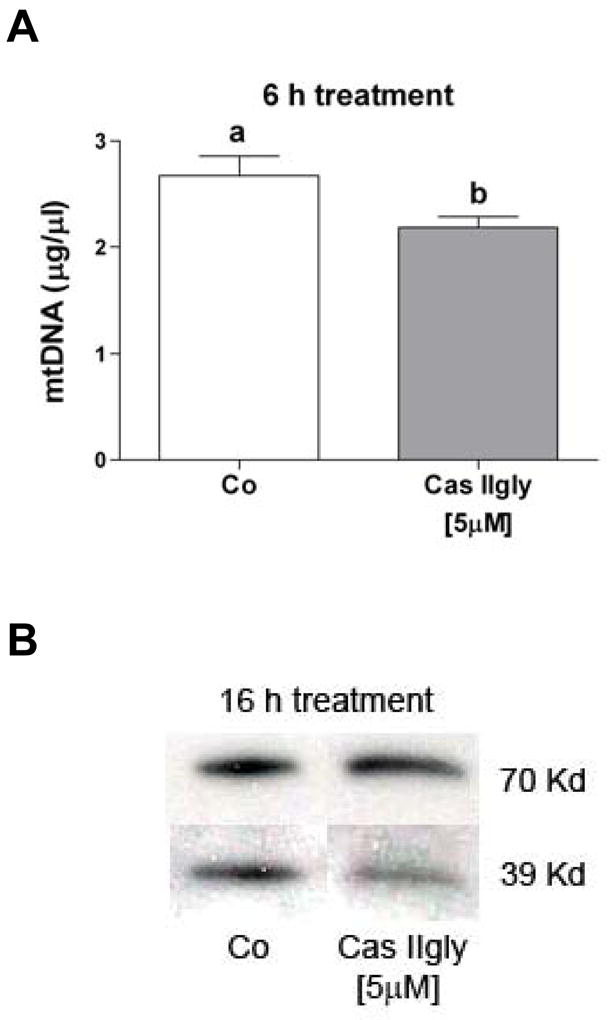
(A) Cas IIgly (5 μM, 6 h) induced mitochondrial DNA (mtDNA) damage in A549 cells, as detected by amplification efficiency assay. Bars with different letters are statistically different from one another (n = 3, P < 0.05). (B) Cas IIgly (5 μM, 12 h) produced an imbalance in the expression of complex I (39 Kd) versus complex II (70 Kd) of the mitochondrial respiratory chain in A549 cells, as shown by immunoblotting. Each figure was representative of three samples and the experiment repeated once.
3.6. Imbalance of the proteins of the mitochondrial respiratory chain
A number of apoproteins of mitochondrial electron transport complexes I, III, IV and V are encoded by mtDNA, whereas complex II is totally encoded by nuclear DNA. mtDNA damage has been associated with an imbalance in the expression of these proteins and an over-production of ROS (Wallace, 1999). H157 and A549 cells were treated for 16 h with 2.5 and 5 μM Cas IIgly, respectively. Mitochondria were isolated, and protein immunoblots demonstrated lower levels of the highly mitochondria encoded respiratory complex I when compared to the nuclear encoded complex II (Fig. 8B), thus showing some evidence of an imbalance. It is worth noting that a similar effect was previously observed using cisplatin in A549 cells, but required a higher concentration and a longer time of treatment (40 μM and 48 h, respectively) (Kachadourian et al., 2007).
3.7. Cas IIgly induces the expression of HO-1
One marker commonly used to detect an antioxidant adaptive response from the cell is the expression of HO-1. As shown by immunoblotting, 6 h treatment with 2.5 μM Cas IIgly induced the expression of HO-1 from non-detectable levels in control samples in H157 cells and higher levels from detectable levels in control samples in A549 cells (Fig. 9).
Figure 9.
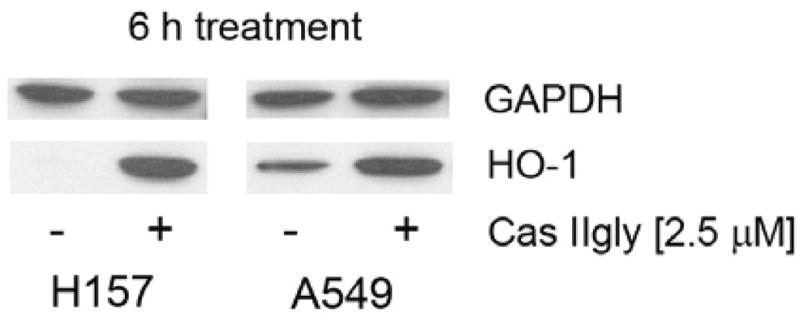
Cas IIgly (2.5 μM, 6 h) induced the expression of heme oxygenase-1 (HO-1, 35 Kd) in H157 and A547 cells, as shown by immunoblotting. GAPDH (40 Kd) was used as loading control. It is worth noting that A549 but not H157 cells showed basal levels of HO-1. Each figure was representative of two samples.
4. Discussion
Previous findings suggest that copper-phenanthroline complexes redox-cycle with thiols and H2O2, resulting in ROS production (Kobashi and Horecker, 1967; Que et al., 1980; Antholine et al., 1985). On the other hand, intracellular GSH depletion eventually induces mitochondrial GSH depletion and has been associated with increased levels of mitochondrial ROS and mitochondrial dysfunction (Armstrong and Jones, 2002). This study demonstrates that Cas IIgly redox-cycles with H2O2 and GSH, and shows a correlation between Cas IIgly-induced cytotoxicity and GSH depletion in two human lung cancer cell lines. It remains unclear whether the unaccounted GSH that was not oxidized to GSSG was converted into higher oxidation states (sulfenic and/or sulfinic acids), formed protein-glutathione adducts, or was used to expel the copper complex from the cell (Ballatori et al., 2005; Steinebach et al., 1994). As previously shown with cisplatin, BSO sensitizes cancer cells to Cas IIgly treatment, thus supporting an important role for oxidative stress. Both cisplatin and copper-phenanthroline complexes are known to interact with DNA, the latter being also able to redox-cycle with GSH. Our results support the notion that GSH under certain conditions can be a substrate for pro-oxidant reactions in the cell. It is worth noting that ascorbic acid has been shown to exacerbate copper toxicity in cell culture models, likely by acting as an electron donor as well (Gaetke and Chow, 2003).
Several studies have been previously devoted to cisplatin-mtDNA interactions, but not, to our knowledge, for copper-phenanthroline-mtDNA interactions. One result of mtDNA damage is an imbalance of the complexes of the respiratory chain, which has been associated with an increased “leakage” of O2•− (Wallace, 1999). This study also shows evidence of early mtDNA damage and a disruption of the mitochondrial respiratory chain. Cas IIgly has been previously shown to interfere directly with the mitochondrial respiratory chain, another effect that could account for an increase in ROS (Marín-Hernández, 2003). However, both delayed ROS burst and mitochondrial depolarization observed by flow cytometry support Cas IIgly-mediated indirect effects, i.e., GSH depletion and a disruption of the mitochondrial respiratory chain due to mitochondrial DNA damage. Given the rapid drop in GSH levels, such delay may be due to the ability of the cells to initially control ROS levels, including O2•− and HO•, until this ability is exhausted. This study shows evidence of at least two new pro-oxidant mechanisms that may be crucial for Cas IIgly-induced cytotoxicity (Fig. 10).
Figure 10.

Working hypothesis. Glutathione (GSH) reacts with Cas IIgly, resulting in reduction of the copper-complex and the formation of the glutathiyl radical (GS•), which can react with another GS• to give oxidized glutathione (GSSG), or with GSH and oxygen to give superoxide (O2•−) and GSSG. The formation of GSSG and O2•− can also result from redox-cycling of Cas IIgly with GSH and oxygen (not shown). Superoxide dismutase (SOD) converts O2•− into hydrogen peroxide (H2O2), which reacts with the reduced Cas IIgly to produce the hydroxyl radical (HO•). HO• can initiate mitochondrial DNA damage which translates in an imbalance of the expression of the proteins of the mitochondrial respiratory chain (MRC) an d, in turn, into more ROS production. Mitochondrial dysfunction would result from both decreased levels of GSH and increased levels of H2O2.
The protective effect of MnTDE-1,3-IP5+ further supports a crucial role for ROS in Cas IIgly-induced toxicity. MnTDE-1,3-IP5+ is a member of a series of porphyrin-based catalytic antioxidants that protect both in vitro and in vivo in a number of oxidative stress models. Manganese porphyrins have been reported to catalyze the dismutation of O2•− and H2O2, scavenge peroxynitrite and inhibit lipid peroxidation (Kachadourian et al., 2004; Ferrer-Sueta et al., 2006; Castello et al., 2008). Another manganese porphyrin (MnTBAP) has previously shown to protect mtDNA from oxidative damage (Milano and Day, 2000). A direct interaction between the two positively charged complexes that would prevent Cas IIgly from redox-cycling is unlikely, but a partial competitive inhibition of Cas IIgly transport into the mitochondria cannot be ruled out.
Previous work suggests that the adaptive response to an oxidative insult varies from one cell line to another (Allen and Balin, 2003; Wang et al., 2006; Kim et al., 2008; Wang et al., 2008). Given the strong pro-oxidant effects of Cas IIgly, H157 cells may be more vulnerable to oxidative damage than A549 cells. The constitutive levels of HO-1 found in A549 but not in H157 cells supports this hypothesis. On the other hand, H157 cells are more resistant to treatment with the tyrosine kinase inhibitor of the epidermal growth factor receptor (EGFR) Gefitinib than A549 cells (Helfrich et al., 2006). Taken together, these results argue in favor of more personalized combinations of drugs that would increase efficacy of treatment and decrease secondary effects based on the tumor phenotype.
Acknowledgments
The authors wish to thank Dr. Sandra S. Eaton and Sam Schroeder (University of Denver) for performing the EPR experiments, and Maria Eleno Bravo (UNAM, Mexico City) for synthesizing Cas IIgly. This work was supported by CONACyT grant 60085 to L. Ruiz-Azuara and NIH grants HL755223, HL84469, ES015678 and ES017582 to B. J. Day. Dr. Day is a consultant for and holds equity in Aeolus Pharmaceuticals that is commercially developing metalloporphyrins as human therapeutic agents.
Abbreviations
- BSO
L-buthionine sulfoximine
- Cas IIgly
casiopeína IIgly
- EPR
electron paramagnetic resonance
- GAPDH
glyceraldehyde 3 phosphate dehydrogenase
- GR
glutathione reductase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HO-1
heme oxygenase 1
- HPLC-EC
HPLC with electrochemical detection
- MnTDE-1
3-IP5+, manganese(III) meso-tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin
- mtDNA
mitochondrial DNA
- LDH
lactate dehydrogenase
- MRP
multi-drug resistant protein
- PCR
direct polymerase chain reaction
- Rh123
rhodamine 123
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alemón-Medina R, Muñoz-Sánchez JL, Ruiz-Azuara L, Gracia-Mora I. Casiopeína IIgly induced cytotoxicity to HeLa cells depletes the levels of reduced glutathione and is prevented by dimethyl sulfoxide. Toxicol In Vitro. 2008;22:710–715. doi: 10.1016/j.tiv.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Allen RG, Balin AK. Effects of oxygen on the antioxidant responses of normal and transformed cells. Exp Cell Res. 2003;289:307–316. doi: 10.1016/s0014-4827(03)00279-9. [DOI] [PubMed] [Google Scholar]
- Antholine WE, Kalyanaraman B, Petering DH. ESR of copper and iron complexes with antitumor and cytotoxic properties. Environ Health Perspect. 1985;64:19–35. doi: 10.1289/ehp.856419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JS. Mitochondria: a target for cancer therapy. Br J Pharmacol. 2006;147:239–248. doi: 10.1038/sj.bjp.0706556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. Faseb J. 2002;16:1263–1265. doi: 10.1096/fj.02-0097fje. [DOI] [PubMed] [Google Scholar]
- Bailey HH, Ripple G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Mahvi D, Schink J, Pomplun M, Mulcahy RT, Wilding G. Phase I study of continuous-infusion L-S, R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst. 2005;89:1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Hammond CL, Cunningham JB, Krance SM, Marchan R. Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol. 2005;204:238–255. doi: 10.1016/j.taap.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Batist G, Schecter RL, Alaoui-Jamali MA. The glutathione system and drug resistance. In: Shilsky R, Milano G, Ratain M, editors. Principles of antineoplastic drug development and pharmacology. Marcel Dekker Inc; New York: 1996. pp. 503–521. [Google Scholar]
- Bernhard D, Schwaiger W, Crazzolara R, Tinhofer I, Kofler R, Csordas A. Enhanced MTT-reducing activity under growth inhibition by resveratrol in CEM-C7H2 lymphocytic leukemia cells. Cancer Lett. 2003;195:193–199. doi: 10.1016/s0304-3835(03)00157-5. [DOI] [PubMed] [Google Scholar]
- Bode AM, Rose RC. Analysis of water-soluble antioxidants by high-performance liquid chromatography with electrochemical detection. In: Packer L, editor. Oxidants and antioxidants, part A, Methods in enzymology. Vol. 299. Academic Press; San Diego: 1999. pp. 77–83. [DOI] [PubMed] [Google Scholar]
- Castello PR, Drechsel DA, Day BJ, Patel M. Inhibition of mitochondrial hydrogen peroxide production by lipophilic metalloporphyrins. J Pharmacol Exp Ther. 2008;324:970–976. doi: 10.1124/jpet.107.132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day BJ, Shawen S, Liochev SI, Crapo JD. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced endothelial cell injury in vitro. J Pharmacol Exp Ther. 1995;275:1227–1232. [PubMed] [Google Scholar]
- Ferrer-Sueta G, Hannibal L, Batinic-Haberle I, Radi R. Reduction of manganese porphyrins by flavoenzymes and submitochondrial particles: a catalytic cycle for the reduction of peroxynitrite. Free Radic Biol Med. 2006;41:503–512. doi: 10.1016/j.freeradbiomed.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Follstad BD, Wang DIC, Stephanopoulos G. Mitochondrial membrane potential differentiates cells resistant to apoptosis in hybridoma cultures. Eur J Biochem. 2000;267:6534–6540. doi: 10.1046/j.1432-1327.2000.01743.x. [DOI] [PubMed] [Google Scholar]
- Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147–163. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, Coldren C, Barón A, Zeng C, Franklin WA, Hirsch FR, Gazdar A, Minna J, Bunn PA. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res. 2006;12:7117–7125. doi: 10.1158/1078-0432.CCR-06-0760. [DOI] [PubMed] [Google Scholar]
- Henkels KM, Turchi JJ. Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res. 1999;59:3077–3083. [PubMed] [Google Scholar]
- Huang HL, Fang LW, Lu SP, Chu CK, Luh TY, Lai MZ. DNA-damaging reagents induce apoptosis through reactive oxygen species-dependent Fas aggregation. Oncogene. 2003;22:8168–8177. doi: 10.1038/sj.onc.1206979. [DOI] [PubMed] [Google Scholar]
- Julian D, April KL, Patel S, Stein JR, Wohlgemuth SE. Mitochondrial depolarization following hydrogen sulfide exposure in erythrocytes from a sulfide-tolerant marine invertebrate. J Exp Biol. 2005;208:4109–4122. doi: 10.1242/jeb.01867. [DOI] [PubMed] [Google Scholar]
- Kachadourian R, Johnson CA, Min E, Spasojevic I, Day BJ. Flavin-dependent antioxidant properties of a new series of meso-N,N′-dialkyl-imidazolium substituted manganese(III) porphyrins. Biochem Pharmacol. 2004;67:77–85. doi: 10.1016/j.bcp.2003.08.036. [DOI] [PubMed] [Google Scholar]
- Kachadourian R, Leitner HM, Day BJ. Selected flavonoids potentiate the toxicity of cisplatin in human lung adenocarcinoma cells: a role for glutathione depletion. Int J Oncol. 2007;31:161–168. [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Kim S, Kim EJ, Park JH, Yang SH, Jeong ET, Park C, Youn MJ, So HS, Park R. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer. 2008;60:47–56. doi: 10.1016/j.lungcan.2007.09.021. [DOI] [PubMed] [Google Scholar]
- Kobashi K, Horecker BL. Reversible inactivation of rabbit muscle aldolase by o-phenanthroline. Arch Biochem Biophys. 1967;161:178–186. doi: 10.1016/0003-9861(67)90022-7. [DOI] [PubMed] [Google Scholar]
- Leal-García M, García-Ortuño L, Ruiz-Azuara L, Gracia-Mora I, Luna-Delvillar J, Sumano H. Assessment of acute respiratory and cardiovascular toxicity of casiopeínas in anaesthetized dogs. Basic Clin Pharmacol Toxicol. 2007;101:151–158. doi: 10.1111/j.1742-7843.2007.00038.x. [DOI] [PubMed] [Google Scholar]
- Leitner HM, Kachadourian R, Day BJ. Harnessing drug resistance: using ABC transporter proteins to target cancer cells. Biochem Pharmacol. 2007;74:1677–1685. doi: 10.1016/j.bcp.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Hernández A, Gracia-Mora I, Ruiz-Ramírez L, Moreno-Sánchez R. Toxic effects of copper-based antineoplastic drugs (Casiopeínas) on mitochondrial functions. Biochem Pharmacol. 2003;65:1979–1989. doi: 10.1016/s0006-2952(03)00212-0. [DOI] [PubMed] [Google Scholar]
- Milano J, Day BJ. A catalytic antioxidant metalloporphyrin blocks hydrogen peroxide-induced mitochondrial DNA damage. Nucleic Acids Res. 2000;28:28968–28973. doi: 10.1093/nar/28.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T, Voigt W, Simon H, Fruehauf A, Bulankin A, Grothey A, Schmoll HJ. Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer Res. 2003;63:513–521. [PubMed] [Google Scholar]
- Nicco C, Laurent A, Chereau C, Weill B, Batteux F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed Pharmacother. 2005;59:169–174. doi: 10.1016/j.biopha.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Que BG, Downey KM, So AG. Degradation of deoxyribonucleic acid by 1,10-phenanthroline-copper complex: the role of hydroxyl radicals. Biochemistry. 1980;18:5987–5991. doi: 10.1021/bi00567a007. [DOI] [PubMed] [Google Scholar]
- Rivero-Müller A, De Vizcaya-Ruiz A, Plant N, Ruiz L, Dobrota M. Mixed chelate complex Casiopeína IIgly binds and degrades nucleic acids: a mechanism of cytotoxicity. Chem Biol Interact. 2007;165:189–199. doi: 10.1016/j.cbi.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67:3364–3370. doi: 10.1158/0008-5472.CAN-06-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweyer S, Soruri A, Heintze A, Radzun HJ, Fayyazi A. The role of reactive oxygen species in cisplatin-induced apoptosis in human malignant testicular germ cell lines. Int J Oncol. 2004;25:1671–1676. doi: 10.3892/ijo.25.6.1671. [DOI] [PubMed] [Google Scholar]
- Steinebach OM, Wolterbeek HT. Role of cytosolic copper, metallothionein and glutathione in copper toxicity in rat hepatoma tissue culture cells. Toxicology. 1994;92:75–90. doi: 10.1016/0300-483x(94)90168-6. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Trejo-Solís C, Palencia G, Zúñiga S, Rodríguez-Ropon A, Osorio-Rico L, Luvia ST, Gracia-Mora I, Marquez-Rosado L, Sánchez A, Moreno-García ME, Cruz A, Bravo-Gómez ME, Ruiz-Ramírez L, Rodríguez-Enriquez S, Sotelo J. Cas IIgly induces apoptosis in glioma C6 cells in vitro and in vivo through caspase-dependent and caspase-independent mechanisms. Neoplasia. 2005;7:563–574. doi: 10.1593/neo.04607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Velsor LW, Kovacevic M, Goldstein M, Leitner HM, Lewis W, Day BJ. Mitochondrial oxidative stress in human hepatoma cells exposed to stavudine. Toxicol Appl Pharmacol. 2004;199:10–19. doi: 10.1016/j.taap.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Wang XG, Hayes JD, Wolf CR. Generation of stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of Nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006;66:10983–10994. doi: 10.1158/0008-5472.CAN-06-2298. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res. 2006;12:5817–5825. doi: 10.1158/1078-0432.CCR-06-1037. [DOI] [PubMed] [Google Scholar]
- Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu BZ, Zhao HT, Kalyanaraman B, Frei B. Metal-independent production of hydroxyl radicals by halogenated quinones and hydrogen peroxide: an ESR spin trapping study. Free Radic Biol Med. 2002;32:465–473. doi: 10.1016/s0891-5849(01)00824-3. [DOI] [PubMed] [Google Scholar]