

Abstract
Epidemiological and clinical studies indicate that elevated circulating level of homocysteine (Hcy) is a risk factor for developing Alzheimer's disease (AD). Dietary deficiency of folate, vitamin B6 and B12 results in a significant increase of Hcy levels, a condition also known as hyperhomocysteinemia (HHcy).
In the present study we tested the hypothesis that a diet deficient for these three important factors when administered to a mouse model of AD, i.e. Tg2576, will result in HHcy and in an acceleration of their amylodotic phenotype.
Compared with Tg2576 mice on regular chow, the ones receiving the diet-deficient for folate, B6 and B12 developed HHcy. This condition was associated with a significant increase in Aβ levels in the cortex and hippocampus, and an elevation of Aβ deposits in the same regions. No significant changes were observed for steady state levels of total APP, BACE-1, ADAM-10, PS1 and nicastrin in the brains of mice with HHcy. No differences were observed for the main Aβ catabolic pathways, i.e IDE and neprilysin proteins, or the Aβ chaperone apolipoprotein E.
Our findings demonstrate that a dietary condition which leads to HHcy may also result in increased Aβ levels and deposition in a transgenic mouse model of AD-like amylodosis. They further support the concept that dietary factors can contribute to the development of AD neuropathology.
Introduction
High level of circulating homocysteine (Hcy), also known as hyperhomocysteinemia (HHcy), has been closely connected to several human diseases, including coronary artery disease, peripheral vascular disease, and stroke (2002; Boushey et al., 1995). In addition to these cardiovascular diseases, HHcy has been recently found to be involved in the development of neurodegenerative diseases such as Alzheimer's disease (AD) (Clarke et al., 1998; Leblhuber et al., 2000; Seshadri et al., 2002). AD is the most common dementia in the seniors and affects more than 5 million people in USA. Although genetic factors such as mutations in amyloid precursor protein (APP) or presenilin 1 (PS1) are sufficient to cause AD, over 97% AD cases are sporadic and other potential-modifiable environmental risk factors seem to be required for its onset (Gandy, 2005).
Previous data have shown that high plasma level of Hcy (> 12μM) can almost doubled the risk of AD development in the elderly (Seshadri et al., 2002), and that this condition represents one modifiable risk factor for AD onset (Chan et al., 2008; Clarke et al., 1998; Flicker et al., 2008; McCaddon et al., 1998; Morris, 2003; Seshadri et al., 2002).
However, a causative role has not been demonstrated yet and negative data have been reported (Luchsinger et al., 2007; Morris et al., 2006). Hcy is a non-protein amino acid, it derives from the methionine metabolism which requires the presence of optimal concentrations of three important cofactors—folate, vitamin B6 and B12. Dietary supplementation of folate, vitamin B6 and B12 reduces Hcy levels, conversely their deficiency can result in HHcy (Morris, 2003). Therefore, understanding the mechanism responsible for the association between HHcy and AD could provide practical means to prevent or reduce the risk of AD development.
Although they remain to be fully elucidated, several potential mechanisms have been proposed to explain the biological links between HHcy and AD pathogenesis. HHcy can induce excitation damage through glutamate receptors (Boldyrev and Johnson, 2007; Lipton et al., 1997); increase oxidative stress (Jacobsen, 2000); alter DNA methylation (Fuso et al., 2005), interfere with DNA repair mechanisms (Kruman et al., 2002) and induce microvascular damage (Troen et al., 2008). The link between HHcy and AD has also been studied by different approaches including crossing a genetic HHcy mouse model with an AD mouse model and showing an increase in amyloid production (Pacheco-Quinto et al., 2006). Zhang et al. reported that by directly injecting homocysteine into animal brain amyloidogenesis was augmented (Zhang et al., 2009). Similar results were also reported by using a dietary intervention to induce HHcy in different AD mouse models (Bernardo et al., 2007; Chan and Shea, 2007; Chan et al., 2009; Fuso et al., 2008; Fuso et al., 2009).
In the present study, we assessed the long term (7 months) effect of a diet deficient of folate, B6 and B12 on the amyloidotic phenotype of an APP transgenic mouse model of AD, i.e. Tg2576. We chose this dietary regimen not only because vitamin B deficiency is a common cause of human HHcy, but also because previous studies have found it effective in elevating homocysteine levels in different mouse models (Fuso et al., 2008; Troen et al., 2003). In addition, Tg2576 mouse develops Aβ pathology only after middle age (10-12 months), providing a good model of the known epidemiological association between chronic mild HHcy and AD in the elderly.
Materials and methods
Tg2576 mice and diet treatments
Animal procedures were approved by the Institutional Animal Care and Usage Committee. Only female transgenic mice over-expressing the human APP with the Swedish mutation (K670N/M671L) (Hsiao et al., 1996) were used in this study. Polymerase chain reaction (PCR) analysis using tail DNA was used to confirm the genotype of all mice. All animals were kept in a pathogen-free environment, on a 12-hour light/dark cycle and had access to food and water ad libitum.
Tg2576 mice were randomized to two different diets: standard rodent chow deficient in folate (<0.2mg/kg), vitamin B6 (< 0.1mg/kg) and B12 (<0.001 mg/kg) (n=6) or standard rodent chow with vehicle (n=7). Diets were custom-made, prepared by a commercial vendor (Harlan Teklad, Madison, WI), and matched for kilocalories (Hofmann et al., 2001). Since the diets were not added with sulfathiazole, a limited amount of folate was produced by gut bacteria in the animals receiving the deficient diet.
Aβ plaques deposition appears in Tg2576 mouse brain around 10-12 months of age, and accumulate to significant levels by 15 months of age (Kawarabayashi et al., 2001). Hence, we started the diet treatment when the mice were 8 month-old (pre-plaque) for 7 months and sacrificed them at the age of 15 months. Animals were perfused intracardially with 0.9% PBS containing 10 mM EDTA. Brain was removed and dissected in two hemibrains by midsagittal dissection: the left hemibrain was used for biochemistry assays; the right one was fixed in 4% paraformaldehyde in 0.1 M PBS (pH 7.6) over night for immunohistochemistry studies.
Immunohistochemistry
Immunostaining analyses were performed as previously reported by our group (Firuzi et al., 2008; Sung et al., 2004a). Tg2576 mice brains were cut in serial 6-μm-thick coronal sections and mounted on Superfrost/plus microscope slides (Fisher scientific, Pittsburgh, PA, USA). The sections were deparaffinized, hydrated, pretreated with formic acid (88%) and subsequently with 5% H2O2 in methanol. After blocking in 2% fetal bovine serum, sections were incubated with primary antibody 4G8 (Covance, Princeton, NJ, USA) overnight at 4°C. After three washing, sections were incubated with a biotinylated anti-mouse secondary antibody (Vector laboratories, Burlingame, CA, USA), then sections developed by using the avidin-biotin complex method (Vector Laboratories, Burlingame, CA, USA) with 3,3′-diaminobenzidine (DAB) as a chromogen.
Light microscopic images from the hippocampus and somatosensory cortex were used to calculate the area occupied by Aβ-immunoreactivity with the software Image-Pro Plus for Windows version 5.0 (Media Cybernetics, Inc., Silver Spring, MD, USA), as previously described (Firuzi et al., 2008). The threshold of optical density that discriminated staining from background was determined, and kept constant for all quantifications. The area occupied by Aβ-immunoreactivity was measured and was divided by the total area of interest to calculate the percentage area occupied by Aβ-immunoreactivity. Analyses were always performed in a coded fashion.
Biochemical analyses
Both cortex and hippocampus were homogenized and sequentially extracted in RIPA and then formic acid (FA), where the RIPA fraction contains the soluble, whereas the FA fraction the insoluble forms of the Aβ peptides, as previously described (Sung et al., 2004b). Sensitive sandwich ELISA tests were performed to measure Aβ1-40 and Aβ1-42 levels (IBL America, Minneapolis, MN, USA). Analyses were always performed in duplicate and in a coded fashion.
Animal blood was collected by cardiac puncture with a 1ml syringe coated with EDTA. Samples were centrifuged at 2000 rpm for 10 minutes at 4°C, and clear upper layer plasmas collected and immediately stored at -80°C until used. The “Abbott Homocysteine assay”, a fluorescence polarization immunoassay, was used to determine the plasma homocysteine levels on the IMX® analyzer (Abbott Laboratories, Abbott Park, IL, USA). This immunoassay requires 50μl plasma from each sample without pretreatment and is based on the highly selective enzymatic conversion of homocysteine to S-adenosyl-L-homocysteine, which is then recognized by a monoclonal antibody (Pfeiffer et al., 1999). Plasma cholesterol and triglycerides levels were determined enzymatically using Sigma reagents (Sigma Chem. Co. St. Louis, MO). 8-iso-Prostaglandin F2α level in cortex RIPA fraction was quantified by an enzyme immunoassay kit, following the protocol provided by the company (Assay Designs, MI, USA).
Immunoblot analyses
RIPA fractions of cortex homogenates were used for immunoblot analyses, as hippocampus homogenates were not enough for all the analyses. Samples were electrophoresed on Tris-glycine polyacrylamide gels and pre-casted gels (Bio-Rad Laboratories, Hercules, CA, USA) and transferred onto nitrocellulose membranes. Antibodies and dilution used in the present study were as followed: anti-APP N-terminal raised against amino acids 66-81 for total APP (22C11; 1:500; Chemicon International, Temecula, CA, USA), anti-sAPPβ (6A1; 2.5 μg/ml, IBL America, Minneapolis, MN, USA), anti-sAPPα (2B3; 2.5 μg/ml; IBL America, Minneapolis, MN, USA), anti-ADAM-10 (1:500, Chemicon International, Temecula, CA, USA), anti-APP C-terminal for CTFs (1:600; EMD Biosciences Inc, La Jolla, CA, USA), anti-BACE-1 (1:400; Prosci Incorporated, Poway, CA, USA), anti-PS1 (1:500; Cell Signaling Technology, Danvers, MA, USA), anti-Nicastrin (1:500; Cell Signaling Technology, Danvers, MA, USA), anti-IDE N-terminal (1:1000; EMD Biosciences Inc, La Jolla, CA, USA), anti-neprilysin (1:150; Santa Cruz biotech. Santa Cruz, CA, USA), anti-apoE (1:100; Santa Cruz biotech.), and anti-β-actin (1:4000; Santa Cruz biotech.). HRP-conjugated secondary antibodies were from Cell Signaling and Pierce Biotechnology (Rockford, IL, USA).
Data analysis
Data analyses were performed using SigmaStat. Statistical comparisons between the different treatment groups were performed by one way ANOVA and Fisher's test post hoc analysis. Values in all figures represent mean ± S.E.
Results
A Folate, vitamin B6 and vitamin B12 deficient diet induced HHcy in Tg2576 mice
To study the effect of HHcy on AD-like amyloidosis, HHcy was induced in an AD mouse model, i.e. the Tg2576 mice, by using a well-established dietary intervention model (Hofmann et al., 2001). Eight-month-old Tg2576 mice were fed with a folate, vitamin B6 and B12 deficient diet (diet group) or vehicle diet (ctrl group) for 7 months. At the end of the diet treatment, body weight, plasma total cholesterol and triglycerides levels were not significantly different between these two groups (Table 1). By contrast, the diet group had a significant higher level of plasma homocysteine than the ctrl group (Fig. 1). Brain levels of 8-iso-prosraglandin F2α were also assayed as a marker of lipid peroxidation (Pratico et al., 2001). We found that diet group had higher 8-iso-prosraglandin F2α levels than the control group, however the difference did not reach statistical significance (Ctrl group: 985.5 ±120.8 pg/mg tissue; Diet group: 1311.6 ±122.0 pg/mg tissue, p<0.1, n = 6 per group).
Table 1.
Effect of seven months exposure of Tg2576 mice to a folate, vitamin B6 and B12-deficient diet on body weight, total plasma cholesterol and triglycerides levels.
| Control (n=7) |
Folate, B6, B12-deficient diet (n=6) |
|
|---|---|---|
| Weight (g) |
30±2.4 | 31±2.1 |
| Total Cholesterol (mg/dL) |
151±25 | 155±30 |
| Triglycerides (mg/dL) |
112±21 | 121±20 |
Results are mean ±S.E.
Figure 1.

Dietary induction of HHcy in Tg2576 mice. Tg2576 mice fed for 7 months with a folate, vitamin B6 and vitamin B12 deficient diet (Diet group, n = 6) had higher level of plasma homocysteine than Tg2576 fed with vehicle diet (Ctrl group, n = 7). Values represent mean ± S.E.M. *P < 0.05.
HHcy elevated Aβ peptide levels in Tg2576 mice brain
Sandwich ELISA quantification was performed to measure the Aβ1-40/42 levels in the extractions from both cortex and hippocampus of the two groups of mice. When compared with control mice, mice with HHcy had higher levels of Aβ peptides. In particular, we observed a significant increase of Aβ1-40 levels in the FA fraction of the hippocampus. Moreover, Aβ1-42 was significantly increased in both RIPA and FA extracted fractions of the cortex (Figure 2).
Figure 2.

Diet-induced HHcy in Tg2576 mice results in increased Aβ peptide levels. RIPA-soluble (RIPA) and formic acid (FA) extractable Aβ1-40 (A, B), and Aβ1-42 (C, D) levels in the cortex (Ctx) and hippocampus (Hippo) from Tg2576 on a folate deficient diet (Diet group, n = 6) or vehicle (Ctrl group, n = 7) were measured by sandwich ELISA. Values represent mean ± S.E.M. *P < 0.05.
HHcy increased Aβ deposition in Tg2576 mice brain
Immunochemical detection of Aβ peptides deposition in the brain sections were performed with 4G8, an anti-Aβ antibody reactive to amino acid residues 17-24. The percentage of area covered by positive immunoreactivity was calculated, as previously described (Firuzi et al., 2008; Sung et al., 2004a). As shown in figure 3, we found that, compared with the control group, the group with HHcy had a significant increase in Aβ immuno-reactivity in both the hippocampus (Ctrl group: 0.61% ± 0.14%; Diet group: 1.27% ± 0.22%, p<0.05) and the somatosensory cortex areas (SSC) (Ctrl group: 0.88% ± 0.16%; Diet group: 1.70% ± 0.30%, p<0.05). Similar results were observed when the number pof amyloid plaques were counted in the same regions (Data not shown).
Figure 3.
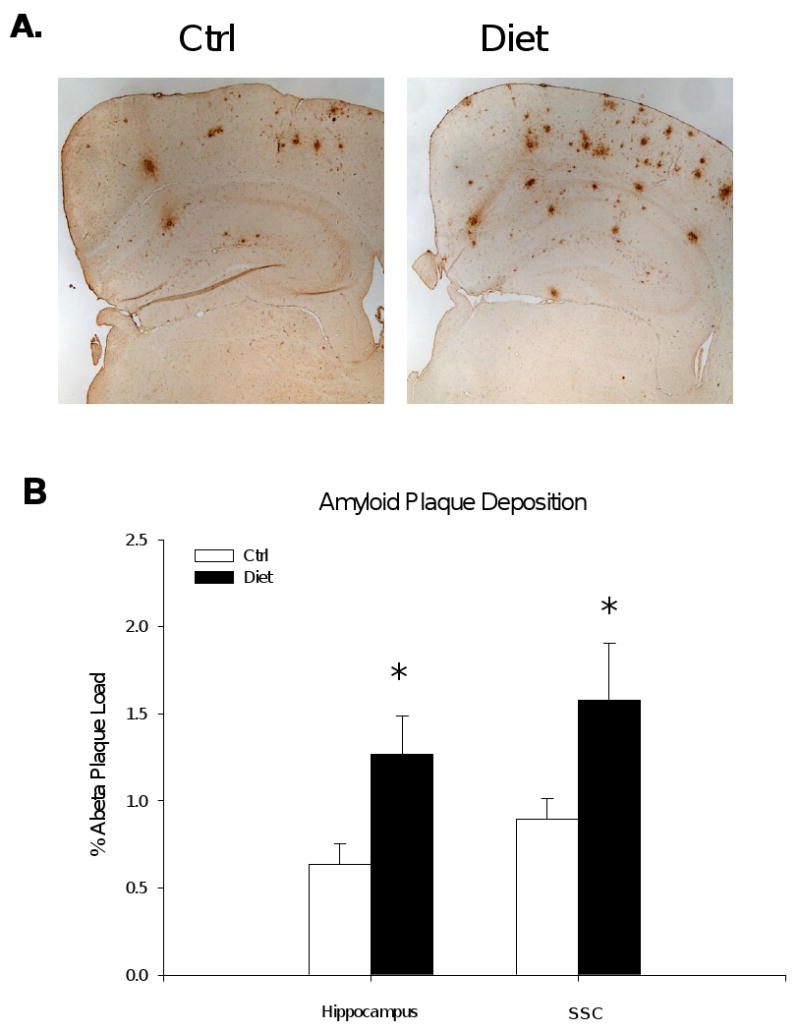
HHcy in Tg2576 mice results in increased Aβ deposition. A) Representative sections of brains of Tg2576 receiving folate deficient diet (Diet group, n = 5), or vehicle (Ctrl group, n = 6) immunostained with 4G8 antibody. B) Quantification of the area occupied by Aβ immunoreactivity in hippocampus and somatosensory cortex (SSC) of Tg2576. Values represent mean ± S.E.M. *P < 0.05.
HHcy and APP metabolism in Tg2576 mice brain
Since we found that diet-induced HHcy resulted in an increased Aβ levels and deposition, we then focused on possible mechanism(s) responsible for this effect. First, we assessed the steady-state levels of APP and its cleavage products. Compared with brain homogenates from the control group, total APP level from the mice with HHcy was not different (Figure 4). Similarly, steady state levels of the β-site APP cleaving enzyme (BACE-1), and sAPPβ (Figure 4), which represent the β-secretase pathway, were unaltered in the diet group when compared with controls. Diet-induced HHcy did not result in any significantly alteration of the α-secretase pathway either, as measured by the sAPPα, C-terminal fragment-α (CTF-α; C83) and ADAM-10 levels, when compared with controls (Figure 4). Further, two major components of the γ-secretase complex, PS1 and Nicastrin, showed no difference between two groups (Figure 4). However, C-terminal fragment-β (CTF-β; C99) level was statistically significant lower in the brains of the diet group (Figure 4). We also analyzed two of the major proteases involved in Aβ degradation, i.e., insulin-degrading enzyme (IDE) and neprilysin (NEP) (Leissring et al., 2003). Steady-state protein levels of IDE and neprilysin measured by western blot were similar between the two groups of animals (Figure 5). The same was also valid for apoE, a chaperone protein which has been involved in the transport of Aβ from the brain into the circulation (Figure 5).
Figure 4.
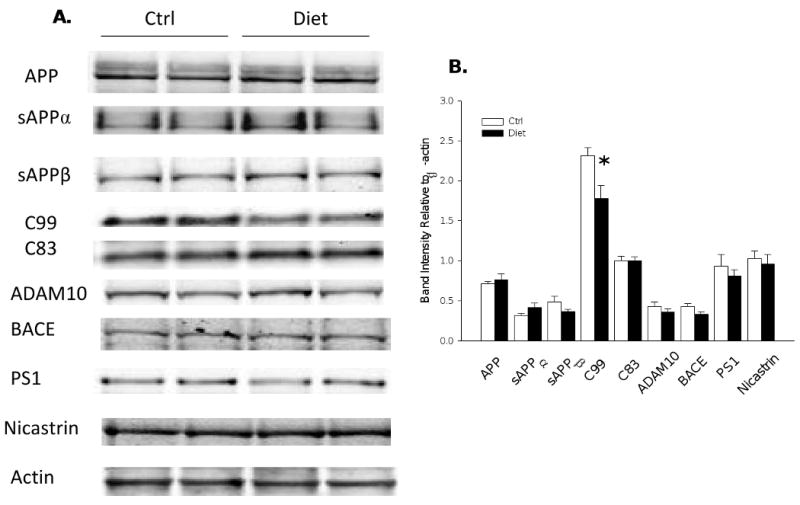
APP metabolism in Tg2576 mice with diet-induced HHcy A) Representative western blots of APP, sAPPα, sAPPβ, CTFs (C99 and C83), ADAM10, BACE1, PS1 and Nicastrin in brain homogenates from Diet group (n = 6) or Ctrl group (n = 7). B) Densitometric analyses of the immunoreactivities to the antibodies shown in panel A (white bars: Ctrl group; black bars: Diet group). Values represent mean ± S.E.M. *P < 0.05.
Figure 5.
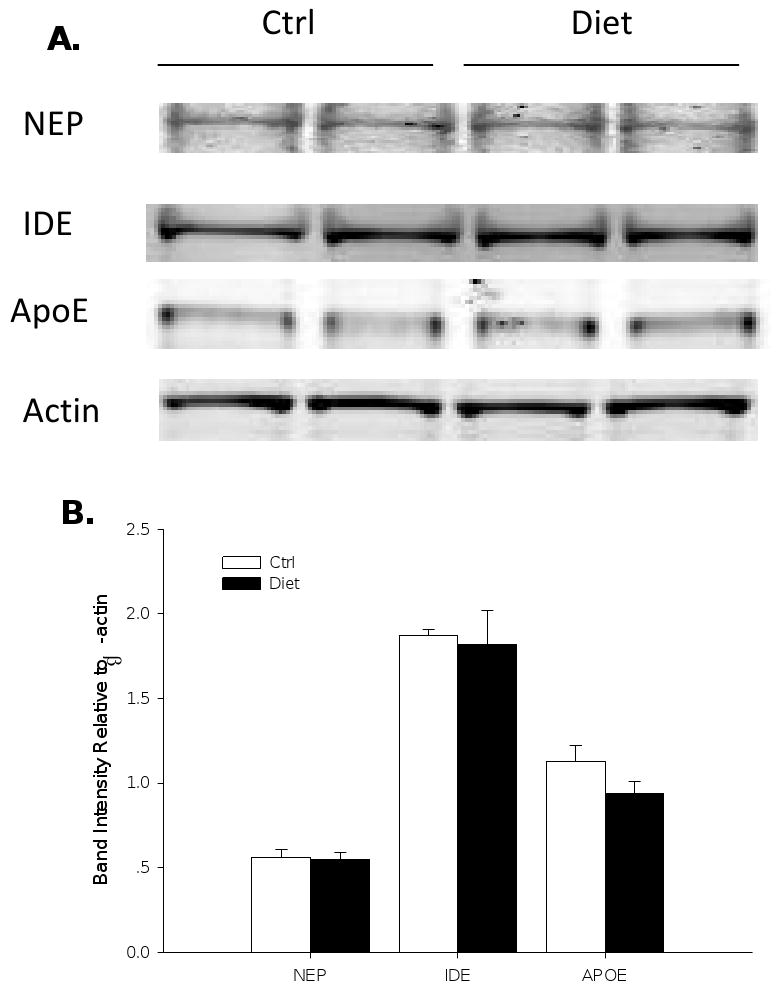
Protein levels of neprilysin (NEP), IDE and apoE in Tg2576 mice with diet-induced HHcy. A) Representative western blots of NEP, IDE and apoE in brain homogenates from Tg2576 on control diet (Ctrl group, n = 7) or folate deficient diet (Diet group, n = 6). B) Densitometric analyses of the immunoreactivities to the antibodies shown in panel A (white bars: Ctrl group; black bars: Diet group). Values represent mean ± S.E.M
Discussion
The current study investigated the effect of diet-induced HHcy on amyloidogenesis in an AD-like mouse model. Plenty of studies have reported the association between HHcy and AD (McCaddon et al., 1998; Quadri et al., 2004; Ravaglia et al., 2005), and longitudinal observations found that the effect of HHcy on AD is independent of several other confounders (Seshadri et al., 2002). However, conflicting results have also been reported (Luchsinger et al., 2007; Morris et al., 2006). Different mechanisms underlying deleterious effect of the Hcy on CNS have been proposed to explain the biological connection between HHcy and AD pathogenesis (Boldyrev and Johnson, 2007; Jacobsen, 2000; Kruman et al., 2002). However, the effect of HHcy on APP metabolism is still not fully elucidated. In this study, HHcy was induced in Tg2576 mice by feeding them with a folate, vitamin B6 and vitamin B12 deficient diet, which is a well-established model to induce HHcy (Hofmann et al., 2001). After 7 months on this diet, Tg2576 had a significant increase in their Hcy levels, which reached about 30μM. These values are lower than the ones reported in other studies with a similar diet (Fuso et al., 2008; Troen et al., 2003), and it is possible that, since our diet had no sulfa drugs added, some folate was formed by gut bacteria in our mice. Interestingly, the relatively mild plasma Hcy increase we observed in our model is within the range of Hcy levels observed in elderly individuals (5.4 to 61.1μM) (Seshadri et al., 2002).
In the current study, diet-induced HHcy associated with a significant increase in the Aβ levels and Aβ deposition in the Tg2576 mice brain. In particular, Aβ1-40 was significantly elevated in the hippocampus, while Aβ1-42 increased in the cortex of the diet group, when compared to control group. These Aβ increases were also confirmed by an immunohistochemical approach, where we found significantly more Aβ deposition in the hippocampus and cortex of the deficient diet-treated Tg2576 mice. In search for possible mechanism of this in vivo effect, we investigated both the APP proteolytic and Aβ catalytic pathways in the mice brains. Protein levels of total APP showed no difference between two groups. Furthermore, both α- and β-secretase metabolic pathways remained unaltered in the diet group as the protein levels of sAPPα, sAPPβ, C83, ADAM10 and BACE1 were the same as for the control group. When we looked at the γ-secretase pathway, we found no difference in the protein levels of two major components of this complex, PS1 and Nicastrin. However, C99 level in the diet group was significant lower than in the control group. Given the fact that APP and sAPPβ remained unaltered, we speculate that the decrease C99 levels is probably due to an elevated γ-secretase activity, secondary to diet-induced HHcy. Interestingly, previous studies found that γ-secretase activity can be modulated without altering its main components protein levels, but by altering its distribution within sub-cellular compartments such as lipid raft (Osenkowski et al., 2008). Thus, our negative findings on any changes in PS1 and Nicastrin protein levels by western blot can not exclude the possible alteration of this secretase activity through ways such as redistribution of this secretase in lipid rafts. Further studies are warranted to address this issue.
To rule out other possible mechanisms responsible for the Aβ changes in the diet group, we analyzed the protein levels of IDE, neprilysin and apoE and find no changes in any of these proteins which are involved in Aβ clearance (Guenette, 2003). Previously, it has been reported that HHcy can stimulate cholesterol metabolism via up-regulation of the HMG-CoA reductase (Li et al., 2002). Since high cholesterol has been shown to increase brain Aβ levels in another animal model of AD (Refolo et al., 2001), it was important for us to check the cholesterol levels in the animals administered with the deficient diet. However, we found no difference between the two groups of mice, suggesting that this factor is not responsible for the elevated Aβ levels and deposition observed in our study. Previous studies have investigated the effect of vitamin B deficiency diets in AD mouse models. Bernardo et al. administered a diet deficient in folate, chlorine and methionine to 16 ∼ 18 months old Tg2576 mice for 6 months, which resulted in cognitive deficits (Bernardo et al., 2007). However, they failed to observe any difference in amyloid aggregation in brain slides by using Thioflavin-S staining, which only detect β sheet structure in amyloid plaques (LeVine, 1999). This fact could explain the difference with our study, where 4G8, which specifically detect Aβ 17-24 amino acid, was used. In another study, 3 week-old TgCRND8 mice were fed with a vitamin B deficiency diet containing 1% sulfa drug for 45-60 days, which resulted in an increase in both Aβ levels and Aβ deposition as observed in the current study (Fuso et al., 2008; Fuso et al., 2009). However, by contrast with our study they found that mRNA and protein levels of both PS1 and BACE were elevated in diet-treated animals. These discrepancies could be explained by the use of two different AD animal models. Unlike Tg2576, the TgCRND8 mouse, which over-expresses APP double mutation, manifests a much rapid development of Aβ deposition (3 versus 12 months) (Chishti et al., 2001). Moreover, compared with our study the plasma Hcy levels reached in the TgCRND8 mice were extremely high (∼ 400μM) after 60 days diet treatment. This difference could also be largely due to the mouse strain difference, because when C57B6 mice received similar treatment as the TgCRND8 mice (considering diet, feeding duration, age, gender etc.), they showed a plasma Hcy levels of approximately 35.2μM, which are close to what we observed in the current study (∼30 μM) (Troen et al., 2008).
Diet-induced HHcy has been associated with different biological effects including oxidative stress, excitotoxcity, and energy metabolism imbalance (Matte et al., 2009; McCully, 2009; Song et al., 2009; Streck et al., 2003a; Streck et al., 2003b; Vogel et al., 2009). Some reports have shown that HHcy may promote Aβ production through increased oxidative stress (Frederikse et al., 1996; Mazur-Kolecka et al., 2004). To this end, we assayed lipid peroxidation levels in the brains of these mice, but despite a trend towards increase in the diet-treated animals, the difference between the two groups did not reach statistical significance.
In summary, our study demonstrates that a folate, vitamin B6, and B12 deficient diet induces HHcy in an AD-like mouse model, and associates with a significant acceleration of their amyloidotic phenotype. Since Aβ plays a central role in AD pathogenesis, our results further support the concept that dietary factors could contribute to the AD neuropathology.
Acknowledgments
This study was supported by a grant from the National Institute of Health, AG-22512 (D.P.), and the Alzheimer's Association (D.P.). We thank Ms. Jennie Meng for her technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. Jama. 2002;288:2015–22. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. Jama. 1995;274:1049–57. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998;55:1449–55. doi: 10.1001/archneur.55.11.1449. [DOI] [PubMed] [Google Scholar]
- Leblhuber F, Walli J, Artner-Dworzak E, Vrecko K, Widner B, Reibnegger G, Fuchs D. Hyperhomocysteinemia in dementia. J Neural Transm. 2000;107:1469–74. doi: 10.1007/s007020070010. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. 2002;346:476–83. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–9. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A, Paskavitz J, Remington R, Rasmussen S, Shea TB. Efficacy of a vitamin/nutriceutical formulation for early-stage Alzheimer's disease: a 1-year, open-label pilot study with an 16-month caregiver extension. Am J Alzheimers Dis Other Demen. 2008;23:571–85. doi: 10.1177/1533317508325093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicker L, Martins RN, Thomas J, Acres J, Taddei K, Vasikaran SD, Norman P, Jamrozik K, Almeida OP. B-vitamins reduce plasma levels of beta amyloid. Neurobiol Aging. 2008;29:303–5. doi: 10.1016/j.neurobiolaging.2006.10.007. [DOI] [PubMed] [Google Scholar]
- McCaddon A, Davies G, Hudson P, Tandy S, Cattell H. Total serum homocysteine in senile dementia of Alzheimer type. Int J Geriatr Psychiatry. 1998;13:235–9. doi: 10.1002/(sici)1099-1166(199804)13:4<235::aid-gps761>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Morris MS. Homocysteine and Alzheimer's disease. Lancet Neurol. 2003;2:425–8. doi: 10.1016/s1474-4422(03)00438-1. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA, Tang MX, Miller J, Green R, Mehta PD, Mayeux R. Relation of plasma homocysteine to plasma amyloid beta levels. Neurochem Res. 2007;32:775–81. doi: 10.1007/s11064-006-9207-7. [DOI] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Schneider JA, Tangney CC, Bienias JL, Aggarwal NT. Dietary folate and vitamins B-12 and B-6 not associated with incident Alzheimer's disease. J Alzheimers Dis. 2006;9:435–43. doi: 10.3233/jad-2006-9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldyrev AA, Johnson P. Homocysteine and its derivatives as possible modulators of neuronal and non-neuronal cell glutamate receptors in Alzheimer's disease. J Alzheimers Dis. 2007;11:219–28. doi: 10.3233/jad-2007-11209. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1997;94:5923–8. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen DW. Hyperhomocysteinemia and oxidative stress: time for a reality check? Arterioscler Thromb Vasc Biol. 2000;20:1182–4. doi: 10.1161/01.atv.20.5.1182. [DOI] [PubMed] [Google Scholar]
- Fuso A, Seminara L, Cavallaro RA, D'Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. J Neurosci. 2002;22:1752–62. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troen AM, Shea-Budgell M, Shukitt-Hale B, Smith DE, Selhub J, Rosenberg IH. B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc Natl Acad Sci U S A. 2008;105:12474–9. doi: 10.1073/pnas.0805350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco-Quinto J, Rodriguez de Turco EB, DeRosa S, Howard A, Cruz-Sanchez F, Sambamurti K, Refolo L, Petanceska S, Pappolla MA. Hyperhomocysteinemic Alzheimer's mouse model of amyloidosis shows increased brain amyloid beta peptide levels. Neurobiol Dis. 2006;22:651–6. doi: 10.1016/j.nbd.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Zhang CE, Wei W, Liu YH, Peng JH, Tian Q, Liu GP, Zhang Y, Wang JZ. Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am J Pathol. 2009;174:1481–91. doi: 10.2353/ajpath.2009.081036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo A, McCord M, Troen AM, Allison JD, McDonald MP. Impaired spatial memory in APP-overexpressing mice on a homocysteinemia-inducing diet. Neurobiol Aging. 2007;28:1195–205. doi: 10.1016/j.neurobiolaging.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Chan A, Shea TB. Folate deprivation increases presenilin expression, gamma-secretase activity, and Abeta levels in murine brain: potentiation by ApoE deficiency and alleviation by dietary S-adenosyl methionine. J Neurochem. 2007;102:753–60. doi: 10.1111/j.1471-4159.2007.04589.x. [DOI] [PubMed] [Google Scholar]
- Chan A, Tchantchou F, Rogers EJ, Shea TB. Dietary deficiency increases presenilin expression, gamma-secretase activity, and Abeta levels: potentiation by ApoE genotype and alleviation by S-adenosyl methionine. J Neurochem. 2009;110:831–6. doi: 10.1111/j.1471-4159.2009.06177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D'Anselmi F, Coluccia P, Calamandrei G, Scarpa S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol Cell Neurosci. 2008;37:731–46. doi: 10.1016/j.mcn.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, Scarpa S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Troen AM, Lutgens E, Smith DE, Rosenberg IH, Selhub J. The atherogenic effect of excess methionine intake. Proc Natl Acad Sci U S A. 2003;100:15089–94. doi: 10.1073/pnas.2436385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Jr, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–83. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–81. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Pratico D. 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer's disease. Faseb J. 2008;22:1169–78. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Pratico D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease. Faseb J. 2004a;18:323–5. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, Trojanowski JQ, Lee VM, Pratico D. Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer's disease. Am J Pathol. 2004b;165:2197–206. doi: 10.1016/s0002-9440(10)63269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer CM, Twite D, Shih J, Holets-McCormack SR, Gunter EW. Method comparison for total plasma homocysteine between the Abbott IMx analyzer and an HPLC assay with internal standardization. Clin Chem. 1999;45:152–3. [PubMed] [Google Scholar]
- Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–7. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Quadri P, Fragiacomo C, Pezzati R, Zanda E, Forloni G, Tettamanti M, Lucca U. Homocysteine, folate, and vitamin B-12 in mild cognitive impairment, Alzheimer disease, and vascular dementia. Am J Clin Nutr. 2004;80:114–22. doi: 10.1093/ajcn/80.1.114. [DOI] [PubMed] [Google Scholar]
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E, Licastro F. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82:636–43. doi: 10.1093/ajcn.82.3.636. [DOI] [PubMed] [Google Scholar]
- Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–40. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenette SY. Mechanisms of Abeta clearance and catabolism. Neuromolecular Med. 2003;4:147–60. doi: 10.1385/NMM:4:3:147. [DOI] [PubMed] [Google Scholar]
- Li H, Lewis A, Brodsky S, Rieger R, Iden C, Goligorsky MS. Homocysteine induces 3-hydroxy-3-methylglutaryl coenzyme a reductase in vascular endothelial cells: a mechanism for development of atherosclerosis? Circulation. 2002;105:1037–43. doi: 10.1161/hc0902.104713. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2001;8:890–9. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- LeVine H., 3rd Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–84. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–70. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Matte C, Mackedanz V, Stefanello FM, Scherer EB, Andreazza AC, Zanotto C, Moro AM, Garcia SC, Goncalves CA, Erdtmann B, Salvador M, Wyse AT. Chronic hyperhomocysteinemia alters antioxidant defenses and increases DNA damage in brain and blood of rats: protective effect of folic acid. Neurochem Int. 2009;54:7–13. doi: 10.1016/j.neuint.2008.08.011. [DOI] [PubMed] [Google Scholar]
- McCully KS. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann Clin Lab Sci. 2009;39:219–32. [PubMed] [Google Scholar]
- Song F, Poljak A, Smythe GA, Sachdev P. Plasma biomarkers for mild cognitive impairment and Alzheimer's disease. Brain Res Rev. 2009;61:69–80. doi: 10.1016/j.brainresrev.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Streck EL, Delwing D, Tagliari B, Matte C, Wannmacher CM, Wajner M, Wyse AT. Brain energy metabolism is compromised by the metabolites accumulating in homocystinuria. Neurochem Int. 2003a;43:597–602. doi: 10.1016/s0197-0186(02)00230-9. [DOI] [PubMed] [Google Scholar]
- Streck EL, Matte C, Vieira PS, Calcagnotto T, Wannmacher CM, Wajner M, Wyse AT. Impairment of energy metabolism in hippocampus of rats subjected to chemically-induced hyperhomocysteinemia. Biochim Biophys Acta. 2003b;1637:187–92. doi: 10.1016/s0925-4439(03)00019-x. [DOI] [PubMed] [Google Scholar]
- Vogel T, Dali-Youcef N, Kaltenbach G, Andres E. Homocysteine, vitamin B12, folate and cognitive functions: a systematic and critical review of the literature. Int J Clin Pract. 2009;63:1061–7. doi: 10.1111/j.1742-1241.2009.02026.x. [DOI] [PubMed] [Google Scholar]
- Frederikse PH, Garland D, Zigler JS, Jr, Piatigorsky J. Oxidative stress increases production of beta-amyloid precursor protein and beta-amyloid (Abeta) in mammalian lenses, and Abeta has toxic effects on lens epithelial cells. J Biol Chem. 1996;271:10169–74. doi: 10.1074/jbc.271.17.10169. [DOI] [PubMed] [Google Scholar]
- Mazur-Kolecka B, Kowal D, Sukontasup T, Dickson D, Frackowiak J. The effect of oxidative stress on amyloid precursor protein processing in cells engaged in beta-amyloidosis is related to apolipoprotein E genotype. Acta Neuropathol. 2004;108:287–94. doi: 10.1007/s00401-004-0890-7. [DOI] [PubMed] [Google Scholar]