

Abstract
The acyloxyalkyl derivatives of a model anti-HBV dinucleotide were synthesized and evaluated as orally bioavailable prodrugs. Our studies have led to the identification of the first orally bioavailable dinucleotide prodrugs for further therapeutic development against the hepatitis B virus (HBV).
Acute and chronic liver infections caused by Hepatitis B virus (HBV) constitute a major worldwide public health crisis affecting nearly 2 billion people including 1.7 million in the US. There are an estimated 350 million chronic carriers of HBV worldwide of which, about one million people die each year from chronic HBV.1 Cirrhotic and liver cancer patients, as well as, significant numbers of liver transplant recipients have a continued need for effective anti-HBV therapy. Although several anti-HBV drugs have been approved for clinical use,2,3 significant unmet medical need exists due to the emergence of antiviral resistance, as well as, dose-limiting toxicities. Hence, there is a need to develop safer and more effective anti-HBV drugs, with novel mechanisms of action that can be used alone, and in combination with other drugs.
We have reported that phosphorothioate dinucleotides and trinucleotides are a new and novel class of anti-HBV compounds with potent activity in vitro and in vivo.4 Our earlier studies, using 35S-labeled compounds in rats had, however, revealed that these compounds have negligible oral bioavailability. Presumably, the hydrophilic nature and negative charge(s) on the internucleotidic backbone of the nucleotide structure hinders their oral absorption. Recently, we had reported5 the design, synthesis, and in vitro evaluations of a few prodrug analogs, including the acyloxyalkylester derivatives,6,7 of the model anti-HBV dinucleotide [Rp,Sp]-3′-dA-ps-U2′OMe (1). In continuing investigations, we carried out additional preclinical studies on two acyloxyalkyl prodrugs 2 and 3 derived from 1. The results are summarized below.
The target prodrug analogs 2 and 3 were synthesized by chemoselective S-alkylation of 1 using the corresponding iodomethyl intermediates (Scheme 1).8 In turn, the iodomethyl acylates were prepared by halogen exchange reaction with the corresponding chloro-compounds.9 The requisite phosphorothioate dinucleotide 1, needed for the preparation of the prodrugs [Rp,Sp]-2 and [Rp,Sp]-3, were synthesized in 8 to 10 mmol scale by solid-phase phosphoramidite chemistry10 on controlled pore-glass (CPG) support using the LOTUS reactor.11,12
Scheme 1.

Synthesis of the dinucleotide prodrugs 2 and 3.
We carried out detailed studies of the in vitro conversion of the prodrugs 2 and 3 to the dinucleotide 1, wherein each prodrug (as Rp, Sp mixture) was exposed to mouse, rabbit, woodchuck and human serum according to previously described procedures.5 In all cases, both isomers of each of the prodrugs 2 and 3 underwent serum-mediated stereospecific conversion13 to the active 1 with only minor amounts of desulfurized product (PO) [corresponding to 1] being observed. It was also gratifying to find that, in each instance, there were no significant rate differences between the individual Rp and Sp isomers of 2 and 3 in their conversion to 1. It is conceivable that the formation of 1 occurs by nucleophilic attack of the enzyme on the ester carbonyl to give 4 (Scheme 2), followed by the interception of the resulting oxyanion by the juxtapositioned phosphoryl group to give the cyclic intermediate 5. In turn, 5 could reorganize to give the trigonal bipyramidal intermediates 6 and 7, which could interconvert by pseudorotation.14 Although in the presumed intermediate 7, the S-acyloxyalkyl group is favorably predisposed to depart from an apical direction [that could have resulted in the formation of desulfurized product of dinucleotide 1], its formation is fortuitously hampered because of the need for considerable reorganization of the initially formed enzyme-substrate complex 6. Consequently, the hydrolytic pathway is directed to occur via 6 to yield the desired 1 with minimal formation of a desulfurized product. Furthermore, it appears that the presence of the 2′-OMe substituent in the dinucleotide structure facilitates hydrolysis of the ester group in each of the isomers with almost equal ease. Presumably, the 2′-OMe substitution in a deoxy-ribofuranose ring that favors a C3′-endo compared to C2′-endo sugar puckering conformation might optimally juxtaposition the ester group in both isomers for enzyme-mediated nucleophilic attack with equal ease. Indeed, in contrast to prodrug analogs 2 and 3, bioreversibility studies of the corresponding derivatives 8-10 derived from the dinucleotides, TpsT, and dA-ps-T (both of which lack the 2′-OMe substituent), revealed that there were significant differences in the rates of hydrolysis of the individual Rp and Sp isomers13 of 8-10 implying thereby that subtle conformational effects are in play in enzyme-mediated hydrolysis of nucleotide prodrugs. The bioconversion of 2 and 3 to 1 is also consistent with broad substrate specificity associated with the ubiquitous esterase enzymes.
Scheme 2.

Proposed mechanism of bioreversibility of the prodrug 3 to the active dinucleotide 1.
The cytotoxicity potential of the prodrugs 2 and 3 were evaluated using a panel of cell lines including MDBK, Vero and HFF using the protocols reported earlier.5 As in the case of the active 1, both prodrugs 2 and 3 had CC50 > 1000 μM thereby demonstrating excellent safety profile.
The prodrug 3 was evaluated for anti-HBV activity in cell-based assays using the HepG2.2.15 cell lines with Lamivudine (3TC) and Adefovir Dipivoxil (ADV) as positive controls (Table 1) as per previously reported assay protocols.4a The compound 3 was evaluated against wild type HBV, as well as, 3TC-, and ADV-resistant mutants - HBV-3TC-R1 to HBV-3TC-R4 and HBV-ADV-R1 respectively (Table 1). As can be seen, 3 was quite active against wild type HBV with potency similar to that of ADV and against all tested Lamivudine-, and ADV-resistant HBV mutants. Similar studies with prodrug 2 are planned.
Table 1.
Anti-HBV activity of the prodrug 3 against wild-type and drug-resistant HBV mutants.
| Virus | Strain | EC50 of 3 (μM) |
EC90 of 3 (μM) |
Control: 3TC (μM) | Control: ADV (μM) | ||
|---|---|---|---|---|---|---|---|
| EC50 | EC90 | EC50 | EC90 | ||||
| HBV | WT | 2.5 | 8.5 | 0.2 | 0.7 | 1.5 | 7 |
| HBV-3TC-R1 | L180M | 2.1 | 8.6 | 5.3 | 20 | 2.1 | 8.2 |
| HBV-3TC-R2 | M204V | 2.4 | 8.3 | >100 | >100 | 1.8 | 7.2 |
| HBV-3TC-R3 | M204I | 3 | 9.2 | >100 | >100 | 2 | 8 |
| HBV-3TC-R4 | LMMV | 3.1 | 9.3 | >100 | >100 | 2.2 | 8.3 |
| HBV-ADV-R1 | N236T | 2.8 | 8.7 | 0.2 | 0.8 | 7.5 | 29 |
An important criterion for oral bioavailability is the requirement for adequate stability of the compounds in gastric fluid before absorption can occur. Given the known susceptibility of nucleosides and nucleotides to acid-catalyzed decomposition, it was not known whether the prodrugs 2 and 3 would have adequate stability in the acidic environment of stomach. Therefore, we evaluated the stability of prodrugs and the dinucleotide 1 in simulated gastric fluid (SGF), as well as, in simulated intestinal fluid (SIF).15,16 It was found that the dinucleotide 1 decomposed rapidly in SGF (t1/2 < 15 min), but was relatively stable in SIF. However, we were gratified to find that both of the S-alkyl prodrugs 2 and 3 displayed high stability in SGF with t1/2 > 1 h, where as in SIF, both the analogs completely converted to 1 in ∼ one hour.17 Our stability data on prodrugs of 1 is consistent with that reported for SATE pronucleotides.18 On the basis of the stability data, coupled with cytotoxicity, and bioreversibility profile, the prodrug analogs 2 and 3 were selected for oral bioavailability studies in vivo.
One of the primary objectives of the oral bioavailability studies was to determine the disposition of the prodrugs and the active dinucleotide 1 in the liver - the target organ for HBV. The disposition of the compounds in the liver was expected to provide a better assessment of the pharmacological bioavailability of the compounds. The analogs 2 and 3 were administered by oral gavage to male Swiss Webster mice. At different time points of 5, 15, 30, and 60 min, the animals were euthanized and blood and tissues were collected. Samples of the blood and liver were processed and subjected to analysis by TLC, HPLC, and MALDI-TOF mass spectrum.19 Typically, in the case of 2, at early time points (5 and 15 min), the presence of the prodrug was detected in plasma and at later time points, mostly the dinucleotide 1 in liver was observed. In the case of 3, only the dinucleotide 1 could be detected in plasma (early time points) and in liver. These in vivo results are consistent with the in vitro bioreversibility studies of the analogs in serum. Independently, Absorption, Distribution, Metabolism and Elimination (ADME) studies were conducted in male and female Sprague-Dawley rats by iv and po administration of 35S-labeled 3. Significant radioactivity was noted in the liver and plasma at different time points with the mean ratio of liver to plasma concentration of radioactivity being as high as 3.2 (iv route) and 3.5 (po route), 1 h after dose administration. Hence, it appears that in general, following their absorption, the acyloxyalkyl prodrugs 2 and 3 are rapidly converted to the active 1 in the serum, which is then distributed into the liver, the target organ. Given that the prodrugs had higher stability in SGF compared to SIF, it is possible that following the oral gavage, their absorption occurs in the upper duodenal region facilitated either by carrier-mediated transport mechanism20 or by passive diffusion. Furthermore, it is likely that esterases mediate the hydrolytic conversion of the prodrugs to 1.
Pharmacodynamic evaluations21 of 2 and 3 were carried out initially at high doses to assess both efficacy, as well as, potential toxicity in the HBV transgenic mouse model. In the initial dose-finding study, the compounds 2 and 3 were administered by oral gavage at a high dose of 300 to 400 mg/kg once daily for 14 days. Analysis of the liver HBV DNA by Southern blot hybridization and PCR revealed that the prodrug analogs 2 and 3 had strong anti-HBV activity (Figure 1, Panel A). The prodrug 3 was slightly more potent than 2. There was no apparent evidence of any toxicity. Subsequently, dose-ranging studies were carried out in the HBV transgenic mouse model using the prodrug 3 at doses of 1, 5, 10, and 100-mg/kg once daily po for 14 days. As shown, dose-dependent reduction of the liver HBV DNA was observed in mice that received the prodrug compared to placebo as evaluated by Southern Blot analysis of the liver of treated animals (Figure 1, Panel B). In all cases, ADV was used as a positive control.
Fig 1.
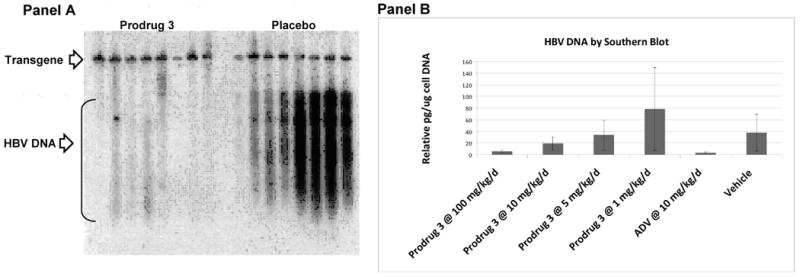
Panel A. Southern blot analysis of HBV DNA of HBV transgenic mice treated with 300 mg/kg of compound 3 daily po for 14 days. Panel B. Plot of liver HBV DNA of HBV transgenic mice treated with 1, 5, 10, and 100 mg/kg of compound 3 for 14 days.
In conclusion, we report here the successful development of orally bioavailable prodrugs of a model anti-HBV dinucleotide. The dinucleotide prodrugs reported herein represent a first-in-class antiviral agent for the treatment of HBV infections. Evaluations of other prodrugs of 1 and toxicology studies are in progress and the results will be published in due course.
Novel acyloxyalkyl dinucleotide prodrugs have been developed as orally bioavailable anti-HBV agents.
Acknowledgments
Support of this research from the National Institutes of Health, under a Research Project Cooperative Agreement Grant Award UO1 AI058270 (RPI, PI) and NIH contract NO1-AI-50036 for (JDM, PI) is gratefully acknowledged. The ADME studies were conducted by SRI International, Menlo Park, California and were supported by NIAID Contract N01-AI-60011 to SRI (JM, PI). JEC and SP contributed equally to the project. We thank Dr. C. Griesberger of PPG fine chemicals for the generous gift of chloromethyl isopropyl carbonate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hepatitis B. Fact sheet WHO/204. Geneva: World Health Organization; Oct, 2000. http://www.who.int/inf-fs/en/fact204.htm. [Google Scholar]
- 2.For reviews see:; (a) Perrillo RP. Semin Liver Dis. 2004;24:23–29. doi: 10.1055/s-2004-828675. [DOI] [PubMed] [Google Scholar]; (b) Lavanchy D. J Viral Hepatitis. 2004;11:97. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 3.Reviewed in:; Iyer RP, Padmanabhan S, Zhang G, Morrey JD, Korba BE. Curr Opin Pharmacol. 2005;5:520. doi: 10.1016/j.coph.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 4.(a) Iyer RP, Jin Y, Roland A, Morrey JD, Mounir S, Korba B. Antimicrob Agents Chemother. 2004;48:2199. doi: 10.1128/AAC.48.6.2199-2205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Iyer RP, Roland A, Jin Y, Mounir S, Korba B, Julander JG, Morrey JD. Antimicrob Agents Chemother. 2004;48:2318. doi: 10.1128/AAC.48.6.2318-2320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Padmanabhan S, Coughlin JE, Zhang G, Kirk CJ, Iyer RP. Bioorg Med Chem Lett. 2006;16:1491. doi: 10.1016/j.bmcl.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 6.Srivastva DN, Farquhar D. Bioorg Chem. 1984;12:118. [Google Scholar]
- 7.Iyer RP, Phillips LR, Biddle JA, Thakker DR, Egan W, Aoki S, Mitsuya H. Tetrahedron Lett. 1989;30:7141. [Google Scholar]
- 8.S-Alkylations were carried out using lyophilized solid 1 with excess of alkylating reagents in aq. acetone or isopropanol. After the completion of reaction, the reaction mixture was concentrated, extracted with hexanes to remove the excess of iodomethyl compounds, lyophilized and purified using column chromatography. Alternatively, the reaction mixture after hexane extraction could be directly taken for HPLC purification. Yields of the purified prodrugs ranged from 44 to 51%. 31P NMR, DMSO-d6, δ ppm: 2, 28.1, 28.9; 3, 27.7, 28.6 ppm.
- 9.Halogen exchange reactions of chloromethyl esters were carried out with excess of sodium iodide (2 eq) in anhydrous acetonitrile at r.t. overnight in the dark. After removal of the solvent, the reaction mixture was extracted between water and DCM, the DCM layer was washed with sodium bisulfite solution (5%), followed by brine, and dried over anhydrous sodium sulfate. The organic layer was concentrated to give the iodomethyl esters; also see:; Iyer RP, Yu D, Ho NH, Agrawal S. Synth Commun. 1995;25:2739. [Google Scholar]
- 10.For a review, see:; Beaucage SL, Iyer RP. Tetrahedron. 1993;49:1925. [Google Scholar]
- 11.(a) Padmanabhan S, Coughlin JE, Iyer RP. Tetrahedron Lett. 2005;46:343. [Google Scholar]; The LOTUS Reactor® is a new chemical reaction chamber designed and built by us for solid-phase synthesis; see:; (b) Iyer RP, Coughlin JE, Padmanabhan S. Org Prep Proc Intl. 2005;37:205. doi: 10.1080/00304940509354949. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Iyer RP, Regan JB, Egan W, Beaucage SL. J Am Chem Soc. 1990;112:1253. [Google Scholar]
- 12.Recently, we have also developed efficient solution-phase synthesis, which provides access to larger quantities of 1 in each batch run (unpublished results).
-
13.Hydrolytic rates of Rp,Sp isomers of prodrug analogs 2-3, and the dinucleotide esters 8-10 were monitored in rabbit
Structure Compd R R1 X B1 B2 Rp:Sp
t = 0Rp:Sp
t = 3 h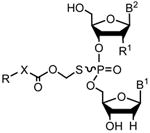
2 
-OCH3 - dA U 47:52 -* 3 
-OCH3 O dA U 49:50 41:58 8 
H - dA T 36:63 1:98 9 
H O dA T 47:52 10:89 10 
H O T T 60:39 1:98 *Complete hydrolysis occurred of both isomers
- 14.For excellent reviews on the concept of pseudorotation in the hydrolysis of cyclic phosphates, see:; Westheimer FH. Acc Chem Res. 1968;1:70. [Google Scholar]; (b) Singleton RJ., Jr J Chem Edu. 1973;50:538. doi: 10.1021/ed050p538. [DOI] [PubMed] [Google Scholar]
- 15.The United States Phamacopeia. 23rd. Vol. 18. The National Formulary; Rockville, MD: 1994. p. 2053. [Google Scholar]
- 16.Stability studies in SGF and SIF: 10 μL of each prodrug (2 mg in 200 μL of DMSO) were diluted with 190 μL of either SGF or SIF (prepared according to Ref. 15). The mixture was incubated at 37 °C in a water-bath for 1 h and at selected time points, each aliquot was treated with 200 μL methanol and adjusted to pH 6.0 by the addition of 500 μL of NH4OAc (0.1 M, pH 7.0) and 85 μL of NaOH (0.1 N). After standing for 5 min, the incubate was centrifuged, the supernatant concentrated in a speed vac, diluted with 300 μL of NH4OAc (0.1 M, pH 7.0) and analyzed by reversed-phase HPLC [Waters Instrument equipped with a 600E gradient controller and a 996 photodiode array detector with Millennium software]. An X-terra MS C-18 2.5 mm, 2.1 × 20 mm column and a gradient of 100% A to 100% B over 30 min of buffer A (0.1M, NH4OAc) and buffer B (80:20, CH3CN:NH4OAc) with a flow rate of 1 mL/min was employed. The retention times (R t) were: Rp,Sp 1, 14.3, and 14.6; Rp,Sp 2, 17.8; and Rp,Sp 3, 18.4 min respectively.
- 17.The decomposition of 1 in SGF was also monitored by 31P NMR For this purpose, initially, the 31P NMR spectrum of 1 in D2O (δ 58.6, 58.8 ppm) was recorded and spectra obtained at different time points following the sequential addition of pepsin and DCl as per the SGF composition in Ref.15. A significant decrease in the peak intensities at δ 58.6, 58.8 ppm was noted after 1h thereby suggesting decomposition of 1 in SGF.
- 18.Shafiee M, Deferme S, Villard AL, Egron D, Gosselin G, Imbach JL, Lioux T, Pompon A, Varray S, Aubertin AM, Van Den Mooter G, Kinget R, Périgaud C, Augustijns P. J Pharm Sci. 2001;90:448. doi: 10.1002/1520-6017(200104)90:4<448::aid-jps1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 19.Plasma and liver samples were processed and analyzed as before.5
- 20.de Vrueh RLA, Smith PL, Lee CP. J Pharmacol Exp Therap. 1998;286:1166. [PubMed] [Google Scholar]
- 21.The pharmacodynamic studies were conducted at USU (JDM, PI). For the pharmacodynamic studies,4b homozygous male and female transgenic HBV mice were used. The mice were originally obtained from Dr. Frank Chisari (Scripps Research Institute, LaJolla, CA) and were subsequently raised in the Biosafety Level 3 (BL-3) area of the AAALAC-accredited USU Laboratory Animal Research Center (LARC). Pre-experiment liver biopsies of mice were obtained and assayed for liver HBV DNA and subsequently block-randomized to the treatment groups (8 animals per group). The compounds were reconstituted with 0.05 M citric acid, pH 2.4 and the final dosing concentrations were such that 0.1 mL was delivered into a 20 g mouse. The animals were dosed orally once daily for 14 days. Following the final dosing, the animals were euthanized and the liver isolated, processed for Southern blot analysis to quantitate liver HBV DNA. Hybridization of the blots was carried out using a [32P]CTP-labeled, HBV genomic probe (digested with Hae III) cloned into the pBluescript plasmid (gift of Dr. Luca Guidotti, The Scripps Institute, LaJolla, CA) overnight at 60°C in a solution of 10% PEG-8000, 0.05 M NaPO4, 0.21 mg/ml salmon sperm DNA, and 7% SDS. The radioactive signals were measured using the phosphorimaging method (Optiquant). An image of the radioactive filter was exposed to a Cyclone™ Storage Phosphor Screen (Perkin Elmer Multisensitive Medium, Type MS PPN 7001723). The exposed screen was transferred to the Cyclone™ drum and read using the 600 dpi setting. The data was stored on a computer, and the density of the bands was measured. The ratio of the viral DNA bands to the transgene band was used to determine the concentration of viral DNA per host DNA. This calculation was based upon the knowledge that there were 1.3 copies of the transgene present per host cell with this line of transgenic mice (personal communication to JDM from F. Chisari). The transgene was used as an internal indicator to calculate the pg of HBV DNA per μg of cellular host DNA. The pg HBV DNA/μg host DNA was calculated.