

Abstract
Rationale
Loss of fibulin-4 during embryogenesis results in perinatal lethality due to aneurysm rupture, and defective elastic fiber assembly has been proposed as an underlying cause for the aneurysm phenotype. However, aneurysms are never seen in mice deficient for elastin, or for fibulin-5, which absence also leads to compromised elastic fibers.
Objective
We sought to determine the mechanism of aneurysm development in the absence of fibulin-4 and establish the role of fibulin-4 in aortic development.
Methods and Results
We generated germline and smooth muscle cell (SMC)-specific deletion of the fibulin-4 gene in mice (Fbln4GKO and Fbln4SMKO, respectively). Fbln4GKO and Fbln4SMKO aortic walls fail to fully differentiate, exhibiting reduced expression of SM-specific contractile genes and focal proliferation of SMCs accompanied by degenerative changes of the medial wall. Marked upregulation of ERK1/2 signaling pathway was observed in the aneurysmal wall of Fbln4GKO and Fbln4SMKO mice and both mutants developed aneurysm predominantly in the ascending thoracic aorta. In vitro, Fbln4GKO SMCs exhibit an immature SMC phenotype with a marked reduction of SM-myosin heavy chain and increased proliferative capacity.
Conclusion
The vascular phenotype in Fbln4 mutant mice is remarkably similar to a subset of human thoracic aortic aneurysms caused by mutations in SMC contractile genes. Our study provides a potential link between the intrinsic properties of SMCs and aneurysm progression in vivo and supports the dual role of fibulin-4 in the formation of elastic fibers as well as terminal differentiation and maturation of SMCs in the aortic wall.
Keywords: Aneurysms, ECM, smooth muscle cells, mouse, elastic fibers
Introduction
Aortic aneurysms are characterized by abnormal enlargements of the aorta caused by global or localized weakness of the vessel wall. Traditionally, aneurysm formation is believed to involve defects in synthesis or assembly of extracellular matrix (ECM) proteins or accelerated degradation of vascular ECM (reviewed in 1, 2). For example, mutations in the fibrillin-1 (FBN1) or type III alpha 1 collagen gene (COL3A1) genes are responsible for Marfan syndrome and vascular Ehlers-Danlos syndrome 3, 4, respectively. Mutations in Fbn1 profoundly affect the formation of microfibrils that surround an elastin core and are required for proper assembly of elastic fibers 5. Genetically-engineered mice provide further evidence that the vascular ECM is the critical determinant of aneurysm development, including an allelic series of Fbn1 mutant mice, and mice homozygous for null alleles of the genes encoding lysyl oxidase, a crosslinking enzyme for elastin and collagen, and ECM proteins such as fibulin-4 (Fbln4) and biglycan (Bgn) 6-9. In addition to the disruption of ECM in the vessel wall, alterations of SMC intracellular contractile proteins have recently been shown to be responsible for subsets of thoracic aneurysms in humans 10-12. Two heterozygous mutations in MYH11 (SM-MHC) were identified in kindreds presenting with thoracic aortic aneurysms and/or aortic dissections (TAADs) and patent ductus arteriosus. Heterozygous missense mutations in ACTA2 (SM α-actin) were also found in 14% of inherited TAADs 12. Thus, the pathogenesis of aortic aneurysm formation may involve abnormalities in homeostasis of vascular ECM proteins or abnormal SMC development, or both.
Fibulin-4 belongs to the fibulin family of ECM proteins consisting of seven known members characterized by tandem repeats of calcium-binding epidermal growth factor (cbEGF)-like motifs and a C-terminal fibulin module 13. In the aorta, fibulin-5 is expressed at one magnitude higher than fibulin-4 and its absence leads to compromised elastic fibers with aggregates of elastin, but does not develop aneurysms. 14, 15. Fbln4-null (Fbln4-/-) mice, in contrast, exhibit a more severe phenotype with perinatal lethality due to rupture of aortic aneurysms and marked disruption of elastic fibers 8. Genetic mutations in the FBLN4 have also been identified in two human cutis laxa patients, both of which exhibited aortic aneurysms 16, 17. Defective elastogenesis was proposed to be an underlying cause of aortic aneuryms in Fbln4-/- mice, however, elastin-null mice (Eln-/-) do not develop aortic aneurysms, rather they exhibit stenosis of the aorta due to subendothelial proliferation of SMCs 18. It is not clear, therefore, whether the phenotypic differences between Fbln4-/- and Fbln5-/- mice are due solely to the difference in severity of elastic fiber defects. It is possible that fibulin-4 has additional function(s) that act independent of, or in concert with, elastogenesis in the developing aorta. Recently, hypomorphic Fbln4R/R mice were shown to contain increased phosphorylated (p)-Smad2, enhanced production of CTGF and collagen fibers, and increased proliferation of SMCs in the aortic wall 19. However, the precise mechanism of upregulation of TGF-β in Fbln4R/R mice or whether it is a primary cause of the vascular phenotype is unclear, especially since SMC-specific ablation of TGF-β receptor type 2 causes aortic aneurysms by down-regulating elastogenic genes 20.
To determine the role of fibulin-4 in aneurysm formation, we generated mice with a germline deletion or vascular cell-specific deletion of Fbln4. SMC-specific, but not endothelial cell (EC)-specific knockout of Fbln4, resulted in the formation of large aneurysms exclusively in the ascending aorta. We found that SMCs failed to fully differentiate in the absence of fibulin-4 in the aorta as evidenced by down-regulation of SMC-specific contractile protein genes and an immature phenotype of Fbln4-null SMCs accompanied by focal hyperproliferation of SMCs and degenerative changes affecting the integrity of the vessel wall. These data highlight a novel role for fibulin-4 in the development of SMCs, and together with its role in the formation of elastic fibers, fibulin-4 functions to protect the aortic wall against aneurysm formation in vivo.
Materials and Methods
Mouse
Generation of conditional knockout mice is described in the expanded Materials and Methods sections online.
Mechanical stretch
Two or 4 × 105 cells were plated on 6-well silicon elastomer-bottomed culture plates covalently coated with type I collagen (Flexcell). After allowing cells to adhere to the plates (6-12 h), the media was changed to serum free and incubated for 24 h to synchronize the cell cycle prior to initiation of stretch stimulation. Cells were subjected to unidirectional cyclic strain (0.2 Hz and 20% strain) for 24 h in a humidified incubator with 5% CO2 at 37°C using FX-4000T Flexcell Tension Plus system (Flexcell International). Cells incubated in static condition served as controls.
Standard protocol was used for immunostaining, Western blotting, qPCR, gelatin zymography, histological and electron microscopic analysis and is available online.
Results
Germline and SMC-specific deletions of Fbln4 in vivo
To investigate aneurysm development in the absence of fibulin-4, we generated a conditional allele of Fbln4 using Cre-loxP technique in ES cells (Online Figure IA). Fbln4neo-loxP mice were mated with transgenic mice expressing the heat stable Flp-recombinase to generate mice homozygous for Fbln4loxP/loxP (Online Figures IB, C). Fbln4neo-loxP mice were also mated with CAG-Cre transgenic mice to delete Fbln4 in germline, and subsequently homozygous mice for null alleles were generated (termed Fbln4GKO). In concordance with the previous reported data 8, all Fbln4GKO mice died in or after parturition due to aneurysm rupture (Online Figure ID) and quantitative RT-PCR (qPCR) confirmed absence of Fbln4 in the aorta of Fbln4GKO mice (Online Figure IE).
Fbln4 is ubiquitously expressed during embryogenesis and is detected in both ECs and SMCs in the aorta (Online Figures IIA, IIB). To determine the cell type responsible for the aneurysm phenotype, we generated EC-specific and SMC-specific knockout mice for Fbln4 by mating Fbln4loxP/loxP mice with transgenic mice expressing Cre-recombinase under a control of the Tie2 promoter and SM22α promoter, respectively 21, 22. EC-specific knockout mice (Fbln4loxP/GKO;Tie2-Cre, termed Fbln4ECKO) were healthy and indistinguishable from control littermates. Histological finding of the aorta was normal (Online Figure III). In contrast, SMC-specific knockout mice (Fbln4loxP/GKO;SM22-Cre, termed Fbln4SMKO) began to die spontaneously at approximately 2 months of age despite absence of embryonic or neonatal lethality. Aortae harvested from adult Fbln4SMKO mice exhibited large aneurysms exclusively in the ascending aorta (Figure 1A). Aneurysms were observed in Fbln4SMKO mice with complete penetrance. The ductus arteriosus was closed and the ligamentous remnant was markedly elongated. qPCR confirmed a significant reduction of Fbln4 transcripts in the ascending aorta where the aneurysms developed, as well as in the descending thoracic aorta where only elongation was evident (Figure 1B). These data indicate that loss of SMC-derived fibulin-4 is responsible for the aneurysm formation and that fibulin-4 derived from other cell types compensated for aneurysm formation during embryonic life and perinatal lethality in Fbln4SMKO mice.
Figure 1.
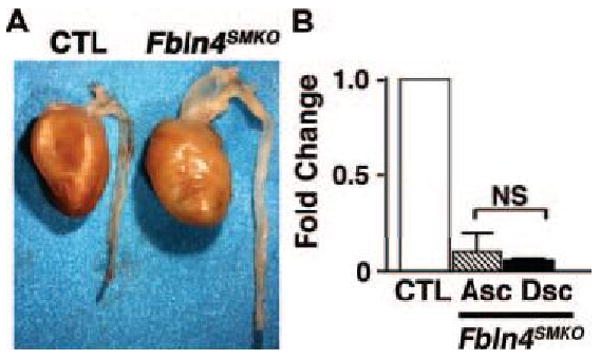
Aneurysms and tortuous aorta in Fbln4SMKO mice. A. Ascending thoracic aortic aneurysm in Fbln4SMKO mouse at P90. B. qPCR analysis of Fbln4 transcripts in the ascending (Asc) and descending (Dsc) thoracic aortae of Fbln4SMKO mice compared with corresponding controls (CTLs). Aortae from 4 different sibling pairs of CTL and Fbln4SMKO were analyzed in which CTL was considered as 1. Data represent means ± SD. NS, not significant.
Compromised elastic fibers, increased wall thickness and disarray of SMCs in the Fbln4GKO aorta
To better understand the pathological processes of aneurysm formation in the absence of Fbln4, we performed a developmental analysis of the ascending aorta in Fbln4GKO embryos. Consistent with a previous observation 8, elastic fibers were clearly detected in the aortic wall of wild-type embryos at E14.5, whereas elastic fibers were decreased in the aorta from Fbln4GKO embryos (Online Figures IVb, d). Despite compromised elastic fibers in the Fbln4GKO aorta, at this timepoint, the overall morphology of SMCs was indistinguishable from the wild-type (Online Figures IVa, IVc). From E15.5, elastic fibers increased progressively in the wild-type aorta and SMCs became spindle-shaped with elongated nuclei (Online Figures IVe-f, IVi-j). In the Fbln4GKO aorta, however, elastic fiber assembly remained defective and SMCs appeared immature with round nuclei (Online Figure IVg-h, IVk-l). Although not reported previously, eosin-positive focal degeneration and marked proliferation of SMCs were observed in aneurysmal lesions of the Fbln4GKO aorta at E17.5 (Online Figure IVo) and E18.5 (arrow in Figure 2b). The degenerative area was devoid of elastic fibers and did not stain with Alcian Blue-PAS, indicating no accumulation of polysaccharides or mucins (Figure 2d and Online Figure IVp, data not shown). Only a small numbers of neutrophils and mononuclear cells were detected around degenerative lesions and some infiltrating through the intima (Online Figure VAa). In advanced lesions with dissection, more infiltrates were observed within the vessel wall (Online Figure VAb). No difference in proMMP-2 or MMP-2 levels was observed between wild-type and Fbln4GKO descending thoracic aortae (Online Figure VB). Interestingly, this focal degeneration of the aortic wall was not detected in embryos younger than E17.5. Since Fbln4GKO mutants die in or after parturition, we further characterized the aortic wall at E18.5. A significant upregulation of p-ERK in ECs and SMCs was observed in the Fbln4GKO aorta (Figure 2f) and GRP78, a marker of the endoplasmic reticulum stress response, was upregulated in SMCs surrounding the degenerative area (Figure 2h). Increased apoptotic cells were also observed in both ECs and SMCs in degenerative areas (data not shown) as well as in advanced aneurysmal lesions (Figure 2j). These observations suggest that aneurysm development in Fbln4GKO mice involved not only defective elastic fibers but also abnormal SMCs with degenerative medial wall changes.
Figure 2.
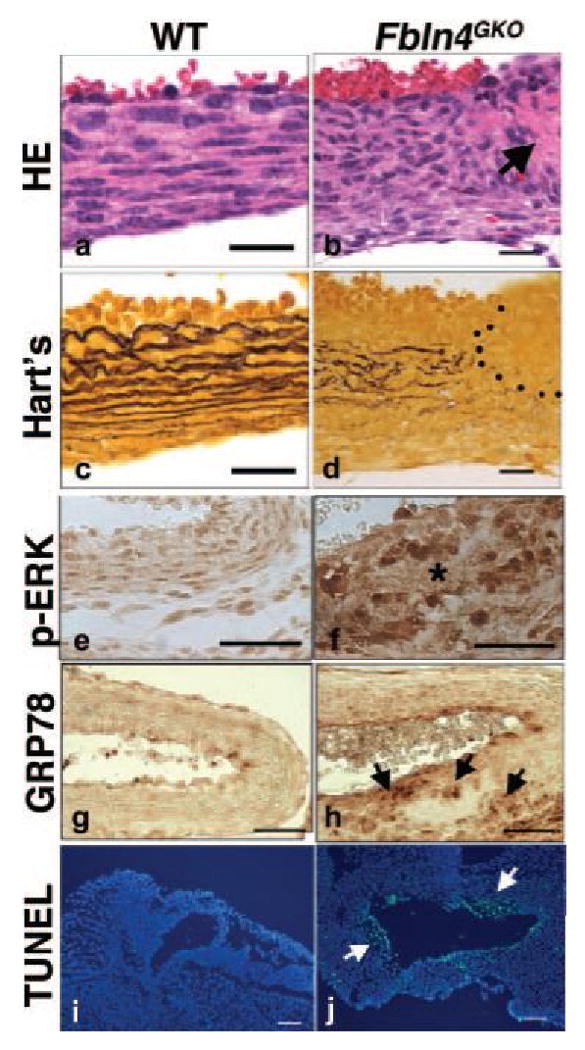
Various pathological changes associated with aneurysms in the Fbln4GKO aorta at E18.5. (a-d) Representative photos of HE and Hart's staining of transverse sections of the ascending aorta from wild-type (a, c) and Fbln4GKO (b, d) embryos (n=5 per genotype). Note focal degeneration of the medial wall in the mutant (arrow in b) devoid of elastic fibers (d). Bars indicate. (e-j) Characterization of aneurysmal lesions (n=3 per genotype). (e, f) p-ERK1/2 levels are increased in the Fbln4GKO aorta (f) compared to wild-type (e). Asterisk in f indicates degeneration of the medial wall. (g, h) Accumulation of GRP78-positive cells around hyaline degeneration in the Fbln4GKO aorta (arrows in h). (i, j) TUNEL staining shows numerous apoptotic cells within the aneurysmal wall, including the endothelial cells in the Fbln4GKO aorta (j). Bars in a-d indicate 20 μm, e-h indicate 40 μm and i-j indicate 100 μm.
Defects in terminal differentiation of SMCs in Fbln4GKO aorta
Histological abnormalities in SMCs of the Fbln4GKO aorta led us to further evaluate the SMC phenotype in Fbln4GKO embryos. Specifically, we examined expression of SMC differentiation markers in wild-type and Fbln4GKO descending thoracic aortae at E16.5. In the wild-type aorta, SM α-actin and SM-MHC, early and late SMC differentiation markers, respectively, were expressed throughout the aortic wall (Figures 3Aa and 3Ac). In the Fbln4GKO aorta, the intensity of both markers was not homogenous along the longitudinal axis of the aorta, particularly for SM-MHC (Figures 3Ab and 3Ad). At E18.5, SM α-actin and SM-MHC levels were increased in both wild-type and mutant aortae. The mutant aorta, however, still exhibited reduced marker expression compared to the wild-type aorta (Figures 3Ai-3Al). To confirm these findings, tissue extracts from the descending thoracic aorta devoid of aneurysm lesions from Fbln4GKO embryos were compared with those of wild-type embryos by Western analysis (Figure 3B). Consistent with tissue immunostaining, we observed significant decreases in SM-MHC and α-SM-actin expression. In addition, a marked increase in MAPK signaling pathways, including p-ERK1/2 and p-MEK1/2, was observed in mutant aortae (Figure 3B). Other components of MAPK pathways were only moderately increased in the mutants.
Figure 3.
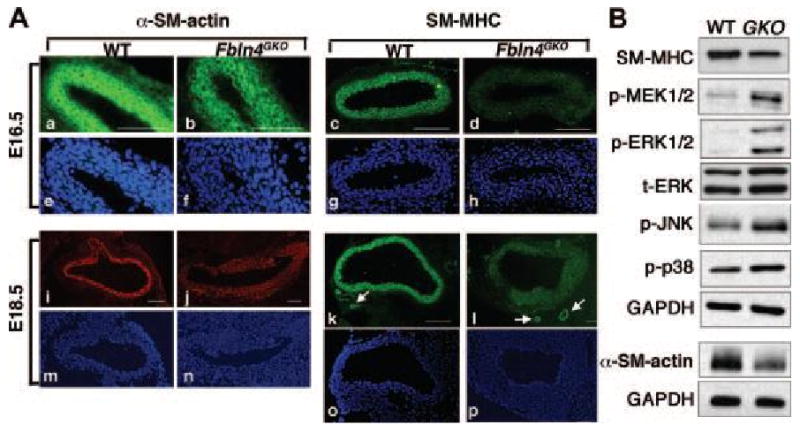
SMCs exhibit terminal differentiation defects in Fbln4GKO aorta. A. Representative transverse sections of the descending aorta from wild-type (n=3) and Fbln4GKO (n=3) embryos immunostained with SM α-actin and SM-MHC at E16.5 and E18.5. Corresponding DAPI staining is shown. At E16.5, when aneurysm formation is not evident, reduced SM α-actin and SM-MHC staining is already observed in the mutant (b, d) compared with the wild-type aorta (a, c). At E18.5, strong staining for SM α-actin and SM-MHC is observed in the wild-type aorta (i, k) whereas expression of both proteins is reduced in the mutants (j, l). Note comparative expression of SM-MHC in small vessels near the aorta in the wild-type and mutant (arrows in k and l). Bars indicate 100 μm. B. Representative Western blots of descending thoracic aorta extracts from E18.5 wild-type (n=3) and Fbln4GKO (n=3) embryos. For α-SM-actin, the experiments were done 2 times using 4 aortae per genotype. SM-MHC is reduced and stress-mediated kinase pathways p-ERK1/2 and p-MEK1/2 are increased in the Fbln4GKO aorta. p-JNK and p-p38 are moderately increased.
Disrupted wall structure and alterations of SMC gene expression in Fbln4GKO aorta
It has been shown that the aorta lacking elastin exhibits increased SMC proliferation and changes in SMC organization 18. We therefore examined aortic ultrastructure at the electron microscopic level in E18.5 embryos. In the wild-type aorta, near continuous elastic laminae were observed throughout the aortic wall, with those closer to the lumen being more completely formed at this time (Figures 4Aa, 4Ac). SMCs maintained close contacts with neighboring cells and developing elastic fibers. In contrast, discontinuous aggregates of elastin were observed in the Fbln4GKO aorta (Figure 4Ab, arrow). Higher magnification showed loose cell-cell associations and very few cell-elastin contacts with increased pericellular spaces in the Fbln4GKO aorta, exhibiting an immature vessel wall (Figure 4Ad).
Figure 4.
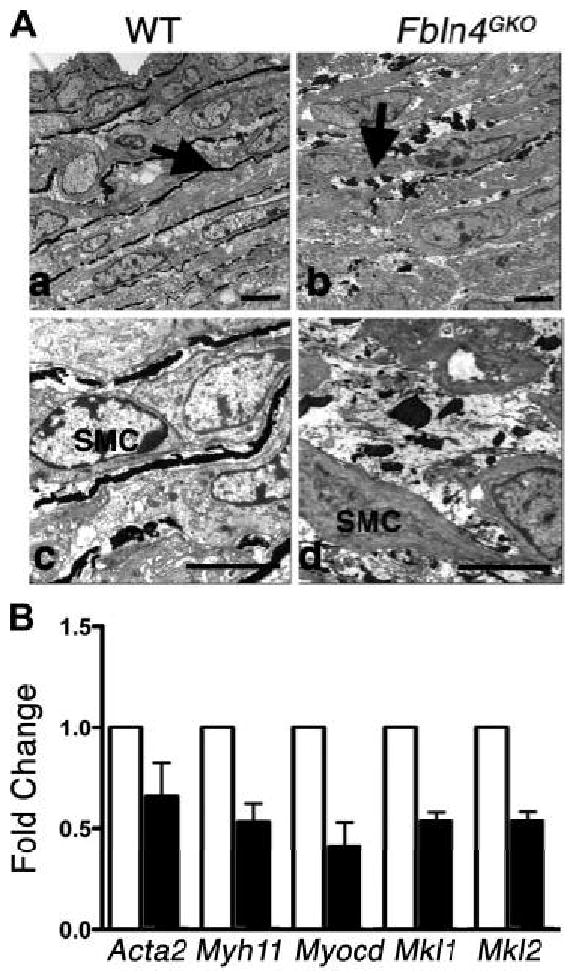
Disorganized elastic laminae and altered expression of SMC marker genes in Fbln4GKO aortae. A. Electron micrographs of E18.5 aortae from wild-type and Fbln4GKO embryos. Elastic fibers (arrow in a) are being formed the wild-type aorta. In the Fbln4GKO aorta, numerous elastin globules are seen in the mutant vessel (arrow in b). At higher magnification, disarray of SMCs and an increased pericellular ECM is observed (d). Bars indicate 5 μm. B. qPCR analysis of SMC-specific genes in the descending thoracic aortae devoid of aneurysm lesions from E18.5 wild-type (designated as 1, white column) and Fbln4GKO embryos (black column). N=4 using 12 embryos per genotype. Bars indicate means ± SD.
To examine if the absence of fibulin-4 affected SMC-specific gene expression in the aortic wall, we performed qPCR analysis using E18.5 descending thoracic aorta from wild-type and Fbln4GKO mice. SMC-specific genes, including Acta2 and Myh11, were downregulated in the mutant aorta (Figure 4B), consistent with the data obtained from Western blot and immunostaining analyses (Figure 3). Interestingly, transcription factors involved in SMC differentiation, including Myocd (myocardin), Mkl1 (myocardin-like 1/MAL/MRTF-A) and Mkl2 (myocardin-like 2/MRTF-B) were also downregulated. Because the lack of intact elastic fibers has been shown to affect SMC phenotype in the Eln-/- aorta, and recombinant tropoelastin induced actin polymerization without changing actin expression in Eln-/- SMCs 23, the immature phenotypes of Fbln4GKO SMCs may be secondary to the loss of intact elastic fibers. To examine this possibility, we evaluated the expression profile of SMC marker genes in Eln-/- aortae. Transcripts for Myocd, Mkl1, Mkl2, Myh11 or Acta2 were not downregulated in Eln-/- aortae (Online Figure VI). Rather, Myocd, Myh11 and Acta2 were upregulated compared to wild-type aortae. Although we cannot completely distinguish a direct or elastic fiber-mediated role of fibulin-4 on differentiation of SMCs in the current experimental system, the distinct transcriptional profile of the Fbln4GKO aorta suggest a unique and/or additional role of fibulin-4 in development of the aortic wall.
Loss of fibulin-4 results in an immature phenotype in Fbln4GKO SMCs
To further investigate the effect of loss of fibulin-4 on SMC phenotype, we isolated SMCs from the aorta of wild-type or Fbln4GKO embryos between E17.5 and E18.5. Morphological analysis with phalloidin staining revealed that stress fibers were decreased and cortical actin staining more prominent in Fbln4GKO SMCs compared to wild-type SMCs (Figure 5A). SM-MHC protein content was markedly reduced in Fbln4GKO SMCs by Western analysis (Figure 5Ac). Separation of SM1 (204 kDa) and SM2 (200 kDa) by SDS-PAGE revealed that loss of SM-MHC was predominantly due to loss of SM1, the predominant myosin heavy chain in the developed aorta. SM α-actin was highly expressed in SMCs and we did not detect any appreciable difference in SM α-actin content between the genotypes by Western blot analysis (data not shown). Next, we examined the capacity of Fbln4GKO cells to proliferate in the absence of serum, a characteristic feature of embryonic-type SMCs 24. Wild-type and Fbln4GKO SMCs were allowed to adhere to the culture dish and the medium was changed to a serum-free medium and grown for 24h. Immunostaining with anti-phosphorylated histone H3 (PH3), a mitosis marker, showed a significant increase in mitotic cells in Fbln4GKO SMCs (Figures 5Bb, 5Bd), whereas positive cells were virtually absent in wild-type SMCs (Figures 5Ba, 5Bc). Proliferation assays performed following stimulation with 20% FBS also exhibited a marked increase in the proliferation of Fbln4GKO cells (Figure 5C).
Figure 5.
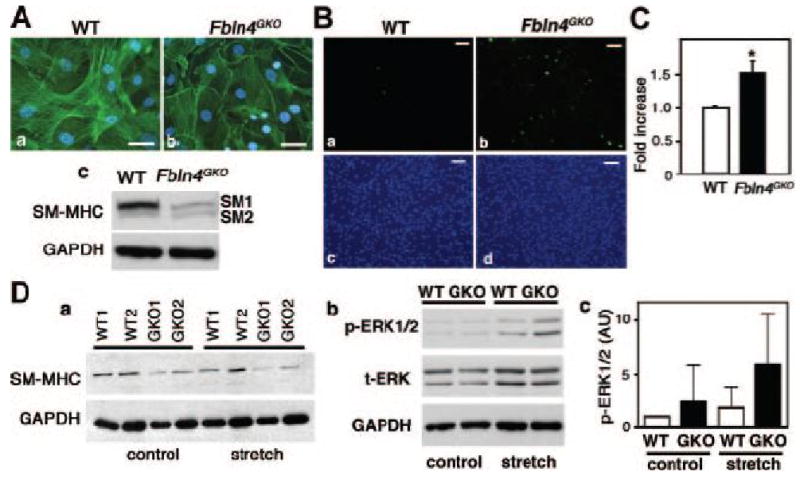
Fbln4GKO SMCs display an embryonic phenotype. A. Actin fibers detected by fluorescein-conjugated phalloidin in wild-type (a) and Fbln4GKO (b) SMCs. Note an overall decrease in actin fibers in Fbln4GKO SMCs. Five captured images were analyzed for each of the three independent experiments. Bars indicate 50 μm. (c) Representative Western blots showing a marked reduction of SM-MHC protein in Fbln4GKO SMCs. B. Immunostaining of wild-type (a, c) and Fbln4GKO SMCs (b, d) grown in serum-free media with anti-PH3 antibody (a, b). The experiment was done twice in duplicates. A significant increase in the number of proliferating cells was observed in Fbln4GKO SMCs. Bars indicate 100 μm. C. Proliferation assays. The experiment was done twice in triplicate. Bars indicate means ± SD. D. a. Cyclic stretch assays. Two × 105 cells (group 1) or 4 × 105 cells (group 2) of wild-type and Fbln4GKO SMCs were subjected to stretch stimulation followed by Western blot analysis with anti-SM-MHC and anti-GAPDH. The experiments were done five times. b. Representative Western blot showing p-ERK1/2 and t-ERK1/2 levels upon cyclic stretch stimulation in wild-type and Fbln4GKO SMCs. c. Densitometric analysis of p-ERK1/2 levels normalized to t-ERK1/2 and GAPDH. Bars are means ± SD (n=5). No statistical significant difference is observed in p-ERK levels.
Compromised contractile forces resulting from the mutations of genes encoding for SMC-specific contractile proteins or cardiac sarcomeric proteins are proposed to cause familial thoracic aortic aneurysms and familial hypertrophic or dilated cardiomyopathy, respectively 25, 26. Therefore, it is possible that Fbln4-null SMCs with reduced expression of SMC contractile proteins may exhibit an abnormal response to mechanical strain compared to wild-type SMCs. To examine this possibility, we performed in vitro stretch assays. Wild-type and Fbln4GKO SMCs were plated on a type I collagen pre-coated elastomer well and subjected to cyclic stretch (0.2 Hz, 20% strain) for 24 h without serum. Cells incubated in a static condition served as controls. Changes in SM-MHC and activation of downstream signaling pathways were evaluated by Western analysis. As Figure 5Da shows, wild-type SMCs maintained SM-MHC levels after the stretch stimulation, however, Fbln4GKO cells showed a marked decrease in SM-MHC levels in both static and stretch conditions compared to wild-type cells (p<0.005). Although a trend indicating a decrease in SM-MHC expression was observed after stretch stimulation in mutant cells by densitometric analysis, no statistical significance was obtained (p=0.056). Cyclic stretch showed a moderate increase in p-ERK in Fbln4GKO SMCs compared to wild-type cells (Figure 5Db), however, statistical significance was not obtained under the given condition (p=0.088, Figure 5Dc). These data suggest that absence of fibulin-4 results in an immature SMC phenotype with compromised level of SM-MHC and altered proliferative response.
Differentiation defect of SMCs and marked upregulation of p-ERK1/2 in the Fbln4SMKO aorta
Finally, we asked if postnatal aneurysms in the Fbln4SMKO mice share the same underlying pathology as those of Fbln4GKO mice. Histological analysis of Fbln4SMKO ascending aorta at P90 revealed focal lesions with a thickened aortic wall, which primarily due to an increase in medial SMC layers (Figures 6Aa, 6Ad). Disarray of SMCs was particularly prominent in the outer two-thirds of the aortic wall in which disorganized elastic fibers were also prominent (Figures 6Ab, 6Ae). Inflammatory infiltrates, including neutrophils and macrophages, were not prominently seen in Fbln4SMKO aortae. An overall increase of collagen fibers was also detected in the mutant aorta (Figures 6Ac, 6Af). Surprisingly, in contrast to the dramatic changes seen in the ascending aorta, the descending thoracic aorta appeared normal without hyperproliferation of SMCs (Online Figure VIId). Elastic laminae were more organized compared to those in the ascending aorta, although discontinuities were more frequently observed in mutants compared to the wild-type aorta (Online Figure VIIe). Masson-Trichrome staining revealed significant increases in collagen fibers in the mutant descending thoracic aorta (Online Figure VIIf).
Figure 6.
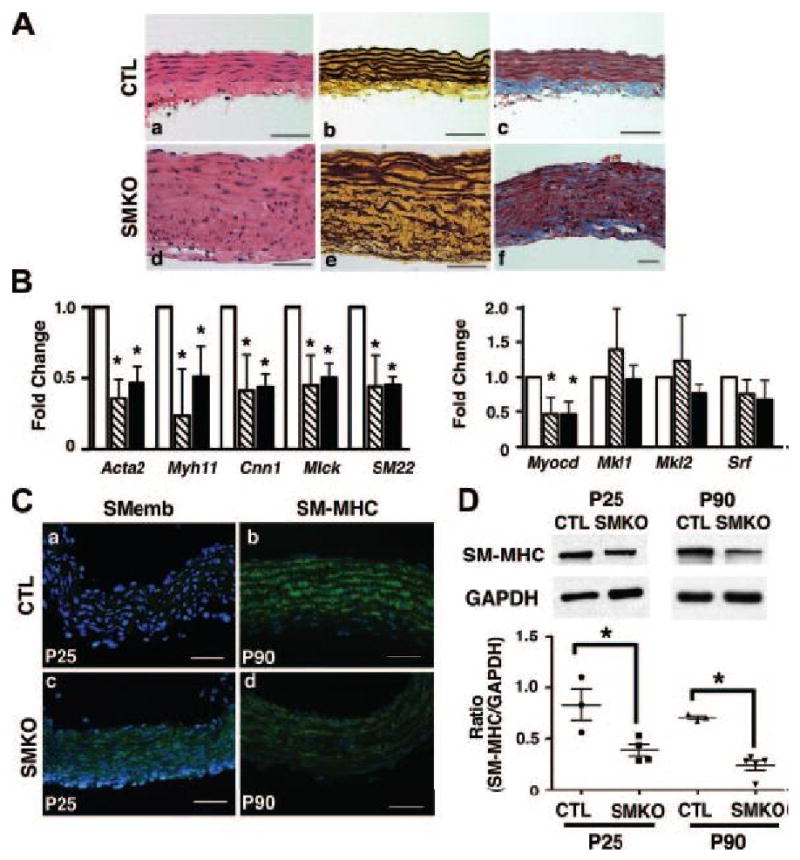
SMC-specific deletion of Fbln4 recapitulates the aortic phenotype in Fbln4-null embryos. A. Histological evaluation of focal proliferative lesions in the Fbln4SMKO aorta at P90. Representative photos of transverse sections of ascending aorta from control (a-c) and Fbln4SMKO (d-f) mice stained with HE (a, d), Hart's (b, e) or Masson-Trichrome (c, f). B. qPCR analysis of SMC specific genes in ascending (stripe column) and descending (black column) thoracic aortae from Fbln4SMKO mice and corresponding controls (white column). Four sibling pairs were used. Bars indicate means ± SD. C. Representative immunostaining of the ascending aorta from control (a, b) and Fbln4SMKO (c, d) mice stained with anti-SMemb (P25) and SM-MHC (P90) antibodies. Note strong expression of SMemb and reduced expression of SM-MHC in the mutant aorta. D. (upper panel) Representative Western blot of the ascending aorta before (P25) and after (P90) the formation of aneurysms. (lower graph) A ratio between SM-MHC to GAPDH in P25 and P90 ascending aortae is determined densitometrically and plotted. Each data point represents a measurement from an ascending aorta at P25 or P90.
To examine if the differentiation defect of SMCs is also present in the Fbln4SMKO aorta, qPCR analysis was performed using ascending and descending thoracic aortae from Fbln4SMKO and control mice at P90. Transcripts for SMC contractile genes were significantly downregulated in the diseased ascending aorta as well as in the descending aorta where no aneurysms were observed (Figure 6B). In addition, Myocd was downregulated in both ascending and descending thoracic aortae. Immunostaining of the ascending aorta at P25 with anti-SMemb, a marker of embryonic SMCs 27, showed faint staining in the wild-type aorta, whereas persistent expression was detected in the mutant aorta (Figures 6Ca and 6Cc). In contrast, immunostaining with anti-SM-MHC detected strong expression in P90 wild-type ascending aorta, whereas a marked reduction of SM-MHC was observed in the mutant aorta (Figures 6Cb and 6Cd). The reduction of SM-MHC levels was further confirmed by Western blot analysis using ascending aortae harvested from P25 and P90 Fbln4SMKO mice (Figure 6D). At both time points, before or after aneurysm development, reduction of SM-MHC was observed in the mutant aorta
Finally, we examined signaling pathways responsible for the proliferative changes of the aneurysmal wall. Upregulation of p-ERK1/2 was observed in the Fbln4SMKO aorta at P25 and P90 by Western blot analysis (Figure 7A). Immunostaining of the P90 ascending aorta revealed strong upregulation of p-ERK1/2 in the mutant SMCs compared to control vessels (Figure 7B). Phosphorylated p38 level was moderately increased in the mutant aorta (data not shown). Levels of p-Smad2/3, p-Smad1/5/8, and CTGF, a downstream target of TGF-β, were comparable between control and Fbln4SMKO aortae examined at P25 and 3-months of age (Online Figure VIII Aa, B). In addition, we did not detect any difference in p-Smad2/3 levels between wild-type and Fbln4GKO aortae by Western blot or immunostaining (Online Figure VIII Ab, data not shown). Taken together, these observations in the Fbln4GKO and Fbln4SMKO aortae suggested that 1) alteration of SMC-specific genes was not simply a consequence of aneurysm formation in the mutant aorta, and 2) the ERK1/2 pathway played a pivotal role in the focal proliferative response of abnormal SMCs in the aneurysmal wall.
Figure 7.
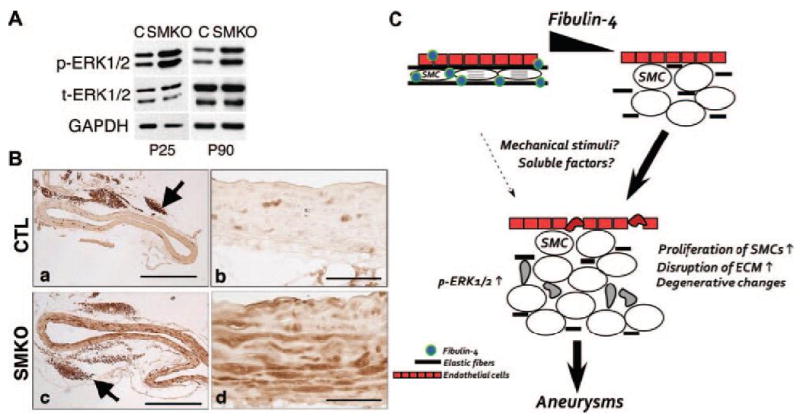
Increased ERK1/2 signaling in the Fbln4SMKO aorta. A. Representative Western blot of ascending aortae from control and Fbln4SMKO mice at P25 and P90. An increase in p-ERK1/2 is already evident at P25. B. Representative immunostaining of P90 ascending aorta from control (a, b) and Fbln4SMKO (c, d) mice stained with p-ERK1/2. Increased level of p-ERK1/2 is observed in SMCs of the Fbln4SMKO aorta. Note strong signal in perivascular adipose tissue from both control and the mutants (arrow in a and c). Bars in (a, c) indicate 500 μm and in (b, d) indicate 50 μm. C. A model illustrating pathological changes in Fbln4 mutant aorta. Absence of fibulin-4 in the aortic wall affects formation of elastic fibers and terminal differentiation of SMCs. Fbln4-null SMCs exhibit an increase in ERK1/2 phosphorylation, possibly mediated by external factors, including mechanical stimuli or soluble factors. Proliferation of SMCs, apoptosis of vascular cells (illustrated by deformed cells) and further destruction of vascular ECM by secondary inflammatory cell response, together contribute to the aneurysm development.
Discussion
Potential contribution of abnormal SMCs to aneurysm development in Fbln4 mutant mice
Deletion of fibulin-4 in a global or SMC-specific manner resulted in a differentiation defect of SMCs with marked down-regulation of SMC contractile genes. Contractile forces in SMCs are generated by cyclic interactions of myosin heavy chain and actin filaments induced by MLCK-mediated phosphorylation of myosin light chains (Reviewed in 28, 29). The mutations in components of the contractile apparatus in SMC such as ACTA2 and MYH11 lead to a focal proliferation of SMCs with disarray, disruption of elastic fibers, and eventual loss of SMCs 10-12. It is also shown that patients with ACTA2 disease haplotype exhibit reduced aortic compliance and increased pulse velocity, altering aortic stiffness 10. Although aortic aneurysms were not observed in genetic ablation of the SM2 isoform of SM-MHC in mice, the mutants did develop an enlarged thin bladder and hydronephrosis and died within 4 weeks of age 30. A concomitant reduction of SM1 was observed in the SM2-/- bladder and an appropriate ratio of SM1 to SM2 was shown to be critical for normal contractility of SMCs. Absence of Acta2 also exhibited a significant decrease in vascular contractility and impaired maintenance of blood pressure and blood flow 31. Thus, quality and quantity of contractile proteins significantly affect contractile functions of SMCs and eventually affect mechanical properties of the aortic wall. Taken together, we suggest that Fbln4 mutant SMCs with reduced expression of contractile proteins and highly synthetic phenotype, together with the abnormal ECM, alter vessel wall stability and results in maladaptation of the vessel wall, providing a potential link to an aneurysm phenotype (Figure 7C).
Fibulin-4 provides a permissive environment for differentiation and maintenance of SMCs
SMC differentiation is largely governed by the myocardin family of transcriptional co-activators (reviewed in 32). Myocardin, Mkl1 and Mkl2 serve as a potent co-factor of SRF and upregulate a variety of SMC contractile genes 33-36. The reduced expression of Myocd, Mkl1, and Mkl2 in the Fbln4GKO mutant aorta indicates a potential mechanism of extracellular regulation of SMC differentiation genes by fibulin-4. Fibulin-4 is located in microfibril bundles which tether elastic fibers to SMCs via integrin-mediated binding at regions of the cell membrane occupied by intracellular membrane-associated dense plaques, anchoring sites for actin filaments 37, 38. Thus, loss of fibulin-4 may disrupt the interaction between elastic fibers and SMCs, leading to the alterations in actin cytoskeleton organization. The actin dynamics between filamentous (F) and globular (G) actin are known to regulate the activity of Mkl1: G-actin binds to cytoplasmic Mkl1 and inhibits translocation of Mkl1 to the nucleus thereby disrupting the interaction between Mkl1 and SRF and downregulating the transcription of SMC contractile genes 39. Our observation that actin filaments were less prominent in Fbln4GKO SMCs compared to wild-type cells is consistent with this hypothesis. A similar disruption of the elastin-SMC connection was reported in a mouse deficient for the polycystic kidney disease 1 (Pkd1) gene, which developed descending aortic aneurysms with hyperproliferative SMCs 40. Ultrastructural studies of Fbn1C1039G/+ aortae also revealed disruption between SMC and elastic fiber connections, leading to activation of SMCs 41. Taken together, fibulin-4 is critical for the maintenance of the permissive and stable environment for SMC differentiation.
Elastic fibers and protection of aortic aneurysms
Our present study, together with other reports, confirmed that a loss of fibulin-4 profoundly affects formation of elastic fibers 8, 19. Various experimental manipulations that disrupt elastic fibers have successfully recapitulated pathological changes of aortic aneurysms in rodents 42, 43. Genetic or pharmacological stabilization of elastin or inhibition of its degradation was shown to attenuate development of abdominal aortic aneurysms 44-46. Although elastic fibers or insoluble elastin have been shown to suppress proliferation and migration of SMCs 23, a causal relationship between disrupted elastic fibers and aneurysm formation during development has not been fully established. Interestingly, mice deficient in the fibrillin-1 gene (Fbn1mgN/mgN) that die from aneurysm rupture within two weeks of age, exhibit maturation defects of the aortic wall and persistent expression of embryonic SMC markers 47. Thus, components of elastic fibers affect SMC phenotypes directly or indirectly during development, a time when SMCs have a high degree of plasticity.
Signaling pathways involved in proliferation of SMCs in the Fbln4 mutant aorta
Hyperproliferation of SMCs is a critical component involved in the alteration of the Fbln4 mutant aorta and is predominantly mediated by the ERK1/2 signaling. A cue for the activation of ERK1/2 may be provided by mechanical stimuli such as increased hemodynamic force and changes in aortic ECM components, paracrine factors or by undefined mechanisms. Cyclic stretch activates MAPK pathway (both ERK and p38) and increases import of nuclear protein as well as transcription of nuclear pore complex 48. Although the causal relationship between cyclic strain and ERK1/2 phosphorylation was not established in Fbln4GKO SMCs using an in vitro system, the observation that aneurysms developed exclusively in the ascending aorta, the region of highest systolic pressure, in Fbln4SMKO mice suggests that mechanical strain may influence SMC function in vivo. Angiotensin II activates the ERK pathway and is also a known mediator of the stretch response in SMCs through local production of angiotensinogen or local conversion of angiotensin I to angiotensin II 49. AT1 receptor blockade inhibits gene transcription and proliferation of SMCs induced by stretch stimulation as well as the hypertrophic response of cardiac myocytes induced by pressure overload 50-52. Upregulation of IGF-1 and phosphorylation of IGF-receptor is also induced by cyclic stretch and lead to the proliferation of venous SMCs 53, 54. Interestingly, a high level of angiotensin II and IGF-1 was observed in primary SMC cultures prepared from a patient with a MYH11 mutation, suggesting the possible involvement of these soluble factors in the proliferative response of SMCs 11. It is also possible that TGF-β activates ERK1/2 pathways by a non-Smad dependent mechanism 55. Thus, Fbln4 mutant mice give us the opportunity to test pharmacological interventions to block specific signaling pathway and attenuate aneurysm formation in vivo.
Limitation of this study
Our current study revealed a critical role for fibulin-4 on differentiation of SMCs and protection from aneurysm development. Since fibulin-4 is a secreted protein, our conditional knockout phenotypes may be partially masked by fibulin-4 supplied by adjacent tissues or remaining production of fibulin-4 within the aortic wall. Fibulin-4 plays a critical role in elastic fiber assembly, therefore, the temporal and causal relationship between defective elastic fibers and alteration of SMC phenotype in Fbln4-null aorta remain to be further investigated. In addition, potential cellular functions of fibulin-4 such as the effect on apoptosis or collagen fiber formation, may be explored.
Supplementary Material
Acknowledgments
We thank Dean Li for Eln+/- mice, James Richardson for assistance with histological analysis, and Patrick Keller and Jesus Acevedo for technical assistance. We thank the Facility for Electron Microscopy Research (FEMR) at McGill University, the Histology Core Laboratory of the University of Texas Southwestern Medical Center for excellent histological preparations, Transgenic Core Facility for blastocysts injection, and Department of Urology for Flexcell unit. We also thank Eric Olson and Masashi Yanagisawa for critical reading of the manuscript.
Source of funding: The work was supported in part by the NIH (R01HL0711 to H.Y. and R01AG028048 to R.A.W), American Heart Association South Central Affiliate (0855200F to H.Y), the National Marfan Foundation (H.Y), and the Canadian Institutes of Health Research (MOP86713 to E.C.D.). E.C.D. is a Canada Research Chair.
Non-standard Abbreviations and Acronyms
- CTGF
connective tissue growth factor
- CTL
control
- ECM
extracellular matrix
- ECKO
endothelial cell-specific knockout
- Eln
elastin
- ERK
extracellular signal –regulated kinase
- Fbln4
fibulin-4
- Fbln5
fibulin-5
- IGF
insulin growth factor
- JNK
Janus kinase
- MEK
mitogen-activated protein kinase kinase and extracellular signal-regulated kinase kinase
- p38
p38 MAP kinase
- SMC
smooth muscle cell
- SMKO
smooth muscle cell-specific knockout
- SM-MHC
smooth muscle myosin heavy chain
- TAAD
thoracic aortic aneurysms and/or aortic dissection
- TGF-β
transforming growth factor-beta
- BMP
bone morphogenetic protein
Footnotes
Disclosure: None.
Novelty and Significance
Fibulin-4 (Fbln4) is a secreted glycoprotein essential for the formation of elastic fibers. Fbln4-null mice develop aortic aneurysms and die at birth from aneurysm rupture. Interestingly, however, mice deficient for the components of elastic fibers, such as elastin or fibulin-5, do not develop aneurysms despite severe elastic fiber defects. These observations led us to hypothesize that fibulin-4 may play crucial roles not only in elastogenesis but also for normal development of the aortic wall. By analyzing Fbln4-null and smooth muscle cell (SMC)-specific Fbln4 knockout mice (Fbln4SMKO), we showed that fibulin-4 is critical for maintenance of a permissive and stable environment for SMC differentiation. Second, we described focal proliferation of SMCs with marked upregulation of phosphorylated (p)-ERK1/2 levels and disruption of the extracellular matrix (ECM) in aneurysm lesions. The aneurysm involving defective SMCs is remarkably similar to a subset of human thoracic aortic aneurysms in which mutations of SMC-specific contractile genes have been identified to be the cause. This new animal model provides opportunities for pharmacological interventions that target the ERK1/2 MAPK signaling pathway for treatment of thoracic aortic aneurysms. In addition, investigations of the mechanism underlying the altered signaling in Fbln4SMKO mice will expand our knowledge on the role of ECM as a modulator of cell signaling.
What is known?
- Fibulin-4 is an elastin binding ECM protein.
- Congenital loss of fibulin-4 results in severe disruption of elastic fibers and aortic aneurysms in mice and humans.
New information contributed by this study?
- Provides evidence that fibulin-4-induced phenotypic alterations of SMCs play a role in aneurysm development.
- Shows that congenital loss of fibulin-4 leads to marked upregulation of phosphorylated (p)-ERK1/2 in SMCs and loss of SMC contractile phenotype in aneurysm lesions.
- Provides a basis for potential ERK1/2 signaling-targeted therapy for ascending aortic aneurysms in humans.
References
- 1.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 2.Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111:816–828. doi: 10.1161/01.CIR.0000154569.08857.7A. [DOI] [PubMed] [Google Scholar]
- 3.Pope FM, Martin GR, Lichtenstein JR, Penttinen R, Gerson B, Rowe DW, McKusick VA. Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc Natl Acad Sci U S A. 1975;72:1314–1316. doi: 10.1073/pnas.72.4.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 1995;4(Spec No):1799–1809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- 5.Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 6.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maki JM, Rasanen J, Tikkanen H, Sormunen R, Makikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin PJ, Chen Q, Horiguchi M, Starcher BC, Stanton JB, Broekelmann TJ, Marmorstein AD, McKay B, Mecham R, Nakamura T, Marmorstein LY. Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol Cell Biol. 2006;26:1700–1709. doi: 10.1128/MCB.26.5.1700-1709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, Young MF, Bianco P. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115:2731–2738. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
- 10.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 11.Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, Guo D, Estrera AL, Safi HJ, Brasier AR, Vick GW, Marian AJ, Raman CS, Buja LM, Milewicz DM. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16:2453–2462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi N, Kostka G, Garbe JH, Keene DR, Bachinger HP, Hanisch FG, Markova D, Tsuda T, Timpl R, Chu ML, Sasaki T. A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J Biol Chem. 2007;282:11805–11816. doi: 10.1074/jbc.M611029200. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross Jr J, Honjo T, Chien KR. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature. 2002;415:171–175. doi: 10.1038/415171a. [DOI] [PubMed] [Google Scholar]
- 15.Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, Olson EN. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 16.Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet. 2006;78:1075–1080. doi: 10.1086/504304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dasouki M, Markova D, Garola R, Sasaki T, Charbonneau NL, Sakai LY, Chu ML. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A:2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 18.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 19.Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MG, Myers L, Maas A, Duncker DJ, Meijers C, Dietz HC, Kanaar R, Essers J. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007;100:738–746. doi: 10.1161/01.RES.0000260181.19449.95. [DOI] [PubMed] [Google Scholar]
- 20.Choudhary B, Zhou J, Li P, Thomas S, Kaartinen V, Sucov HM. Absence of TGFbeta signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115–121. doi: 10.1002/dvg.20466. [DOI] [PubMed] [Google Scholar]
- 21.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 22.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 23.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 24.Cook CL, Weiser MC, Schwartz PE, Jones CL, Majack RA. Developmentally timed expression of an embryonic growth phenotype in vascular smooth muscle cells. Circ Res. 1994;74:189–196. doi: 10.1161/01.res.74.2.189. [DOI] [PubMed] [Google Scholar]
- 25.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 26.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 27.Kuro-o M, Nagai R, Nakahara K, Katoh H, Tsai RC, Tsuchimochi H, Yazaki Y, Ohkubo A, Takaku F. cDNA cloning of a myosin heavy chain isoform in embryonic smooth muscle and its expression during vascular development and in arteriosclerosis. J Biol Chem. 1991;266:3768–3773. [PubMed] [Google Scholar]
- 28.Ogut O, Brozovich FV. Regulation of force in vascular smooth muscle. J Mol Cell Cardiol. 2003;35:347–355. doi: 10.1016/s0022-2828(03)00045-2. [DOI] [PubMed] [Google Scholar]
- 29.Arner A, Lofgren M, Morano I. Smooth, slow and smart muscle motors. J Muscle Res Cell Motil. 2003;24:165–173. doi: 10.1023/a:1026001513928. [DOI] [PubMed] [Google Scholar]
- 30.Chi M, Zhou Y, Vedamoorthyrao S, Babu GJ, Periasamy M. Ablation of smooth muscle myosin heavy chain SM2 increases smooth muscle contraction and results in postnatal death in mice. Proc Natl Acad Sci U S A. 2008;105:18614–18618. doi: 10.1073/pnas.0808162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schildmeyer LA, Braun R, Taffet G, Debiasi M, Burns AE, Bradley A, Schwartz RJ. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. Faseb J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- 32.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, Proweller A, Epstein JA, Parmacek MS. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest. 2008;118:515–525. doi: 10.1172/JCI33304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Chang S, Qi X, Richardson JA, Olson EN. Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Mol Cell Biol. 2006;26:5797–5808. doi: 10.1128/MCB.00211-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh J, Richardson JA, Olson EN. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc Natl Acad Sci U S A. 2005;102:15122–15127. doi: 10.1073/pnas.0507346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 38.Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- 39.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 40.Hassane S, Claij N, Lantinga-van Leeuwen IS, Van Munsteren JC, Van Lent N, Hanemaaijer R, Breuning MH, Peters DJ, DeRuiter MC. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol. 2007;27:2177–2183. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 41.Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 42.Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michel JB. Elastase-induced experimental aneurysms in rats. Circulation. 1990;82:973–981. doi: 10.1161/01.cir.82.3.973. [DOI] [PubMed] [Google Scholar]
- 43.Chiou AC, Chiu B, Pearce WH. Murine aortic aneurysm produced by periarterial application of calcium chloride. J Surg Res. 2001;99:371–376. doi: 10.1006/jsre.2001.6207. [DOI] [PubMed] [Google Scholar]
- 44.Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008;102:e73–85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 45.Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isenburg JC, Simionescu DT, Starcher BC, Vyavahare NR. Elastin stabilization for treatment of abdominal aortic aneurysms. Circulation. 2007;115:1729–1737. doi: 10.1161/CIRCULATIONAHA.106.672873. [DOI] [PubMed] [Google Scholar]
- 47.Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis A, Hazeki N, Keene DR, Sakai LY, Ramirez F. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281:8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng J, Du J. Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin-like growth factor-1 receptor. Arterioscler Thromb Vasc Biol. 2007;27:1744–1751. doi: 10.1161/ATVBAHA.107.147371. [DOI] [PubMed] [Google Scholar]
- 49.Hosokawa H, Aiuchi S, Kambe T, Hagiwara Y, Kubo T. Mechanical stretch-induced mitogen-activated protein kinase activation is mediated via angiotensin and endothelin systems in vascular smooth muscle cells. Biol Pharm Bull. 2002;25:1588–1592. doi: 10.1248/bpb.25.1588. [DOI] [PubMed] [Google Scholar]
- 50.Ruiz-Ortega M, Lorenzo O, Ruperez M, Konig S, Wittig B, Egido J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: molecular mechanisms. Circ Res. 2000;86:1266–1272. doi: 10.1161/01.res.86.12.1266. [DOI] [PubMed] [Google Scholar]
- 51.Li Q, Muragaki Y, Ueno H, Ooshima A. Stretch-induced proliferation of cultured vascular smooth muscle cells and a possible involvement of local renin-angiotensin system and platelet-derived growth factor (PDGF) Hypertens Res. 1997;20:217–223. doi: 10.1291/hypres.20.217. [DOI] [PubMed] [Google Scholar]
- 52.Malhotra R, Sadoshima J, Brosius FC, 3rd, Izumo S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes In vitro. Circ Res. 1999;85:137–146. doi: 10.1161/01.res.85.2.137. [DOI] [PubMed] [Google Scholar]
- 53.Standley PR, Obards TJ, Martina CL. Cyclic stretch regulates autocrine IGF-I in vascular smooth muscle cells: implications in vascular hyperplasia. Am J Physiol. 1999;276:E697–705. doi: 10.1152/ajpendo.1999.276.4.E697. [DOI] [PubMed] [Google Scholar]
- 54.Richard MN, Deniset JF, Kneesh AL, Blackwood D, Pierce GN. Mechanical stretching stimulates smooth muscle cell growth, nuclear protein import, and nuclear pore expression through mitogen-activated protein kinase activation. J Biol Chem. 2007;282:23081–23088. doi: 10.1074/jbc.M703602200. [DOI] [PubMed] [Google Scholar]
- 55.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.