

Abstract
The clustering of voltage-gated sodium channels at the axon initial segment (AIS) and nodes of Ranvier is essential for the initiation and propagation of action potentials in myelinated axons. Sodium channels localize to the AIS through an axon-intrinsic mechanism driven by ankyrin G, while clustering at the nodes requires cues from myelinating glia that interact with axonal neurofascin186 (Sherman et al., 2005; Dzhashiashvili et al., 2007; Yang et al., 2007). Here, we report that in zebrafish mutants lacking Schwann cells in peripheral nerves (erbb2, erbb3, and sox10/colorless), axons form numerous aberrant sodium channel clusters throughout their length. Morpholino knockdown of ankyrin G, but not neurofascin, reduces the number of sodium channel clusters in Schwann cell-deficient mutants, suggesting that these aberrant clusters form by an axon-intrinsic mechanism. We also find that gpr126 mutants, in which Schwann cells are arrested at the promyelinating stage (Monk et al., 2009), are deficient in the clustering of neurofascin at the nodes of Ranvier. When Schwann cell migration in gpr126 mutants is blocked, there is an increase in the number of neurofascin clusters in peripheral axons. Our results suggest that Schwann cells inhibit the ability of ankyrin G to cluster sodium channels at ectopic locations, restricting its activity to the AIS and nodes of Ranvier.
Introduction
The proper localization of voltage-gated sodium channels in axons is essential for normal neural function (Salzer et al., 2008). In myelinated axons, sodium channels are clustered in the short, unmyelinated gaps (nodes of Ranvier) that occur between the myelinated segments (internodes). This clustering of sodium channels at the nodes is essential for the rapid, saltatory conduction of action potentials that is characteristic of myelinated axons (Sherman et al., 2005). Sodium channels are also clustered at the base of the axon [the axon initial segment (AIS)], and this localization is required for the initiation of action potentials in many neurons (Khaliq and Raman, 2006; Palmer and Stuart, 2006).
Recent work describes two related, but distinct, mechanisms by which sodium channels form clusters in peripheral axons. In the first mechanism, the myelinating glia (Schwann cells) present a ligand to discrete loci on the surface of underlying axons. This ligand stimulates the clustering of axonal neurofascin, which in turn recruits sodium channels to the nascent cluster via ankyrin G. This “neurofascin-dependent” mechanism is thought to be responsible for the clustering of sodium channels at the nodes of Ranvier (Eshed et al., 2005; Sherman et al., 2005; Dzhashiashvili et al., 2007). In the second mechanism, ankyrin G forms clusters in the absence of glial input. Clustered ankyrin G then separately recruits sodium channels and neurofascin. This “axon-intrinsic” mechanism is believed to initiate clustering of sodium channels at the AIS only (Dzhashiashvili et al., 2007; Yang et al., 2007).
While the importance of glia in establishing sodium channel clusters at nodes of Ranvier is well established, no study has examined axonal sodium channels in the complete absence of glia in vivo. In the zebrafish, mutants for erbb2, erbb3, and sox10 lack Schwann cells in peripheral nerves (Kelsh and Eisen, 2000; Lyons et al., 2005; Pogoda et al., 2006). Here, we report the unexpected finding that numerous abnormal sodium channel clusters form throughout the length of nerves that lack Schwann cells. Morpholino studies provide evidence that these abnormal clusters require ankyrin G, but not neurofascin, implying that the axon-intrinsic mechanism of clustering that normally functions at the AIS can act ectopically in the absence of Schwann cells. We also find that neurofascin clusters at the nodes of Ranvier are severely reduced in gpr126 mutants, in which Schwann cells associate with axons but arrest at the promyelinating stage (Monk et al., 2009); this result suggests that Schwann cells stimulate clustering at nodes at the onset of myelination in zebrafish, as has been shown in mammals (Salzer et al., 2008). Surprisingly, removal of Schwann cells from peripheral nerves actually increased the number of clusters present in gpr126 mutants, providing evidence that Schwann cells inhibit clustering of node molecules at inappropriate locations. Based on these data, we propose a new role for Schwann cells in restricting axon-intrinsic sodium channel clustering to the AIS. This inhibitory function complements the well established role of myelinating glia in promoting cluster formation at the nodes of Ranvier.
Materials and Methods
Zebrafish stocks.
The erbb2st61, erbb3st48, and gpr126st49 mutant lines were isolated in genetic screens for defects in myelinated axons (Lyons et al., 2005; Pogoda et al., 2006; Monk et al., 2009). The clst3 and Tg(FoxD3:GFP)17 lines have been described previously (Kelsh and Eisen, 2000; Gilmour et al., 2002).
Antibodies and immunofluorescence.
The following antibodies and dilutions were used: mouse anti-acetylated tubulin (Sigma; 1:1000), mouse anti-panNavCh (Sigma; 1:500), rabbit anti-FIGQY (a gift from M. Rasband, Baylor College of Medicine, Houston, TX; 1:1000), rabbit anti-tyrosine hydroxylase (Millipore Bioscience Research Reagents; 1:500), purified rabbit anti-ankyrin G (see below; 1:2000), purified guinea pig anti-extracellular neurofascin (see below; 1:20).
To raise antibodies against ankyrin G, a region of ank3b, one of two duplicate genes encoding ankyrin G in zebrafish (corresponding to nucleotides 2437-3252 of a predicted ank3b cDNA, accession XM_695014) was amplified by RT-PCR from adult zebrafish brain RNA. In this region, which corresponds to part of the spectrin-binding domain, the predicted Ank3a and Ank3b proteins are >80% identical. The resulting cDNA was ligated in-frame downstream of the maltose-binding protein (MBP) encoding region of pMAL–c2X (New England Biolabs). Purified fusion protein was used to raise antibodies in rabbits (Covance Immunology Services). The resulting immune serum was incubated with purified MBP that had been conjugated to Affigel (BioRad) to separate anti-MBP from the immune serum. Anti-MBP-depleted immune serum was then incubated with MBP–ankyrin G fusion protein conjugated to Affigel. Bound anti-ankyrin G was washed by standard procedures, then eluted with 0.2 m glycine, pH 2.0, in 150 mm NaCl and immediately neutralized with 0.1 volumes of 2 m Tris, pH 8.5. BSA was added to 30 mg/ml, and the antibodies were dialyzed against TBS. The purified antibodies detected multiple bands on immunoblots of adult brain, likely corresponding to different ankyrin G isoforms (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Antibodies versus extracellular neurofascin were raised and purified as described for anti-ankyrin G above with the following exceptions. A region of neurofascin encoding part of the mucin-like domain (corresponding to nucleotides 3340–3666 of the neurofascin cDNA, accession FJ669144) was amplified by RT-PCR from adult zebrafish brain RNA and ligated in frame into pMAL–c2X. Purified MBP–mucin domain fusion protein was used to immunize guinea pigs, and anti-extracellular neurofascin was purified from immune serum by incubating with MBP–mucin domain-conjugated Affigel.
Immunofluorescence was performed as described previously (Voas et al., 2007), except that before incubation with anti-ankyrin G or anti-extracellular neurofascin antibodies, the skin of embryos was peeled away from the body after fixation to facilitate antibody penetration. All fluorescent images were taken using a Zeiss LSM 5 Pascal confocal microscope.
Morpholino oligonucleotides.
Gene Tools synthesized the following antisense morpholino oligonucleotide (MO) sequences: ank3a MO: CTCTCTCTTCCCACCCGTACCTTTT; neurofascin MO: AAGGTTTCATCCATTCTCACGTTGG.
The ank3a MO is predicted to hybridize to the exon/intron boundary that follows the first exon of the neural-specific transcript of ank3a (Kordeli et al., 1995). The neurofascin MO is predicted to hybridize to the exon/intron boundary that follows the sixth exon of neurofascin. MO were diluted in water and injected into embryos from erbb2 st61/+ intercrosses at the one-cell stage. For ank3a injections, 2 ng of MO was injected per embryo. For neurofascin MO, 1.5 ng was injected per embryo for the sodium channel and neurofascin assays (see Fig. 4). For transmission electron microscopy (TEM) analysis, 2.5 ng of neurofascin MO was injected (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Control embryos were injected with water alone. The total volume injected per embryo was 1 nL in all cases.
Figure 4.

Sodium channel clustering in Schwann cell-deficient peripheral nerves is inhibited by knockdown of ankyrin G but not neurofascin. A, Embryos from erbb2 st61/+ heterozygous intercrosses were injected with water or with MO against the neural-specific transcript of ank3a or with MO against neurofascin. The average number of sodium channel clusters per PLLn is shown at 72 hpf (error bars indicate ±1 SD). The p values for unpaired t test (single-tailed) comparisons are shown. One PLLn was scored per embryo, and the number of embryos scored from left to right is 18, 20, 20, 15, 20, and 21. B–D, Water-injected wild-type sibling (B), nfasc MO-injected wild-type sibling (C), and nfasc MO-injected erbb2st61 mutant (D) at 72 hpf, immunolabeled with anti-panNavCh (green) and anti-extracellular neurofascin (red). Neurofascin is shown alone in B′–D′. E, F, Wild-type 72 hpf embryos injected with water (E) or neurofascin MO (F) labeled with anti-acetylated tubulin (red) and anti-FIGQY (recognizes intracellular epitope of neurofascin, green).
Transmission electron microscopy.
Embryos were prepared for TEM at 3 d postfertilization (dpf), essentially as described previously (Lyons et al., 2008). Images were collected using a JEOL TEM1230.
Western blotting.
To make crude lysate, an adult brain was ground in the presence of protease inhibitors, sample buffer, and reducing agent. The lysate was then passed through a 27-gauge needle, boiled for 5 min, and centrifuged. SDS-PAGE and Western blotting were performed by standard procedures. The purified rabbit anti-ankyrin G antibody was used at a dilution of 1:2000.
AG1478 drug treatment.
AG1478 (CalBiochem) was applied at 17 h postfertilization (hpf) to embryos from gpr126 st49/ + heterozygote intercrosses at a final concentration of 4 μm in embryo medium with 1% DMSO. The medium plus AG1478 was changed daily until 4 dpf. Mock treatment was performed by applying embryo medium with 1% DMSO only.
Results
Abnormal clusters of axonal proteins form in the absence of Schwann cells
From two screens for mutants with defects in myelinated axons, mutant alleles of erbb2 and erbb3 were isolated (Lyons et al., 2005). In these mutants, proliferation and migration of Schwann cells along growing axons is disrupted, causing a lack of immature and myelinating Schwann cells in peripheral nerves (Lyons et al., 2005; Pogoda et al., 2006). Previous work in zebrafish has shown that sodium channel clustering in the PNS normally only occurs in myelinated axons, while unmyelinated axons have diffusely localized sodium channels (Voas et al., 2007). By immunofluorescence, we found that abundant clusters of sodium channels form in the posterior lateral line nerve (PLLn) of erbb2 and erbb3 mutants at 5 dpf (Fig. 1 A–C). Unlike sodium channel clusters in the wildtype, clusters in mutants are asymmetric and elongated and tend to occur in groups within the nerve rather than being evenly distributed (Fig. 1 A–C). Also, the total number of clusters in mutants is reduced compared to the wildtype (Fig. 2 A). Most of these clusters do not appear to occur at the AIS, because they are distributed throughout the length of the nerve in the mutants (Fig. 2 B). The neuronal cell bodies that give rise to the PLLn remain tightly grouped at the ganglia in mutants, as they do in the wildtype (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), indicating that the majority of clusters occur distant from the axon initial segments of the PLLn. At the AIS of erbb2 mutants, sodium channel clustering is similar to that seen in wild-type siblings (Fig. 1 I,J), indicating that the abnormal clustering phenotype is restricted to axonal regions that are distal to the AIS. These data demonstrate that in the absence of Schwann cells, peripheral axons form aberrant clusters of sodium channels at a distance from the AIS.
Figure 1.
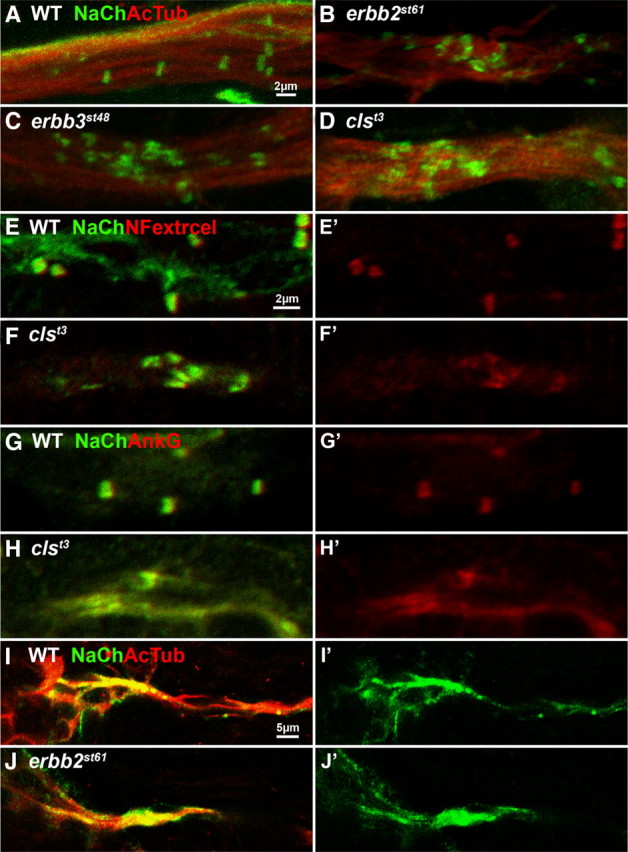
In the absence of Schwann cells, peripheral axons form aberrant clusters of sodium channels, NF186, and ankyrin G. A–D, Whole-mount immunofluorescent labeling of sodium channels (anti-panNavCh; green) and axons (anti-acetylated tubulin; red) in wild-type (A), erbb2st61 (B), erbb3st48 (C), and clst3 (D) mutants at 5 dpf reveals the presence of clusters in the PLLn of Schwann cell-deficient mutants. E, F, Immunofluorescent labeling of sodium channels (green) and the extracellular domain of neurofascin (red) in the PLLn of clst3 mutants and wild-type siblings. G, H, Sodium channels (green) and ankyrin G (red) in the PLLn of clst3 mutants and wild-type siblings. I, J, Immunofluorescently labeled sodium channels (green) and acetylated tubulin (red) at the AIS of multiple PLLn axons in wild-type (I) and erbb2st61 (J) embryos at 3 dpf. E′–J′, Single-channel images corresponding to E–J.
Figure 2.

In the absence of Schwann cells, peripheral axons form numerous sodium channel clusters throughout their length. A, The total number of sodium channel clusters per PLLn is shown from 2 to 5 dpf for erbb2st61 mutants (red) and wild-type siblings (blue). Error bars indicate ±1 SD. For each data point, five PLLn were scored from different fish. B, The positions of sodium channel clusters in the PLLn of one erbb2st61 mutant (red) and one wild-type sibling (blue) at 5 dpf were recorded using the underlying somitic boundaries as landmarks. The position of the PLLn ganglion, which is found near the origin of the first somite, is indicated. Each data point represents the number of PLLn clusters that overlie each one-fifth of a somite. Similar results have been observed in erbb3st48 and in clst3 mutants (data not shown).
To determine if the clustering phenotype in these mutants reflects the absence of Schwann cells, or if the loss of a specific erbb2/3 function causes this phenotype, we examined another mutant that lacks Schwann cells in peripheral nerves. The zebrafish colorless mutant (cls; the ortholog of mammalian sox10) is also strongly deficient in the production of Schwann cells (Kelsh and Eisen, 2000; Gilmour et al., 2002). As in erbb2 and erbb3 mutants, cls mutants also form abnormal clusters of sodium channels in the PLLn (Fig. 1 D). The clusters in cls mutants have the same defects in morphology and distribution as erbb2 and erbb3 mutants (Fig. 1 D). From these data, we conclude that aberrant sodium channel clusters form in axons of mutants that lack Schwann cells.
Sodium channels in axonal clusters are known to colocalize with several other axonal molecules. Two of these molecules in particular, ankyrin G and neurofascin, are thought to play critical roles in the assembly of sodium channel clusters (Salzer et al., 2008). We found that sodium channel clusters in erbb2, erbb3, and cls mutants also colocalize with clusters of ankyrin G and neurofascin (Fig. 1 E–H) (data not shown). Given the importance of ankyrin G and neurofascin in sodium channel clustering in mammals, their presence in sodium channel clusters in these mutants suggests that ankyrin G and neurofascin may contribute to cluster initiation in Schwann cell-deficient peripheral nerves.
The absence of Schwann cells in the PLLn of cls, erbb2, and erbb3 mutants has been previously described using a number of criteria (Kelsh and Eisen, 2000; Gilmour et al., 2002; Lyons et al., 2005; Pogoda et al., 2006). To further examine the ultrastructure of the mutant nerves, we performed TEM on the PLLn of clst3, erbb2st61, and erbb3st48 mutants and wild-type siblings at 3 dpf. As expected, all three mutants show the complete absence of Schwann cells associated with the PLLn, including immature and myelinating Schwann cells (Fig. 3 A–D). Consistent with previous reports (Gilmour et al., 2002; Lyons et al., 2005; Pogoda et al., 2006), axons are present, but sometimes defasciculated, in the mutants (Fig. 3 B–D). The mutant nerves are frequently located in the skin in cls, erbb2, and erbb3 mutants (Fig. 3 F) (data not shown), instead of immediately beneath the skin as occurs in the wildtype (Fig. 3 E). Although the axons appear to have normal ultrastructure, the number of axons is reduced in cls, erbb2, and erbb3 mutants (Fig. 3 G).
Figure 3.
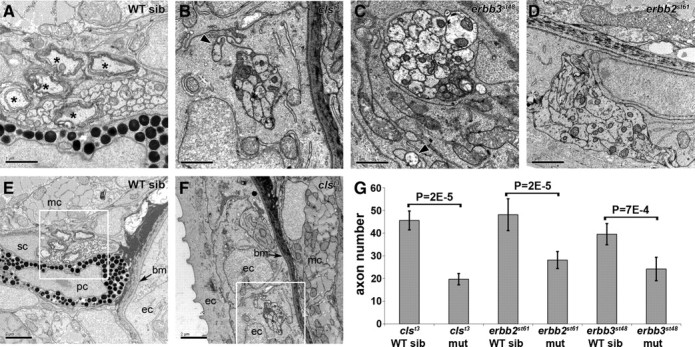
clst3, erbb2st61, and erbb3st48 mutants lack Schwann cells and have fewer PLLn axons. A, TEM of the PLLn from a wild-type sibling at 3 dpf reveals the presence of several myelinated axons (asterisks). B–D, clst3 mutant (B), erbb3st48 mutant (C), and erbb2st61 mutant (D) embryos at 3 dpf completely lack myelin and recognizable Schwann cells. It is common for axons from these mutant nerves to become defasciculated (arrowheads in B and C). E, In the wild type, the PLLn is normally located beneath the skin, adjacent to muscle cells. The basement membrane (arrow) separates the PLLn from the epidermal cells of the skin. F, In Schwann cell-deficient embryos, the PLLn is frequently located superficial to the basement membrane. Scale bars: A–D, 1 μm; E, F, 2 μm. Boxed areas in E and F indicate the region shown in A and B, respectively. G, Quantification of axon number in wild-type siblings and clst3, erbb2st61, and erbb3st48 mutants. The p values for unpaired t test comparisons (2-tailed) are shown. The following number of nerves were scored: clst3 sibling (sib) − 5 nerves from 3 embryos; clst3 mutant (mut) − 7 nerves from 4 embryos; erbb2st61 sib − 8 nerves from 5 embryos; erbb2st61 mut − 7 nerves from 4 embryos; erbb3st48 sib − 11 nerves from 6 embryos; and erbb3st48 mut − 5 nerves from 3 embryos. Error bars represent ±1 SD. mc, Muscle cell; sc, Schwann cell; pc, pigment cell; ec, epidermal cell; bm, basement membrane.
The PLLn contains both afferent and efferent axons (Ghysen and Dambly-Chaudière, 2004). To determine if the aberrant clusters found in the PLLn of Schwann cell-deficient mutants occur preferentially in afferent or efferent axons, we labeled erbb2st61 mutants and wild-type siblings with anti-neurofascin, and anti-tyrosine hydroxylase, which labels efferent axons in the zebrafish PLLn (Faucherre et al., 2009). We found that in both mutants and wild-type siblings, most neurofascin clusters are not associated with tyrosine hydroxylase-positive axons (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), suggesting that these clusters most frequently occur on afferent axons.
Clustering of sodium channels in Schwann cell-deficient peripheral nerves depends upon ankyrin G but not neurofascin
Ankyrin G has essential functions in the clustering of sodium channels at both the AIS and the nodes of Ranvier. To investigate the role of ankyrin G in clustering sodium channels in zebrafish, we used an MO to disrupt the function of the ank3a gene, one of two genes encoding ankyrin G in zebrafish. We injected one-cell stage embryos from intercrosses of erbb2 st61/+ heterozygotes with an MO that targets the neural-specific transcript of ank3a (Kordeli et al., 1995). We found that the number of sodium channel clusters per PLLn in MO-injected wild-type animals was 49% of that seen in water-injected wild-type controls shortly after cluster formation begins at 72 hpf (Fig. 4 A). We also found that ank3a MO-injected embryos have variably reduced levels of sodium channel clustering at the AIS (compare supplemental Fig. S4A,B to supplemental Fig. S4D, available at www.jneurosci.org as supplemental material), which is consistent with ankyrin G's role in recruiting sodium channels to the AIS. At 80 hpf, the morphology of ank3a MO-injected embryos did not differ from that of water-injected controls (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). The total number of axons and the number of myelinated axons in the PLLn did not differ between MO-injected embryos and water-injected embryos (supplemental Fig. S6A,B,D,E, available at www.jneurosci.org as supplemental material), indicating that there is no developmental delay in ank3a MO-injected embryos. Sodium channel clusters were not completely eliminated in ank3a MO-injected embryos, perhaps because of the activity of the duplicate gene ank3b or because the effective concentration of MO is reduced by 72 hpf, when clusters are readily assayed. These results indicate that ankyrin G is necessary for the formation of sodium channel clusters, and thus, its function is conserved between zebrafish and mammals.
We next asked if ankyrin G is required for the formation of clusters in peripheral nerves that lack Schwann cells. In MO-injected erbb2 mutants, we found only 40% of the sodium channel clusters per PLLn that are found in water-injected mutants (Fig. 4 A). These data provide evidence that ankyrin G is also required for the formation of clusters in Schwann cell-deficient peripheral nerves, suggesting that clustering in these mutants occurs by one of the known mechanisms that requires ankyrin G.
To determine if clustering in Schwann cell-deficient nerves occurs through the neurofascin-dependent mechanism, we injected erbb2 mutants and wild-type siblings with an MO that targets the neurofascin gene. The MO did not affect gross morphology compared to water-injected controls (supplemental Fig. S5A,C, available at www.jneurosci.org as supplemental material), and the number and myelination of PLLn axons were unaffected in neurofascin MO-injected embryos (supplemental Fig. S6A,C–E, available at www.jneurosci.org as supplemental material). As expected, the neurofascin MO showed no effect on the ability of sodium channels to cluster at the AIS (supplemental Fig. S4C,D, available at www.jneurosci.org as supplemental material). At other positions in the axons, however, sodium channel clusters were reduced: at 72 hpf, the number of clusters per PLLn in MO-injected wild-type embryos is 46% of that in water-injected, wild-type controls (Fig. 4 A). This result is consistent with the observation that neurofascin is required for clustering at nodes of Ranvier in peripheral nerves (Sherman et al., 2005). Neurofascin was undetectable by immunofluorescence in the remaining sodium channel clusters of morpholino-injected embryos (Fig. 4 B–F), which may indicate that even low levels of neurofascin are sufficient to initiate some clustering at nodes of Ranvier in wild-type embryos. In contrast to wild type, morpholino-injected erbb2 mutant embryos showed no reduction in sodium channel clustering compared to water-injected erbb2 mutants (Fig. 4 A). From our morpholino analysis, we conclude that clustering of sodium channels in Schwann cell-deficient peripheral nerves requires the function of ankyrin G but not neurofascin. Thus, the mode of clustering in these Schwann cell-deficient nerves resembles the axon-intrinsic mechanism that normally functions at the axon initial segment.
Schwann cells restrict axon-intrinsic sodium channel clustering
The foregoing results provide evidence that an axon-intrinsic mechanism drives clustering throughout the length of peripheral nerves in the absence of Schwann cells (Figs. 2 B, 4 A). These data suggest that Schwann cells may have a role in restricting axon-intrinsic clustering to the AIS. To test this hypothesis, we examined how clustering of axonal molecules is affected in zebrafish gpr126 mutants, in which Schwann cells associate with and migrate along axons normally (Fig. 5 A,B), then arrest at the promyelinating stage (Pogoda et al., 2006; Monk et al., 2009). The average number of neurofascin clusters in the PLLn of gpr126 mutants is reduced by 11-fold compared to wild-type siblings (mean = 22 with SD = 7, and mean = 245 with SD = 30, respectively) (Fig. 5 C,D,G). Moreover, the average number of clusters in the gpr126 mutant PLLn is fourfold lower than that found in the Schwann cell-deficient erbb2 mutant nerves (mean = 22 with SD = 7, and mean = 88 with SD = 9, respectively) (Fig. 5 G). These data demonstrate that peripheral nerves that lack Schwann cells have more clusters than peripheral nerves with Schwann cells that are arrested at the promyelinating stage. The marked reduction of neurofascin clusters in gpr126 mutants supports the conclusion that Schwann cells arrested at the promyelinating stage can inhibit axon-intrinsic clustering but cannot initiate clustering of neurofascin at nodes of Ranvier.
Figure 5.
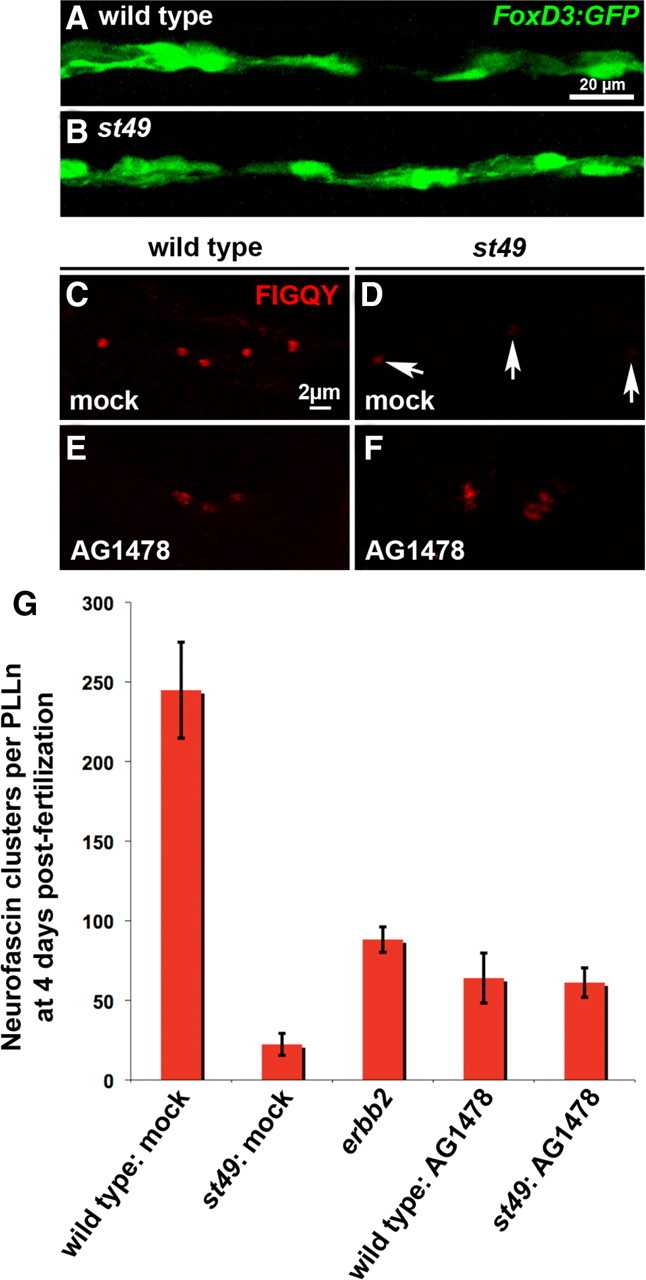
Schwann cells arrested at the promyelinating stage inhibit neurofascin clusters. A, B, Schwann cells labeled by a FoxD3:GFP transgene are present in wild-type siblings (A) and gpr126st49 mutants at 5 dpf (B). C–G, Neurofascin clusters were scored in the PLLn of mock-treated wild-type siblings (C, G), mock-treated gpr126st49 mutants (D, G), AG1478-treated wild-type siblings (E, G), and AG1478-treated gpr126st49 mutants (F, G) using anti-FIGQY at 4 dpf. Arrows in D mark weakly labeled clusters in the gpr126 mutant. In G, one PLLn was scored per embryo, and the number of embryos scored from left to right is 5, 5, 5, 12, and 11.
An important issue in the interpretation of the gpr126 mutant phenotype is the possibility that peripheral nerves in these mutants may not be competent to form clusters of axonal molecules. To determine if gpr126 mutant axons can form clusters when Schwann cells are eliminated, we used the ErbB kinase inhibitor AG1478 to block Schwann cell proliferation and migration in gpr126 mutants. When applied before Schwann cell precursors migrate from the cranial ganglia, AG1478 blocks the migration of Schwann cells along peripheral nerves, producing an erbb2/3-like phenotype (Lyons et al., 2005). We applied 4 μm AG1478 to gpr126 mutants and wild-type siblings from 17 hpf to 4 dpf and then assayed the formation of axonal clusters in the PLLn. Similar to erbb2 and erbb3 mutants, AG1478-treated wild-type fish also form clusters of neurofascin in the PLLn (Fig. 5 E). The mean number of neurofascin clusters in AG1478-treated gpr126 mutants is also comparable to that seen in erbb2 mutants (61 with SD = 9, and 88 with SD = 9, respectively) (Fig. 5 F,G). Importantly, AG1478 treatment increases the average number of neurofascin clusters in gpr126 mutants by 2.8-fold (Fig. 5 G). These experiments demonstrate that the presence of Schwann cells arrested at the promyelinating stage inhibits the formation of neurofascin clusters in gpr126 mutants.
Discussion
Our analysis shows that numerous axonal clusters of sodium channels, neurofascin, and ankyrin G are present throughout the length of peripheral nerves that lack Schwann cells. This analysis was possible in zebrafish, because the Schwann cell-deficient erbb2, erbb3, and cls/sox10 mutants survive for several days beyond the onset of sodium channel clustering. Mammalian mutants for Erbb2, Erbb3, and Sox10 are also strongly deficient in Schwann cells, but these mutants die before sodium channel clustering can be observed (Riethmacher et al., 1997; Morris et al., 1999; Britsch et al., 2001). Several viable mammalian mutants with myelination defects have been analyzed for effects on the clustering of axonal proteins. These mutants generally show a direct positive relationship between the presence of myelin and number of sodium channel clusters (for review, see Poliak and Peles, 2003). However, most of these examples are of hypomyelinating or demyelinating models, which typically have extensive contact between Schwann cells and axons at the time when sodium channel clusters are formed. A notable exception is the severe dystrophic mouse, which has a mutation in laminin α2 (Lama2dy). In Lama2dy mutants, large sections of peripheral nerves up to 10 or more millimeters in length lack Schwann cells (Bradley and Jenkison, 1973). By immunofluorescence and immuno-electron microscopy, Deerinck et al. (1997) found clusters of sodium channels in regions of peripheral nerves from Lama2dy mutant mice that were devoid of Schwann cells. In these Schwann cell-deficient regions of peripheral nerves, sodium channels cocluster with ankyrin G, and the clusters have elongated and asymmetric morphologies much like those we observe in zebrafish Schwann cell-deficient mutants (Fig. 1) (Deerinck et al., 1997). The formation of clusters in Schwann cell-deficient regions of Lama2dy peripheral nerves suggests that Schwann cells have a conserved function in the inhibition of ectopic axon-intrinsic clustering in zebrafish and mammals. This function may allow myelinating Schwann cells to inhibit inappropriate sodium channel clusters at the internode.
The observation that sodium channel clusters, albeit abnormal ones, are present in mutants deficient in Schwann cells (Deerinck et al., 1997; this manuscript) appears to contrast with studies that detected no sodium channel clusters in axons of cultured mammalian dorsal root ganglion (DRG) neurons. For example, Eshed et al. (2005) found that DRG axons in the absence of Schwann cells do not form clusters of neurofascin or sodium channels unless the neurofascin-binding domain of the Schwann cell ligand Gliomedin is added to the culture. This discrepancy may indicate that there are important differences between experiments performed in culture and those performed in vivo. Nonetheless, culture studies and in vivo analyses in mouse and zebrafish all support an essential role for cues from myelinating Schwann cells in establishing the sodium channel clusters that are characteristic of the mature node of Ranvier (Salzer et al., 2008; this manuscript).
Previous work has shown that Schwann cells normally promote sodium channel clustering at nodes, and here we demonstrate that Schwann cells inhibit axon-intrinsic clustering outside of nodes and the AIS (summarized in supplemental Fig. S7, available at www.jneurosci.org as supplemental material). It is possible that these seemingly contradictory functions reflect a dual role for Schwann cell-derived cues in the regulation of ankyrin G. While ankyrin G is known to initiate sodium channel clustering at the AIS, it also plays an essential role in the neurofascin-dependent pathway that functions at the nodes of Ranvier (Dzhashiashvili et al., 2007). The biochemical function of ankyrin G is similar at the AIS and at nodes, so perhaps the primary effect of clustering neurofascin at nodes of Ranvier is to activate the axon-intrinsic mechanism of sodium channel clustering at discrete loci. This convergence of extrinsic and intrinsic factors may ensure the rapid reorganization of axonal proteins so that there is no loss of axonal function during the transition from continuous to saltatory modes of conduction.
In conclusion, we have shown that in the absence of Schwann cells, axonal proteins form clusters in an ankyrin G-dependent manner. Additionally, Schwann cells must transition from the promyelinating stage to the myelinating stage before they stimulate the normal clustering of sodium channels that is characteristic of the node of Ranvier. The presence of axonal clusters in the absence of Schwann cells reveals a role for Schwann cells in the inhibition of axon-intrinsic mechanisms of clustering.
Footnotes
This work was supported by grants to W.S.T. from the National Multiple Sclerosis Society (RG3943-A-2) and the National Institutes of Health (NIH; NS050223). M.G.V. was supported by a fellowship from the NIH. T.D.G. and A.R.R. were supported by an NIH training grant. We thank Matthew Rasband for providing the FIGQY antibody and members of our laboratory for helpful discussion and comments on this manuscript.
References
- Bradley WG, Jenkison M. Abnormalities of peripheral nerves in murine muscular dystrophy. J Neurol Sci. 1973;18:227–247. doi: 10.1016/0022-510x(73)90009-9. [DOI] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerinck TJ, Levinson SR, Bennett GV, Ellisman MH. Clustering of voltage-sensitive sodium channels on axons is independent of direct Schwann cell contact in the dystrophic mouse. J Neurosci. 1997;17:5080–5088. doi: 10.1523/JNEUROSCI.17-13-05080.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL. Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol. 2007;177:857–870. doi: 10.1083/jcb.200612012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed Y, Feinberg K, Poliak S, Sabanay H, Sarig-Nadir O, Spiegel I, Bermingham JR, Jr, Peles E. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron. 2005;47:215–229. doi: 10.1016/j.neuron.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Faucherre A, Pujol-Martí J, Kawakami K, López-Schier H. Afferent neurons of the zebrafish lateral line are strict selectors of hair-cell orientation. PLoS One. 2009;4:e4477. doi: 10.1371/journal.pone.0004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghysen A, Dambly-Chaudière C. Development of the zebrafish lateral line. Curr Opin Neurobiol. 2004;14:67–73. doi: 10.1016/j.conb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Gilmour DT, Maischein HM, Nüsslein-Volhard C. Migration and function of a glial subtype in the vertebrate peripheral nervous system. Neuron. 2002;34:577–588. doi: 10.1016/s0896-6273(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Kelsh RN, Eisen JS. The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development. 2000;127:515–525. doi: 10.1242/dev.127.3.515. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270:2352–2359. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Pogoda HM, Voas MG, Woods IG, Diamond B, Nix R, Arana N, Jacobs J, Talbot WS. erbb3 and erbb2 are essential for Schwann cell migration and myelination in zebrafish. Curr Biol. 2005;15:513–524. doi: 10.1016/j.cub.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Naylor SG, Mercurio S, Dominguez C, Talbot WS. KBP is essential for axonal structure, outgrowth and maintenance in zebrafish, providing insight into the cellular basis of Goldberg-Shprintzen syndrome. Development. 2008;135:599–608. doi: 10.1242/dev.012377. [DOI] [PubMed] [Google Scholar]
- Monk KR, Naylor SG, Glenn TD, Mercurio S, Perlin JR, Dominguez C, Moens CB, Talbot WS. A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science. 2009;325:1402–1405. doi: 10.1126/science.1173474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- Palmer LM, Stuart GJ. Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci. 2006;26:1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoda HM, Sternheim N, Lyons DA, Diamond B, Hawkins TA, Woods IG, Bhatt DH, Franzini-Armstrong C, Dominguez C, Arana N, Jacobs J, Nix R, Fetcho JR, Talbot WS. A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev Biol. 2006;298:118–131. doi: 10.1016/j.ydbio.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- Salzer JL, Brophy PJ, Peles E. Molecular domains of myelinated axons in the peripheral nervous system. Glia. 2008;56:1532–1540. doi: 10.1002/glia.20750. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Voas MG, Lyons DA, Naylor SG, Arana N, Rasband MN, Talbot WS. alphaII-spectrin is essential for assembly of the nodes of Ranvier in myelinated axons. Curr Biol. 2007;17:562–568. doi: 10.1016/j.cub.2007.01.071. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ogawa Y, Hedstrom KL, Rasband MN. betaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol. 2007;176:509–519. doi: 10.1083/jcb.200610128. [DOI] [PMC free article] [PubMed] [Google Scholar]