

Abstract
Microscopy has been one of the most direct and powerful tools since the beginning of biological research. Continued advances such as confocal and two-photon fluorescence microscopy and fluorescent proteins now make imaging useful at a variety of spatial scales (molecules, circuits, cells, tissues, and even whole embryos) and temporal scales (<seconds to days). Zebrafish is uniquely poised to benefit from these continued technological improvements because of its inherent suitability for both imaging and genetics. Here we present an approach called “in toto imaging”. The goal of in toto imaging is to image and track every single cell movement and division that forms a tissue or organ. This approach is powerful for understanding how cell lineage, shape changes, and movements control the morphogenesis of a tissue. When used with transgenic lines, in toto imaging can be used to “digitize” data at single cell level over time from a living organism. This quantitative, digitized data can then serve as the basis for forming models of how biological circuits orchestrate developmental processes.
Keywords: Confocal microscopy, two-photon, in toto imaging, in vivo, timelapse, zebrafish, image analysis, fluorescent, GFP, live cell imaging
1. Introduction
In the relatively short period of their use, zebrafish have already proven their power in the field of developmental biology. Their small size and transparency is also allowing zebrafish researchers to ask developmental questions at higher levels of resolution, namely understanding how development works at a cellular level. This single-cell precision has been one of the real hallmarks of research on C. elegans allowing it to become a powerful model. We feel that zebrafish can match this ability yet in a vertebrate system. We also feel that the natural progression of many current approaches such as development, genetics, genomics, and imaging is to become more and more quantitative and systematic in our understanding of how the biological circuits encode by the genome orchestrate the transformation of an egg into an embryo, and that zebrafish is the best model system in which to integrate these various approaches. Towards this goal we have developed an approach called in toto imaging that seeks to extract quantitative cellular and molecular data from intact developing zebrafish embryos in a high-throughput and systematic fashion. The goal of in toto imaging is to extract complete cell lineages of how tissues and organs are formed, but also to annotate these lineages with quantitative molecular data suitable for making predictive models of development. In toto imaging is pretty technically challenging so here we break down the steps of in toto imaging into labeling, mounting, image acquisition, and image analysis to try to make this approach more widely useable.
2. Materials
10× Tricaine
Make a 0.1% solution of Tricaine in egg water. It will be very acidic.
add 1M Tris pH8.0 to adjust the tricaine solution to 10mM Tris. It will still be a little acidic
slowly add 0.1M NaOH to the tricaine solution to bring it to a final pH of 7.0.
store in dark at 4C for up to 1 week.
300% Danieau buffer
174 mM NaCl
2.1 mM KCl
1.2 mM MgSO4
1.8 mM Ca(NO3)2
15 mM HEPES
pH 7.6
*Use at 30% for raising dechorionated embryos
3. Methods
The steps of in toto imaging can be roughly divided into: labeling, mounting, image acquisition, and image analysis. In toto imaging is technically very difficult (barely possible) for most tissues so it is essential that these four steps be considered together and that all the steps are optimized as a whole. For example, the image analysis is only possible if the images were acquired at high enough signal-to-noise, spatial, and temporal resolution, and this is only possible if the embryos were labeled and mounted well. Thus, if you are having trouble in the last step of image acquisition, it could be due to the first step of labeling not being optimized. For in toto imaging to work, every step must be perfected together, not just gotten by.
3.1 Labeling
There are 2 types of labels used for in toto imaging—segmentation markers and expression markers. Segmentation markers are fluorescent labels that allow all the cells to be individually recognized, tracked, and relevant subcellular compartments to be identified. Expression markers are fluorescent labels that mark some other piece of data pertinent to the specific experiment at hand such as a particular gene expression pattern. For some experiments such as lineage analysis, an expression marker is not required. For segmentation markers, we currently use a combination of a membrane-localized fluorescent protein in one color and a histone-fused fluorescent protein of another color. The histone fusion marks chromatin allowing nuclei to be tracked as cells move. Nuclei tend to be easier to track than whole cells since they have a simpler shape and are more separated. Importantly, the histone fusion also stays localized to chromatin during mitosis allowing daughter nuclei to be linked with their mother. For some tissues such as the neural tube, the nuclei can appear touching even in very thin optical sections making automatic segmentation more difficult. The membrane localized fluorescent protein is useful in such cases because this signal can be used to split touching nuclei. The membrane label is also nice in that it highlights tissue morphology well and along with the histone label allows the nuclear, cytoplasmic, and membrane subcellular compartments to be identified.
Each marker must be a different color fluorescent protein, should be as bright as possible, and should be well separated spectrally. If no expression marker is needed, we currently use histone-EGFP and membrane-mCherry. If an expression marker is used, then the expression marker is done with citrine and the segmentation markers are histone-cerulean and membrane-Cherry. It is important to consider the effects of the “tail” of fluorescent emission and the relative levels of expression of each marker. For example, if the segmentation markers are expressed at very high levels, bleed though into the expression marker channel may be a problem unless it is expressed at similar levels or is well separated spectrally.
The simplest method for labeling with segmentation markers is using RNA injection. This approach allows very high levels of label and thus bright signals to be achieved. Since RNA takes a few hours to get expressed and is degraded after a few days, it is only useful if you are interested in the period of development from 5 hpf to 60 hpf. For earlier or later stages, a transgenic source of segmentation markers should be used.
Labeling using RNA injection:
RNA for histone2B-EGFP and membrane-mCherry can be made using in vitro transcription from the plasmids pCS-H2B-EGFP and pCS-membrane-mCherry available from the author. These plasmids should be linearized with NotI, purified using a Qiagen nucleotide removal spin column, and used as template for in vitro transcription with the Ambion mMessage Machine kit. In vitro transcribed RNA can be purified using a Qiagen RNA cleanup column and quantitated on a NanoDrop spectrophotometer. RNA integrity can also be assessed using gel electrophoresis.
The amount of RNA injected should be determined by titration such that the maximal level of labeling that allows normal development is achieved. Make up injection solutions in the range of 20 ng/ul, 40 ng/ul, and 80 ng/ul of RNA individually and together. Inject these solutions into batches of 1-cell stage embryos. Allow these embryos to develop until 24 hpf and check for any signs of developmental retardation or malformation compared with uninjected embryos. Also check embryos for levels of fluorescence using a fluorescent dissecting microscope. Comparing different batches can be done easier if they are photographed under the same conditions on a fluorescent dissecting microscope and these images compared side by side. The combination of RNA concentrations that gives the brightest labeling in both channels yet still gives normal development should be determined and used subsequently. This is typically 40 ng/ul of each RNA.
Labeling embryos for in toto imaging: Setup male and female fish in mating cages separated by a divider the night before. The next morning remove the dividers and let the fish mate for 10–15 minutes before collecting eggs. It is useful to collect the eggs soon after mating so they can be injected early. Image analysis is much easier when the cells are evenly and uniformly labeled. To achieve uniform labeling, inject eggs between 10 and 25 minutes after fertilization (in the early to middle part of the 1-cell stage). It is also best to inject into the single blastomere or blastodisc rather than into the yolk.
Raise embryos to the appropriate stage. Discard any embryos that are not morphologically normal. Screen remaining embryos on a fluorescent dissecting microscope in both channels. Look for embryos with bright, uniformly distributed labeling with the segmentation markers. Discard any embryos that are dim or have patchy/mosaic expression of the segmentation markers. Select the 5 to 10 best embryos for mounting and transfer to a fresh Petri dish.
3.2 Mounting
Mounting is a critically important step to achieving high quality images. The goals of mounting are: the embryos stays healthy and develops normally; the tissue of interest is positioned such that it can be viewed as directly as possible (through the least amount of overlying tissue) throughout the timelapse; and the tissue of interest is positioned as close to the coverslip as possible (important for using high numerical aperture, short working distance objectives). Zebrafish is often touted as being superior for imaging because of the transparency of its embryos. While certainly true, the suitability of zebrafish embryos for mounting is as important. The first goal of normal development is relatively easy to achieve for zebrafish embryos since they are aquatic, normally develop freely outside their mother, and can readily develop outside their chorion. The last two goals of getting the cells of interest as close to the objective as possible and with the least amount of intervening tissue is made easier in zebrafish embryos by their relatively small size. The exact details of mounting may vary depending on the tissue being studied, but the above goals will still apply.
We present below a general approach for mounting we call megamounts or embryo arrays. These mounts are available for both dorsal and lateral views and allow most parts of the embryo to be imaged continuously across most of embryonic development. Embryo arrays are agarose mounts made from a plastic template. The plastic template creates “embryo shaped” wells in defined positions in the agarose that allow the embryo to develop in a defined orientation (lateral or dorsal). Embryos can develop normally in the mounts for at least 3 days and be continuously imaged during this time.
Tricaine solutions should be made fresh weekly from powder that is <6 months old. The 10× solution must be adjusted to pH7.0. The final working concentration should be empirically determined by testing different concentrations to find the range that prevents movement but permits normal development. The working concentration is normally ~0.013%. If it is much higher, then your tricaine is probably bad and you should buy more.
The templates for the mounts are made from Lucite machined with a 500um bit on a CNC mill to contain the features as shown on the designs for the dorsal or lateral mounts.
Make 100ml of 0.7% agarose in 30% Danieau by dissolving 0.7g agarose in 100ml media by microwave.
Let the agarose cool to 65C.
-
For use on an upright microscope: Add 1ml of 10× tricaine to a 60mm × 15mm Petri dish. Add 9 ml of molten agarose to dish and mix with the tricaine. Gently lay the template onto the molten agarose. You can avoid trapping air bubbles by laying one edge down first. Let set for 1hr.
For use on an inverted microscope. Prepare a coverslipped bottom Petri dish by epoxying a large coverslip to the bottom of a 60mm Petri dish that has had a window cut out of its bottom using a flame heated scalpel. Apply 0.5ml of 0.7% agarose in embryo media with 1× tricaine to the coverslip. Place the template on top and center it. Put a weight (I use a glass vial filled with sand) on top of the template to hold it against the coverslip. Let set 1hr.
Add 10ml of 30% Danieau with 1× tricaine. Let set for 30’ (this helps loosen the template and makes sure the agarose is well set)
Gently remove the template from the agarose by slowly prying up the tabbed edge using forceps. For inverted mounts, it is useful to gently press on the bottom of the coverslip from below with a Kimwipe such that the coverslip and the agarose will slightly bow. Since the template won’t bow, it will become loose from the agarose.
Mounting the embryos: For lateral mount the embryos can be mounted anytime after shield stage. For dorsal mounts, anytime after epiboly. Manually dechorionate embryos in embryo media using sharp forceps.
Transfer the embryos to the mounting chamber. It is easiest to see the wells of the mounting chamber using oblique trans-illumination.
Under a dissecting scope, position 1 embryo in each well using a hairloop. Orient the embryos such that the animal pole is pointing where the head should go and the shield or somties is pointing where the back should go. With lateral mount this is most easily accomplished by first positioning the embryos animal side up in the circular depression within each well. The embryos can then be rotated about their animal-vegetal axis such that the shield is toward the side it should be. The embryos can then be rotated along the D-V axis so that the animal pole is pointing where it should be. With the dorsal mounts, the embryos do not spin easily within the hemispherical depression in each well so the embryos should be dropped into the hemisphere already in the proper orientation. This is most easily accomplished by positioning the embryos in the groove just posterior to the hemisphere with their animal side up and their dorsal facing the posterior end of the well. The embryos can then be rolled forward into the hemisphere such that they undergo a quarter rotation as they fall into the hemisphere resulting in their dorsal side being up and their animal pole facing the anterior of the well.
Coverslipping: A coverslip is placed within the square created by the template in order to hold the embryos in place. A coverslip should be used that is 1mm smaller than the width of the template such that it barely fits within the square depression created by the template (25mm square). The coverslip should be prewetted by soaking it in embryo media for 10 minutes to reduce is hydrophobicity. Properly applying the coverslip without moving the embryos requires practice, especially with the lateral mounts where the embryos can move much more easily. To apply the coverslip, rotate the mount such that the tab that sticks off the square in the agarose depression is to your right. Pick up the wetted coverslip with a forcep in your right hand. Under a dissecting scope, position the left edge of the coverslip in the bottom left edge of the agarose square depression. Lower the right edge (the side being gripped by the forceps) of the coverslip to the surface of the media such that the edge is right at the surface so that the coverslip is half sunk and half floating. At this point, the coverslip should be partly submerged with its left edge in position under the media along the left edge of the agarose depression and its right edge stuck to the surface of the media / floating. You can the release the coverslip and it should stay in this half sunk / half floating position. You should be able to position the coverslip in this position without disturbing the embryos and you can even reposition some of the embryos if you’re careful.
-
Lowering the coverslip: Now take a pair of forceps in both hands. Put the closed point of the forceps in your right hand under the floating edge of the coverslip with your fingers resting on the edge of the Petri dish for support. With the forceps in you left hand press the floating edge of the coverslip under the media such that it sinks onto the forceps in your right hand. Very carefully and slowly lower the forceps in your right hand down and into the agarose such the coverslip will be lowered into the square. Slowly withdraw the forceps through the tabbed area (it is ok to pit the agarose in this area). Check all 4 corners of the coverslip to make sure that they are within the square agarose depression and that the coverslip is lying flat against the agarose.
Lowering the coverslip can also be done using a micromanipulator. After the coverslip is in the half floating / half sunk arrangement, position a pulled glass pipet attached to an xyz micromanipulator under the floating edge of the coverslip. Using forceps press the floating edge under the surface so that it sinks onto the pulled glass pipet. Using the micromanipulator, slowly lower the coverslip.
It is now ready to image! For an upright compound scope, the objectives can be dipped into the media in the Petri dish. For an inverted compound scope, a drop of water can be placed on a water immersion objective and the objective raised to meet the coverslip as normal. For long-term imaging on a dissecting scope, the Petri dish can be covered with a watch glass to prevent evaporation and the embryos observed through the watch glass or it can be covered with a Pertri dish lid containing a coverslip “skylight” for imaging.
3.3 Image Acquisition
After the zebrafish have been mounted the next step is to acquire images. For tracking all the cells in a tissue it is necessary to capture a complete volume of the tissue of interest (an xyz image) and to repeat this over time to create an xyzt image set. Capturing volumetric images of tissues requires the use a confocal or multiphoton microscope. We typically use a laser-scanning confocal for imaging (see Note 1). To segment cells in 3d (across the z-axis) you need high z-resolution (1 um), which requires thin optical sections and a close spacing between optical sections (see Note 2) which in turn requires the use of high numerical aperture objectives (see Note 3). Tracking cells as they move and divide also requires high temporal resolution (~2 minutes). Achieving high signal to noise, high spatial resolution, and high temporal resolution requires a difficult balancing act. It is easy to achieve one at the expense of another but can be challenging to achieve all these things and a healthy embryo at the same time.
Actually acquiring all the images can also be difficult due to the sheer number of images and the amount of disk space they require. An in toto image set can be >100Gb so it is much easier if not essential that these images be stored as multiple files rather than one huge file. It is also quite useful to be able to adjust the imaging parameters (e.g. laser power, gain, location of z-stack) during image acquisition to account for photobleaching and drift of the embryo. Some image acquisition routines that come with microscopes block out user interaction during acquisition.
We have developed an image acquisition macro for Zeiss microscopes called Megacapture that automates the collection of in toto image sets. This macro allows you to set the number of time points, time interval, number of z-sections, and z-interval. It saves each xy image as a separate file which makes file management of huge image sets easier, and can also optionally save images compressed saving valuable disk space. Megacapture can also be used with a motorized stage to automate tiling across embryos and for capturing multiple embryos in a row/column format such as with embryo arrays. And finally, Megacapture allows the image acquisition parameters to be altered during an experiment and separately records the imaging parameters for each image. It can be downloaded from the software section of digitalfish.org.
Install the proper objective onto the microscope (see Note 3).
Prewarm the microscope to 28C for 2hrs prior to starting imaging. The use of an incubator box that encloses the entire microscope is preferable because it is easier to maintain a uniformly controlled temperature which is essential for proper embryonic development and to prevent focal drift caused by thermal expansion/contraction.
Mount embryos as discussed above and transfer the dish to the microscope. Your microscope should have a stage insert capable of securely holding a Petri dish. For an inverted microscope, add water to the objective and then place the Petri dish on top with your embryo directly over the front lens of the objective. For an upright microscope, the 30% Danieau in the Petri dish can serve as the immersion media.
Find your embryo. This can be done using bright field and manually adjusting the focus while looking through the oculars until the embryo is in focus. If you mounted multiple embryos, scan through all the embryos to find the one that is mounted best and has the brightest and most uniform labeling with the segmentation markers. Center the tissue of interest in the field of view.
Setup your image acquisition parameters. We have been successful on a Zeiss 510 with the following: 25mW 488nm laser for exciting EGFP set to a power of <5%, 5 mW 543nm for mCherry set to a laser power of <15%, the emission filter for EGFP set to 500–530nm band pass, the emission filter for mCherry set to a 600nm long pass, and both pinholes set to 1 Airy unit. We do “single tracking” which means both channels are captured simultaneously. It is more difficult to get cleanly separated and balanced channels using single tracking but it allows the imaging to be done twice as quickly. We also use bidirectional scanning in which pixels are captured on both the forward and backward pass of the laser. This can cause aliasing artifacts but also doubles the speed of acquisition. We use 1024×1024 pixel images and scan at the maximal scan rate which results in a speed of just under 1 second per xy image. The gain and background should be adjusted such that the full dynamic range of the signal is captured—the gain should be increased until a small percentage of pixels in the brightest parts of the image have maxed out and the background should be set such that only pixels in area where there is no signal have a minimal value. A pixel depth of 8-bits is sufficient to capture the informational content of fluorescent images taken under these condition due to their inherent “shot noise” (Poisson noise).
Through the microscope control software, set the top and bottom limits of your z-stack. To achieve a 2 minute time resolution with a 1 second frame rate, you are limited to about 100 images per z-stack after some time for microscope movement and saving images is accounted for. At 1um z-spacing, this gives a maximal z-stack size of 100um. In setting the upper and lower limits of your z-stack you should bracket your tissue of interest by several microns to allow for focal drift of the microscope and normal morphogenetic movements. You should also be able to predict the direction and amount of any tissue movements caused by morphogenesis and set the block of space that you image such that your tissue grows within this space.
Click start on Megacapture and hope for the best. Watch the first few z-stacks get captured and make sure they are being saved properly.
“Babying” the microscope. To play it safe, it is a good idea to check on the imaging every hour or two throughout the timelapse. It might be necessary to add immersion water to make up for any evaporation. On an upright microscope, water can just be added to the Petri dish with a pipet. On an inverted microscope, water can be added between the objective and the coverslipped bottom Petri dish using a flame-pulled P1000 (blue) plastic pipet tip. Laser power and gain can be adjusted to make up for any photobleaching/degradation of the segmentation markers or changes in the expression levels of expression markers. The xy stage position and z-stack limits can also be adjusted to account for morphogenetic movement. Ideally, all these adjustments should be made in the short gap in between time points.
At the end of the timelapse, make sure all the images are safely stored to hard drive or DVD. Clean the objective front lens with fresh water, blot dry, and store safely. You can optionally unmount the embryo that was imaged by carefully removing the upper coverslip and then raise the embryo long enough to convince yourself that it is healthy and did not suffer any ill effects from imaging. Catch up on sleep before proceeding.
3.4 Image Analysis
The goal of image analysis for in toto imaging is to make biological sense of the huge amount of image data. There are two important components to image analysis. The first is visualization. In toto imaging generates huge image sets that are 4-dimensional. There is a basic need just to be able to picture how the tissue is moving and changing shape within the large image sets over the course of the experiment. The second component of image analysis is the actual quantitative analysis. This comes down to converting the pixel-based data that is in the images into cell-based data that is useful for a biologist. This process of recognizing and demarcating cells, cell tracks, and cell lineages within images is called “segmentation”. The enormity of the number of images and cells makes manual segmentation unfeasible. Automatic segmentation is the process in which a computer program automatically parses an image set to recognize and demarcate objects of interest. It has been a very active area of research in computer science and artificial intelligence for several decades because it turns out that a human’s ability to recognize objects in their surroundings is quite complex and very difficult to mimic with a computer. Cell segmentation in particular can now be done fairly routinely for well isolated cells such as those grown in flat culture and imaged in 2d. However, automatic segmentation still poses a significant challenge for densely packed cells in real tissue and for performing segmentation in 3d and in 4d as is needed for in toto imaging.
We have invested a lot of effort into developing a software package called GoFigure to address the image analysis needs of in toto imaging. GoFigure allows for visualization of large 4-dimensional image sets. It allows the user to work with image sets larger than the physical memory of the computer (which is typically 30× smaller) by providing different methods of dynamically subsetting the data. The default is to load all the z-sections for the current time point to form an xyz subset and all the timepoints for the current focal plane to form an xyt subset. GoFigure can also load for example every other time point or z-section, downsample the images in xy, or crop the loaded images along the z-axis or time axis to facilitate working with large image sets.
GoFigure allows segmentation to be done manually as well as automatically using several different segmentation algorithms. GoFigure breaks down segmentation into several steps corresponding to the different dimensions of the image set: namely, figures correspond to the 2d objects such as sections through cells, meshes correspond to 3d objects such as a whole cell, tracks correspond to 4d objects such as the movement of a cell over time, and lineages represent the branched trees that represent cell divisions. GoFigure provides different algorithms for automatically generating each of these levels of segmentation.
Download and install GoFigure. GoFigure currently runs on Windows but we will soon release a cross-platform version of GoFigure that will run on Windows, Mac, and Unix. GoFigure can be downloaded from the software section of digitalfish.org.
Import images into GoFigure. GoFigure provides several methods for importing images. If images were captured using MegaCapture, then they can be automatically imported with all of their proper dimensions and image capture settings into GoFigure. GoFigure can also import collections of raw images that have their coordinates in z and time indicated in their filenames using a wild card in the name. We are currently developing import filters for Zeiss and other format image collections.
Visualize your image set to ensure you captured the complete volume and complete time window. Areas where the embryo moved can be identified as discontinuities in the xz or yz computed cross-sections.
Manually segment ~10 representative cells. This can be done using the pencil tool and drawing outlines around each cell using the mouse.
Select these cells by clicking on them and use them to train the automatic segmentation tool. GoFigure uses the shape and size of what cells should look like to help guide the segmentation process.
Click on the automatic segmentation button to segment other cells in the image set. Depending on the segmentation algorithm this procedure may need to be repeated to build progressively higher dimension segmented objects.
Manually edit the automatically segmented objects. Automatic segmentation saves a great deal of time but it can make both false positives and false negatives so the results should be verified by a well trained human eye.
Once the data is segmented, it is possible to perform cell based quantitative analysis. The cell size, fluorescent content, location, speed, shape, and many other parameters is displayed in the “list views” and can be used to generate interactive histograms and scattergrams within GoFigure
Figure 1.

Design for a lateral embryo array template
Figure 2.

Design for a dorsal embryo array template.
Figure 3.

Correct initial position for shield stage embryos in lateral mounts. The vegetal pole should be pointing ~20 degrees above the horizontal and ~20 degrees out of the page
Figure 4.
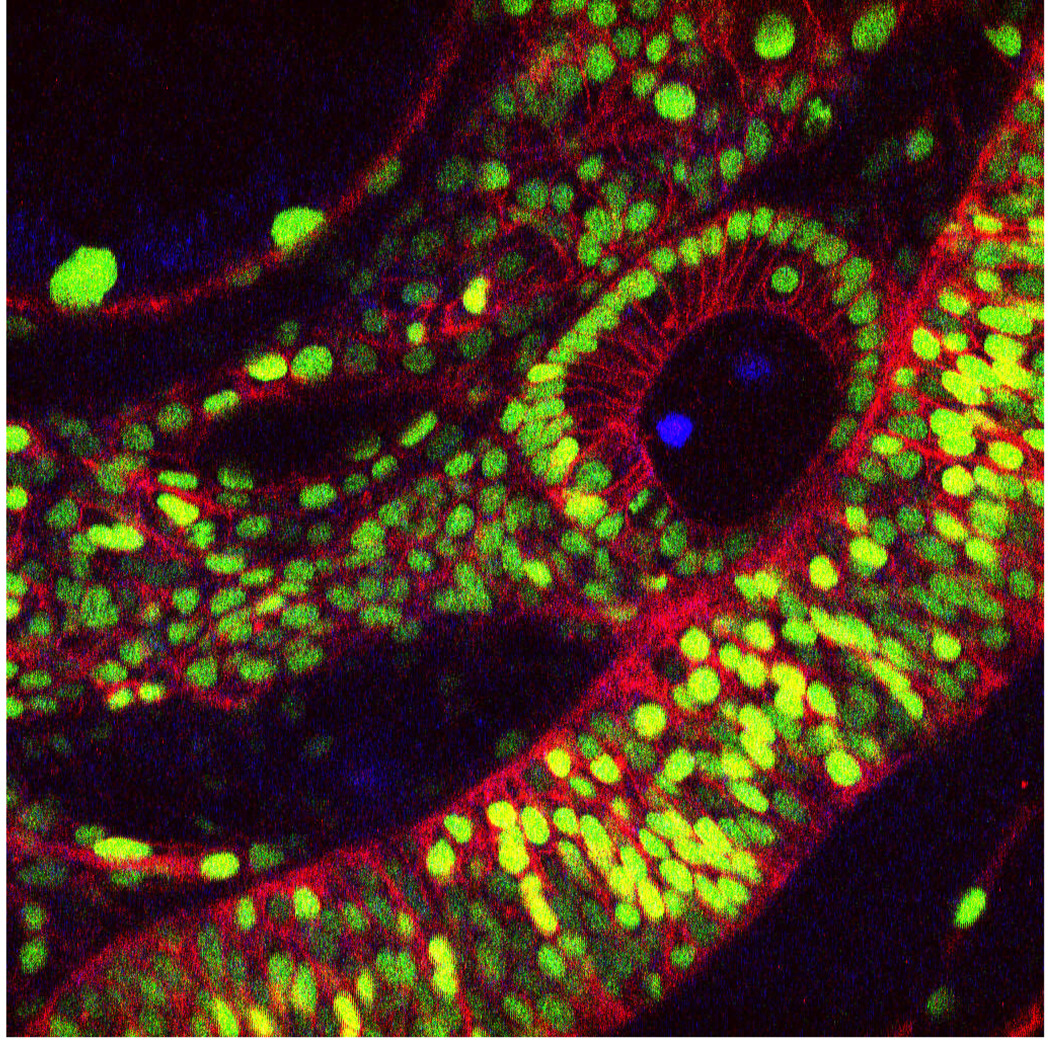
A single xy image extracted from a very large in toto image to show a typical image. This image is a dorsal-lateral view of the otic vesicle and hindbrain. Nuclei are marked in green with H2B-EGFP and cell membranes are marked in red with a membrane localized mCherry.
Footnotes
A spinning disc confocal allows too much cross talk between pinholes with a widely labeled specimen such as is used in in toto imaging causing image degradation. Two-photon microscopy can be used but it is more expensive, more difficult to maximally excite multiple fluorescent proteins simultaneously (since there is typically just a single tunable laser line), and the resolution is twice as bad (since the wavelength is twice as long). Laser scanning confocal can typically achieve good enough quality images for cell tracking to a depth of 100um. For deeper tissues, two photon may be preferable. Two photon also can provide less photobleaching and photodamage, which are typically much less of a problem in zebrafish than other systems with properly done confocal imaging.
Achieving thin optical sections requires the use of a tight pinhole (we aim for 1 Airy unit) and a high numerical aperture objective (>1.0). The use of a high numerical aperture objective is absolutely imperative. While lateral resolution is inversely proportional to the NA, axial resolution is inversely proportional to the square of the NA. In practice, resolution is often limited by signal to noise ratio (SNR) in addition to diffraction, but NA also helps here since signal is directly proportional to the square of the NA. Optical section spacing should ideally be set to half of the optical section thickness to achieve proper sampling across the z-axis according to Nyquist criteria. However, in practice there is often just not enough time in between time-points to capture that many z-sections so we often settle for a z-section spacing of 1um.
There are only a few objectives that are really suitable for in toto imaging. A suitable objective should have an NA of >1.0, enough working distance to reach the cells being studied (200–600um), and be water immersion. Zeiss makes two 40× objectives that meet these criteria—the 40× C-Apochromat 1.2NA (290um) and the 40× LD C-Apochromat 1.1 NA (600um working distance). Zeiss, Leica, and Olympus have also all come out with 20× objectives with NAs of 0.95 to 1.05 and long working distances.