

Abstract
Since the late 1980s, intrathecal (IT) analgesic therapy has improved, and implantable IT drug delivery devices have become increasingly sophisticated. Physicians and patients now have myriad more options for agents and their combination, as well as for refining their delivery. As recently as 2007, The Polyanalgesic Consensus Conference of expert panelists updated its algorithm for drug selection in IT polyanalgesia. We review this algorithm and the emerging therapy included. This article provides an update on newly approved as well as emerging IT agents and the advances in technology for their delivery.
Keywords: Intrathecal drug delivery, Ziconotide
Introduction
Since the late 1980s, intrathecal (IT) analgesic therapy has increasingly become an accepted alternative to standard medical management for the treatment of persistent pain [1]. Because drug doses are much lower when applied directly to the IT space compared with oral or parenteral application, side effects and drug toxicities are often much less. With the development of implantable IT drug delivery devices, patients may benefit from continuous IT administration of analgesics for years without fear of infection or dislodgment. Newer sophisticated implanted IT drug delivery systems and drug development allow clinicians to further improve analgesia and limit adverse effects through combination therapy and precise control over delivery [2•] (Table 1). Clinicians may take advantage of the wide variety of available agents and devices to tailor treatment regimens to individual patients.
Table 1.
2007 polyanalgesic algorithm for intrathecal therapies
| Treatment category | Intrathecal drugs |
|---|---|
| First-Line drugs | Morphine |
| Hydromorphone | |
| Ziconotide | |
| Second-Line drugs | Fentanyl |
| Morphine/hydromorphone + ziconotide | |
| Morphine/hydromorphone + bupivacaine/clonidine | |
| Third-Line drugs | Clonidine |
| Morphine/hydromorphone/fentanyl/bupivacaine + clonidine + ziconotide | |
| Fourth-Line drugs | Sufentanil |
| Sufentanil + bupivacaine + clonidine + ziconotide | |
| Fifth-Line drugs | Ropivacaine |
| Buprenorphine | |
| Midazolam | |
| Meperidine | |
| Ketorolac | |
| Sixth-Line drugs | Experimental agents |
| Gabapentin | |
| Octreotide | |
| Neostigmine | |
| Adenosine | |
| Xen2174 | |
| ZGK 160 |
(Data from Deer et al. [2•])
Three Polyanalgesic Consensus Conferences of expert panelists have convened since 2000 for the purpose of formulating an algorithm for drug selection in IT polyanalgesia. These guidelines were published in 2000, 2003, and 2007 and were based on “best evidence” and expert opinion, and included guidelines for utilizing an IT drug selection algorithm [2•, 3, 4], the results of surveys given to clinicians in the field [2•], and directives for future studies [5].
This article provides an update on ziconotide, a recently approved IT agent, as well as new and emerging agents. New and emerging technology is also discussed.
First-Line Drugs: Morphine, Ziconotide, Hydromorphone
The 2007 Polyanalgesic Consensus Conference labeled morphine, ziconotide, and hydromorphone as first-line therapy for IT application. Currently, only morphine and ziconotide have approval from the US Food and Drug Administration (FDA) for IT use. Hydromorphone has been included as first-line therapy due to its widespread clinical use and safety profile.
Ziconotide is an N-type calcium channel antagonist that is effective for the treatment of neuropathic, nociceptive, and mixed neuropathic/nociceptive pain. Ziconotide blocks the N-type calcium channel located on the presynaptic terminal of dorsal horn C fibers. By blocking calcium entry into the presynaptic nerve terminal, ziconotide prevents the release of neurotransmitters into the synapse. Morphine, which acts on the µ-opioid receptor, is linked to this calcium channel via a G protein–coupled mechanism (Fig. 1). Whereas the inhibition of the N-type calcium channel by ziconotide is direct, the inhibition of the calcium channels by morphine is indirect and partial for several reasons. First, not all µ receptors are linked to N-type calcium channels. In addition, tolerance functionally uncouples the µ receptor and the calcium channel. Due to the direct action of ziconotide on the calcium channel, tolerance does not occur [6]. Ziconotide is a nonopioid analgesic approved by the FDA for IT delivery in 2004. The efficacy of ziconotide in providing pain relief has been demonstrated in several trials, prompting its addition to the 2007 Polyanalgesic Consensus Conference panel list of level 1 drugs [2•, 7–10]. The panel recommended ziconotide as a viable alternative for patients who cannot tolerate IT administration of morphine and/or hydromorphone [2•].
Fig. 1.
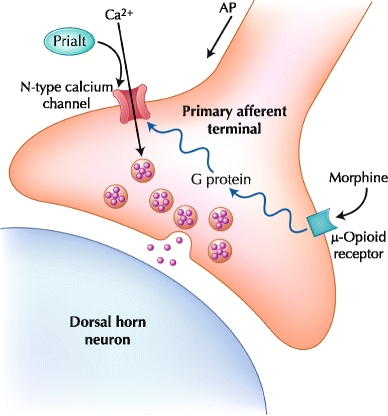
A nerve terminal of a primary afferent nociceptor is depicted, which is stimulated by noxious stimuli in peripheral tissues such as the skin or joints. Transmitters (eg, glutamate and substance P) released from these neurons stimulate secondary neurons in the dorsal horn of the spinal cord, which send the noxious signal to the brain. AP—action potential. (Courtesy of Elan Corporation)
Ziconotide’s structure has implications for its therapeutic use. It is relatively large compared with other analgesics (eg, molecular weight is 2,639 Da compared with 285 Da for morphine and 180 Da for aspirin); thus, diffusion is relatively slower. At physiological pH, ziconotide is permanently charged in contrast to other analgesics. Permanently charged drugs penetrate into tissues slower than drugs that alternate between charged and uncharged forms at physiological pH. For example, morphine has one ionizable amino group, so its freebase (neutral, uncharged) form and its salt (positively charged) form are in equilibrium. The freebase readily permeates hydrophobic tissue barriers, and the salt readily diffuses in aqueous spaces of tissues. Therefore, morphine distributes relatively efficiently into the spinal cord and brain when administered orally or parenterally. Three disulfide bonds wrap ziconotide into a molecular ball. This and the modified N and C termini confer resistance to cerebrospinal fluid (CSF) peptidases. Thus, clearance of ziconotide from CSF is by the bulk flow of CSF, not by metabolism in the CSF. The elimination half-life of IT ziconotide from CSF in humans averaged 4.6 h. The methionine amino acid is susceptible to oxidation by molecular oxygen. Therefore, strategies to limit exposure to oxygen are instituted. For example, the drug solution is sparged with nitrogen (bubbled with N2 gas to drive off O2) before the vial is capped for packaging, storage, and shipping. Other drugs (eg, morphine, hydromorphone, baclofen) can increase the rate of oxidation of ziconotide. Oxidized ziconotide is less active than ziconotide, so a pump containing a combination of ziconotide and another medication may need to be refilled more often. For this reason, the package insert for ziconotide does not recommend using the drug in combination with other drugs. However, a vast amount of clinical experience is surfacing on the use of ziconotide in combination with a variety of drugs with no adverse events reported. However, there are only two publications on the combination of morphine and ziconotide. In one study, ziconotide was added to morphine in 26 patients on a stable IT morphine regimen [11]. There was a mean 14.5% reduction in pain and a 14.3% reduction in analgesic use at week 5; however, response was highly variable. In a second study, morphine was added to 25 patients on stable IT ziconotide therapy [12]. This resulted in a mean 26.3% reduction in pain and a 49.1% reduction in opioid consumption at week 4. These studies suggest that there is a synergism between morphine and ziconotide.
There were two initial double-blind, placebo-controlled studies on ziconotide, one in malignant pain and one in nonmalignant pain. Both studies were fast titration studies with rapid increases in ziconotide dose over 5–10 days. The studies resulted in dramatic percent changes in pain in the ziconotide group over placebo (53.1% vs 18.1% in the malignant study, and 31.2% vs 6% in the nonmalignant study); however, in both studies there was a much higher discontinuation rate in the ziconotide group [8, 10]. This prompted a double-blind, placebo-controlled slow titration study in which the ziconotide was titrated at a slower rate over 3 weeks. Although the percent change in pain in the ziconotide group versus placebo was not as dramatic (14.4% vs 6.4%), it still reached statistical significance. In addition, there was a much lower discontinuation rate and better tolerability [7]. These studies demonstrated that ziconotide has a dose-dependent effect, which can be titrated to analgesia while minimizing side effects. In addition, the studies also showed a significant effect on the clinical global impression of pain control and satisfaction, as well as a significant improvement in sleep.
Adverse reactions have not been reported following accidental overdoses of ziconotide into the IT space or tissue pocket as in overflow, system fracture, or pump miss on refill [13]. IT ziconotide may be abruptly discontinued without major withdrawal symptoms in patients who do not tolerate the drug because of adverse effects [10]. However, despite having demonstrated efficacy, ziconotide is associated with several adverse effects. The most frequent adverse effects of IT ziconotide in descending order are memory impairment, dizziness, nystagmus, speech disorder, nervousness, somnolence, and abnormal gait [14]. Other adverse effects reported in clinical trials and case studies include (but are not limited to) elevated creatinine kinase levels, sedation, nausea, headache, lightheadedness, depression, confusion, ataxia, and emotional distress, with certain symptoms possibly correlated with the rate of infusion [2•]. Although some adverse effects correlate with infusion rate, they have not been found to be dose-dependent [2•, 14]. Several cases of accidental overdoses of ziconotide have occurred. In all cases the patients had normal respirations and cardiovascular function and recovered spontaneously [15].
IT ziconotide should be initiated at no more than 2.4 µg per day (0.1 µg/hr) and titrated to patient response. The dosage may be increased by up to 2.4 µg per day (0.1 µg/hr) at intervals no more than two to three times per week up to a recommended maximum of 19.2 µg per day (0.8 µg/hr). The effective dose of ziconotide for analgesia is variable. The package insert does not specify the length of titration. The full analgesic effect of a new dose of ziconotide may not be fully realized for several days following a dose change. Clinical experience suggests that ziconotide should be started at a lower dose than 2.4 µg per day, titrate the dose approximately once per week, and use an overall titration period longer than 3 weeks. Faster titration is not recommended due to increased adverse events and should only be used if there is an urgent need for analgesia that outweighs the risk to patient safety.
Second- Through Fourth-Line Therapy: Fentanyl, Bupivacaine, Clonidine, Sufentanil
Because of improved efficacy and reduced adverse effects such as granuloma formation, fentanyl and drug mixtures, including bupivacaine and clonidine, are second-line drugs; clonidine as a single drug is third-line [13]. Sufentanil is ranked fourth-line and has also been shown to reduce risk of granuloma formation [13].
Clonidine binds to pre- and postsynaptic α2 receptors in the dorsal horn. Activation of these receptors depresses presynaptive C-fiber transmitter release and hyperpolarizes the postsynaptic membrane through the Gi-coupled potassium channel [16]. Clonidine is FDA approved for epidural use in cancer pain only based on a large clinical trial in 85 cancer patients [17]. However, it is most commonly used intrathecally for the treatment of chronic neuropathic pain based on extensive preclinical literature and clinical experience. Clonidine is generally well tolerated, with hypotension, bradycardia, and sedation occurring with overdose.
Bupivacaine is rarely used as a single agent and most commonly used in combination with an opioid. Doses less than 20 mg per day are generally well tolerated. Higher doses usually result in tachyphylaxis and can lead to sensory and motor disturbances, as well as urinary retention and orthostatic hypotension [18].
Fentanyl and sufentanil are highly lipophilic µ-opioid agonists. There are no reports or studies in the literature on the use of fentanyl and sufentanil for chronic spinal drug delivery. However, there is a lot of experience with the acute delivery of these agents for postoperative and obstetric pain. Fifteen micrograms of sufentanil delivered acutely has been shown to be well tolerated with anesthetic properties equivalent to 5% lidocaine [19]. Unlike morphine and hydromorphone, which are highly hydrophilic, fentanyl and sufentanil are highly lipophilic with a rapid onset when delivered via acute blousing. This “kinetic-based potency” makes these agents attractive when using the Personal Therapy Manager or treating breakthrough pain (see “Future Directions—New Technologies” for discussion).
Fifth-Line Therapy: Ropivacaine, Buprenorphine, Midazolam, Meperidine, Ketorolac
Buprenorphine is a partial µ-opioid receptor agonist and κ-opioid receptor antagonist that improves pain when administered intrathecally [20]. Although buprenorphine is a partial µ agonist, its high affinity for the µ receptor leads to a preferential agonist effect. Because it is an antagonist at the κ receptor, it may minimize the dysphoric effects seen with full agonists that reach the supraspinal regions [21].
Midazolam is a benzodiazepine-receptor agonist that is a potential adjunctive therapy to IT opioid treatment. Preclinical and clinical investigations have found midazolam to contribute to pain reduction without major adverse effects [22]. However, rat studies reveal possible neurotoxicity [23]. Furthermore, the commercially available formulation of midazolam available in the United States contains a preservative; therefore, midazolam should not be used in patients with noncancer pain unless they are at end of life [2•].
The cyclooxygenase (COX) inhibitor ketorolac has been investigated for IT delivery with promising results. COX is released at the spinal level in response to acute pain and peripheral inflammation, contributing to central sensitization [24]. Therefore, IT delivery of COX inhibitors theoretically would reduce pain and central sensitization. Animal data suggest that IT delivery of COX inhibitors can do just that [24]. Safety studies in healthy volunteers have not identified any adverse neurological or pain regulatory effects [25, 26].
There are no studies or reports in the literature on the use of ropivacaine and meperidine for IT delivery. However, both agents have been used extensively for the treatment of postoperative pain. Meperidine has been used via acute blousing as an anesthetic agent with good tolerability [27].
Sixth-Line/Experimental Agents: Gabapentin, Octreotide, Corpeptide, Neostigmine, Adenosine, XEN2174, AM336, XEN, ZGX160
The potential to improve safety and efficacy of treatment drives the development of new therapeutic agents. By expanding the available choices of IT agents, clinicians are afforded the opportunity to further individualize treatment regimens to meet patient needs. However, until safety may be thoroughly investigated, new agents should be used with caution. Although not all FDA approved for IT administration, the agents categorized as first- through fifth-line are generally accepted as safe and effective for IT use in cancer and noncancer pain. Experimental agents should only be used in terminal patients.
Gabapentin is a γ-aminobutyric acid analogue with FDA approval for the treatment of epilepsy and postherpetic neuralgia. Although gabapentin’s exact mechanism of action is unknown, it is thought to bind to the α2δ subunit of voltage-gated calcium channels resulting in the inhibition of glutamate release in the spinal dorsal horn [28] or to activate the noradrenergic system after nerve injury [2•, 29]. Gabapentin delivered intrathecally in various rat models of pain demonstrate a very favorable analgesic profile [29].
Gabapentin has saturable transport both in the gut and at the blood–brain barrier, making it a poor oral and systemic agent [30]. Given the vast amount of preclinical studies supporting a spinal mechanism together with its poor absorption and blood–brain barrier penetration, the IT delivery of gabapentin is very attractive. Gabapentin is not yet FDA approved for IT use in humans; however, a phase 2 trial is in progress.
Octreotide is a synthetic octapeptide of the growth hormone somatostatin [31]. Because somatostatin is located in the substantia gelatinosa and mitigates nociception, octreotide has been investigated for similar effect. In a preclinical study of rats with chronic constriction injury of the sciatic nerve, IT octreotide reduced the behavioral effects of thermal hyperalgesia [32]. In a dog model, IT octreotide infusions of 40 µg per hour were not neurotoxic [31]. Human use, though limited, has also not revealed neurotoxicity. IT octreotide administered to patients with cancer pain chronically over 5 years reduced pain without adverse side effects [33]. One prospective, double-blind human study of IT octreotide at doses of 405–650 µg per day did not reveal any neurotoxicity or adverse side effects [31]. However, a need for frequent pump refills may be evidence of potential for tolerance [2•].
Adenosine is an endogenous purine nucleoside that modulates many physiological processes, including those in the heart and central nervous system. Adenosine is an endogenous ligand that acts at four types of spinal receptors to modulate pain transmission [34]. Animal models show that systemic opioids increase spinal adenosine release [35]. IT adenosine does not inhibit acute pain [36] and has not been found effective in reducing postoperative pain when delivered 30 min prior to anesthesia [37]. Clinically, low-dose IT adenosine is more effective at reducing allodynia and hyperalgesia than spontaneous pain [38]. Increasing evidence suggests that IT adenosine can be effective in the treatment of neuropathic pain in humans [2•, 39–41]. Although preclinical studies have not found adenosine to be neurotoxic in rat spinal cord, further investigation into adenosine’s neurotoxic profile is warranted before the drug can be recommended for clinical use [2•].
Xen2174 is a chi-conopeptide derived from the venom of the marine cone snail Conus marmoreus [42]. Like tricyclic antidepressants, chi-conopeptides inhibit the norepinephrine transporter [42], making them an attractive potential treatment for chronic neuropathic pain. Unlike tricyclic antidepressants, chi-conopeptides are highly selective for the norepinephrine transporter, and thus less likely to cause side effects. A study in rats with either a chronic constriction injury of the sciatic nerve or an L5/L6 spinal nerve injury comparing Xen2174 with tricyclic antidepressants and clonidine found IT Xen2174 to reduce allodynia [43]. The antiallodynic, antihyperalgesic, and antinociceptive effect of IT Xen2174 may be due to upregulation of descending noradrenergic inhibition in the dorsal horn [42].
CGX-1160 is a conopeptide-based drug that produces analgesia through activation of the neurotensin receptor type 1 (NTR1) [42]. The mechanism of NTR1-induced antinociception is unknown. The biotechnology company Cognetix, Inc. has been developing CGX-1160 for IT use and was granted an Orphan Drug designation for use in neuropathic pain associated with spinal cord injury by the FDA in 2005 [42]. A phase 1b clinical trial at Brigham and Women’s Hospital in Boston found CGX-1160 to be safe and effective for chronic intractable pain in a small group spinal cord–injured patients [42].
Resiniferatoxin is an investigational drug that desensitizes primary dorsal root ganglion neurons [44]. Resiniferatoxin is a potent capsaicin analog that has been found to produce analgesia in animal studies [44]. A phase 1, nonrandomized, open-label, uncontrolled clinical trial of IT resiniferatoxin in advanced cancer patients with severe pain is currently underway to determine the effect of treatment in humans [45].
P-Saporin is a neurotoxin that selectively destroys cells containing neurokinin-1 receptor neurons [46]. Because neurokinin-1 receptor neurons transmit pain signals from the spinal dorsal horn to the brain, their destruction decreases pain signaling [46]. Animal studies have demonstrated reduction in pain-related behaviors without long-lasting toxicity or adverse effects [46, 47]. P-Saporin is currently being evaluated for IT use in cancer patients with chronic intractable pain [47].
Discussion on the Question of Efficacy of Chronic Spinal Drugs
With the exception of ziconotide, there are no prospective randomized controlled trials on the other agents used for chronic IT therapy. From the reports in the literature, it is difficult to draw conclusions on the efficacy of this therapy due to many deficiencies, including 1) lack of psychological assessment, 2) no mention of the methods used to screen patients for responsiveness to intraspinal drug therapy, 3) no control groups, randomization, or blinding, 4) no definition of the pain syndrome, 5) no standardization of the methods used to assess outcome, 6) no standard protocols for selecting, increasing, or changing the drug used for intraspinal drug therapy, and 7) the studies to date are short to intermediate follow-up. In all fairness, the criticisms of these studies are inherent to the nature of spinal drug delivery in that it is a highly invasive therapy that makes it difficult to study using randomized controlled trials.
One large study compared spinal drug delivery with comprehensive medical management (CMM) of cancer pain [48]. This study randomly assigned 202 patients to an implantable drug delivery system (IDDS) or CMM. Clinical success was defined as ≥ 20% reduction in pain scores or equal scores with a ≥ 20% reduction in toxicity. More IDDS patients achieved success and more IDDS patients achieved ≥ 20% reduction in both pain and toxicity. Although there was a nonsignificant change in mean pain score between groups, the IDDS patients had a significantly greater change in toxicity scores. IDDS patients also had improved survival, with 53.9% survival at 6 months compared with 37.2% in the CMM group.
Combination Spinal Drug Therapies
There are several persuasive reasons to assume that the codelivery of agents with different mechanisms of action may be therapeutically advantageous. First, many clinical pain states are a composite of several mechanisms (eg, acute afferent drive from the injured site that leads to a persistent facilitated state and the expression of long-term persistent changes in nervous system function after nerve injury). In such states, it is reasonable to presume that an agent with defined mechanisms of action may modulate one component, but not all components of the systems that underlie the specific pain state. Second, there is strong evidence that the several agents may not display cross tolerance (eg, µ and ∂ and µ and α2) and attenuate the concurrent development of tolerance otherwise associated with an equipotent dose of a single drug when given alone [49]. Finally, even for pain states that are mediated by the same mechanism, agents that act on different elements of the systems may display nonlinear interactions that enhance the therapeutic ratio. Numerous studies have emphasized that spinally delivered agents may show positively synergistic interactions with regard to modulation of nociceptive processing [50]. In the case of the interaction between spinal µ and α2 agonists, enhanced nociception can result without an attendant increasing respiratory depression or hypotension [51].
Given the numerous pain pathways located in the dorsal horn cells (various receptor systems and ion channels), spinal drug delivery lends itself to a vast improvement in efficacy and tolerability. Clinical experience has supported this observation, as combination therapy is widely used. Although there is a vast amount of literature on the safety of single agents in animal toxicity studies, there is none on the safety of drug combinations. An extensive discussion of safety evaluation for spinally delivered drugs can be found elsewhere [52]. The increased use of IT drug combinations has led to a proliferation of compounding pharmacists used to prepare such mixtures. In addition, this has led to the frequent use of higher concentrations of drugs to increase the time between pump refills, which has led to increasing reports of catheter tip granuloma formation (see discussion below). When using higher concentrations, close observation of the patient is required with documentation of a detailed neurological examination and attention to new neurological symptoms or increase in pain. Also, close communication between the compounding pharmacist and physician is necessary. The physician’s role is to monitor the patient for efficacy, safety, and tolerability, while the compounding pharmacist must evaluate the mixtures for pyrogenicity and solubility. Close communication between the physician and pharmacist as well as close monitoring of the patient can provide the patient with the vast benefits of drug combination therapy.
Future Directions—New Technology
New IT technologies aim to improve both safety and efficacy. IT drug delivery devices are becoming increasingly compatible with agents and admixtures. Programmable delivery systems have also been evolving to address the intra-and inter-patient pain variability. Technology that can effectively and safely treat breakthrough pain is a continued goal. As programming technology advances, however, space for infusate storage decreases and implantable pumps become more bulky. Increased power requirements from bolus options and programming features also limit battery life, necessitating more frequent surgical battery exchange. Pumps that require less energy and rechargeable systems are a relatively new approach to this problem. A piezoelectric membrane pump has been developed that reduces both bulk and energy requirement [53]. Technology behind rechargeable spinal cord stimulators may be eventually applied to IT pumps [13]. Finally, less fragile pump materials may have a future in reducing drug delivery system damage, breakdown, and failure [13]. By broadening the available options of drug delivery, innovations in IT therapy may reach new subsets of patients.
A number of new pump systems have recently been developed to improve IT delivery. The Prometra Programmable Pump System (InSet Technologies, Inc., Mount Olive, NJ) is an investigational, fully implantable device designed to improve precision in drug delivery. The system utilizes a positive pressure design with gate-controlled administration of morphine into the 20-mL fixed-volume control chamber to ensure dosing accuracy [54]. This system allows a flow rate of up to 28 mL per day. It weighs 150 g, has a diameter of 71 mm, is 20 mm thick, and is comprised of highly durable, mostly immobile parts to promote usage for more than 10 years [54]. A clinical trial of the Prometra pump conducted in individuals with intractable chronic cancer pain or chronic nonmalignant pain demonstrated an overall 97.5% dose accuracy rate [55–57] and statistically significant improvements in visual analogue scale score, numeric rating scale score, and Oswestry disability index assessments from baseline to 1 year [58]. FDA approval of the Prometra pump is expected shortly based on the pump’s ability to achieve preestablished end points for accuracy, efficacy, and safety at desirable flow rates.
The MedStream Programmable Infusion System (Codman and Shurtleff, Inc., a Johnson & Johnson Co., Raynham, MA) is another implantable device currently under investigation. This implantable drug delivery system offers improved catheter technology and pump durability. Its SureStream intraspinal catheter has been designed to resist kinking and tearing. The MedStream ceramic drive system maintains the infusion rate without motors, gears, or rotating parts [59].The pump has either a 20- or 40-mL reservoir, and measures 76 mm by 21.6 mm (20 mL) or 28.2 mm (40 mL) [59]. The MedStream is currently pending FDA approval, although it is available for sale in several European countries [59].
The Medallion (Advanced Bionics, Valencia, CA) is an implantable drug delivery system awaiting FDA approval for study in humans. The Medallion system focuses on safety improvements with the creation of a negative pressure reservoir. Should a break in the system occur, medication would not leak out of the reservoir into the body. Furthermore, the negative pressure draws medication from the syringe during pump refills rather than requiring syringe plunger manipulation [60]. A pressure sensor is present to detect flow resistance. The Medallion, with either a 20- or 40-mL pump, measures 2.6 in. in diameter and is 0.72 in. thick across the flat portion of the can [60]. Both sizes include a sutureless connector, a radio-opaque IT catheter, and are compatible with an 8-year battery at 0.5 mL per day [60]. The Medallion clinical trials will assess the system’s pressure sensor, in addition to the overall safety and efficacy of the device [60].
The Personal Therapy Manager (PTM, Medtronic, Inc., Minneapolis, MN) is a compatible handheld device to be used with an existing IT drug delivery system. This technology is designed to address breakthrough pain by allowing patients to self-administer a bolus dose of infusate. The PTM was approved by the FDA in 2005 for concomitant use with the SynchroMed II pump [61]. Maximum bolus dosing is specified in order to avoid administrative overdose. The device also records usage, pre- and post-bolus pain scores, and technical events, as well as calculates predicted refill requirements based on background infusion and bolus doses [13, 62, 63]. Patients using the device have reportedly experienced a 29% reduction in pain, improved quality of life, and reduced need for supplemental analgesics, and there have been no reported major adverse events associated with its use [62]. The PTM is only compatible with the newer SynchroMed pumps and is not covered by many managed care organizations or Medicare. The out-of-pocket cost for patients is estimated at $500.
Concerns
As recently described by Coffey et al. [64], IT opioid drug infusion is not a completely harmless technique. They found higher than expected mortality among noncancer pain patients treated with IT opioid infusion attributable to inadvertent drug overdose. Incorrect programming, pump malfunction or fracture, and complications with pump refill all pose risk of drug overdose. Likewise, abrupt withdrawal or otherwise improper dose titration is a potential adverse outcome of improper system management.
Patient characteristics may also increase complication risk and interfere with IT drug delivery. Myriad comorbidities, including systemic infection risks, skin breakdown or localized infection, spinal metastatic disease, bleeding diathesis, poor nutritional status, or severe psychiatric disturbance may make IT therapy undesirable and even unsafe. IT drug delivery in noncancer patients or nonterminal cancer patients has potential for greater complication risk simply due to length of treatment. As new technology and pharmaceutical options arise, clinicians must continue to exert care over patient selection.
Catheter-tip granulomas are another potential source of adverse outcome with IT drug delivery. IT inflammatory masses are relatively common. Sixty-three percent of responding physicians recently polled in an online survey reported experience with granulomas in their patients [2•]. All agents used in implantable infusion systems except fentanyl and sufentanil have been associated with granuloma formation [2•]. Clinical research suggests that dose escalation and increases in concentration can prompt the development of IT granulomas. Although usually innocuous, IT granulomas may lead to catastrophic neurologic dysfunction and permanent paralysis [65]. Signs suggestive of granuloma formation include loss of analgesic effect and new and progressive neurologic symptoms [66]. Small granulomas that are identified early require only minimally invasive therapy such as cessation of drug administration through the affected catheter and awaiting gradual granuloma shrinkage, whereas larger masses require surgical removal [66]. Vigilant monitoring for signs of possible inflammatory mass formation will assist in early diagnosis, and may avoid surgical removal and neurologic damage. When possible, catheter tip placement in the lumbar thecal sac and use of lowest possible opioid doses will lower the risk of granuloma formation [66].
Finally, surgical complications such as infection and bleeding pose risk of complications. There are no controlled trials on surgical implantation technique [13]. Future investigation into optimal perioperative care, percutaneous approach, and surgical technique to improve outcome and reduce complications is warranted.
Conclusions
Since the first clinical implant of an infusion pump IT drug delivery to treat chronic pain, there have only been two agents that have achieved FDA approval. If IT therapy were to be limited to these two agents, the efficacy of this therapy would be inadequate. Therefore, the vast majority of the clinical experience has been with the off-label use of a variety of agents with different mechanisms of action. This has led to the widespread use of this therapy to treat both cancer and noncancer pain. The success of the clinical trials with ziconotide demonstrate that this therapy can be studied using randomized controlled trials. Hopefully, this will open the door for more agents to reach the market such as gabapentin, which is currently in phase 2 trials.
There remains a lot unknown about IT therapy for the treatment of chronic pain. The delivery methods for IT therapy are fairly straightforward; however, the pharmacology is quite complex. The polyanalgesic consensus panel has provided detailed guidance over the past decade. Future directions should focus on gathering data on the efficacy and safety of this therapy. Given the complexities and costs of randomized controlled trials in the area of pain medicine, large multicenter registries are probably the best way of achieving this goal. Until this is achieved, physicians will continue to rely on guidelines created from limited data and their own clinical experience.
Acknowledgments
Disclosure
Dr. Mark S. Wallace has received research support from Medtronic, Inc.
No other potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Erin F. Lawson, Email: erlawson@ucsd.edu
Mark S. Wallace, Email: mswallace@ucsd.edu
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.Smith HS, Deer TR, Staats PS, et al. Intrathecal drug delivery. Pain Physician. 2008;11(2 Suppl):S89–S104. [PubMed] [Google Scholar]
- 2.• Deer T, Krames ES, Hassenbusch SJ, et al.: Polyanalgesic Consensus Conference 2007: recommendations for the management of pain by intrathecal (intraspinal) drug delivery: report of an interdisciplinary expert panel. Neuromodulation 2007, 10:300–328. This is the most recent consensus guidelines, a result of a consensus meeting of top experts on spinal drug delivery. [DOI] [PubMed]
- 3.Bennett G, Burchiel K, Buchser E, et al. Clinical guidelines for intraspinal infusion: report of an expert panel. PolyAnalgesic Consensus Conference 2000. J Pain Symptom Manage. 2000;20:S37–S43. doi: 10.1016/S0885-3924(00)00202-5. [DOI] [PubMed] [Google Scholar]
- 4.Hassenbusch SJ, Portenoy RK, Cousins M, et al. Polyanalgesic Consensus Conference 2003: an update on the management of pain by intraspinal drug delivery-report of an expert panel. J Pain Symptom Manage. 2004;27:540–563. doi: 10.1016/j.jpainsymman.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Bennett G, Deer T, Du Pen S, et al. Future directions in the management of pain by intraspinal drug delivery. J Pain Symptom Manage. 2000;20:S44–S50. doi: 10.1016/S0885-3924(00)00201-3. [DOI] [PubMed] [Google Scholar]
- 6.Pirec V, Laurito CE, Lu Y, Yeomans DC. The combined effects of N-type calcium channel blockers and morphine on A delta versus C fiber mediated nociception. Anesth Analg. 2001;92:239–243. doi: 10.1097/00000539-200101000-00046. [DOI] [PubMed] [Google Scholar]
- 7.Rauck RL, Wallace MS, Leong MS, et al. A randomized, double-blind, placebo-controlled study of intrathecal ziconotide in adults with severe chronic pain. J Pain Symptom Manage. 2006;31:393–406. doi: 10.1016/j.jpainsymman.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Staats PS, Yearwood T, Charapata SG, et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- 9.Wallace MS, Rauck R, Fisher R, et al.: Intrathecal ziconotide for severe chronic pain: safety and tolerability results of an open-label, long-term trial. Anesth Analg 2008, 106:628–637, table of contents. [DOI] [PubMed]
- 10.Wallace MS, Fisher R, Byas-Smith M, et al. Intrathecal ziconotide in the treatment of chronic nonmalignant pain: a randomized, double-blind, placebo controlled clinical trial. Neuromodulation. 2006;9:75–86. doi: 10.1111/j.1525-1403.2006.00055.x. [DOI] [PubMed] [Google Scholar]
- 11.Wallace MS, Kosek PS, Staats P, et al. Phase II, open-label, multicenter study of combined intrathecal morphine and ziconotide: addition of ziconotide in patients receiving intrathecal morphine for severe chronic pain. Pain Med. 2008;9:271–281. doi: 10.1111/j.1526-4637.2007.00355.x. [DOI] [PubMed] [Google Scholar]
- 12.Webster LR, Fakata KL, Charapata S, et al. Open-label, multicenter study of combined intrathecal morphine and ziconotide: addition of morphine in patients receiving ziconotide for severe chronic pain. Pain Med. 2008;9:282–290. doi: 10.1111/j.1526-4637.2007.00356.x. [DOI] [PubMed] [Google Scholar]
- 13.Ilias W, Todoroff B. Optimizing pain control through the use of implantable pumps. Med Dev Evid Res. 2008;1:41–47. doi: 10.2147/mder.s3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webster LR, Fisher R, Charapata S, Wallace MS. Long-term intrathecal ziconotide for chronic pain: an open-label study. J Pain Symptom Manage. 2009;37:363–372. doi: 10.1016/j.jpainsymman.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 15.Prialt (ziconotide intrathecal infusion) package insert: Manufactured by Elan Pharmaceuticals, Dublin, Ireland.
- 16.Bouchenafa O, Livingston A. Autoradiographic localisation of alpha 2 adrenoceptor binding sites in the spinal cord of the sheep. Res Vet Sci. 1987;42:382–386. [PubMed] [Google Scholar]
- 17.Eisenach JC, DuPen S, Dubois M, et al. Epidural clonidine analgesia for intractable cancer pain. The Epidural Clonidine Study Group. Pain. 1995;61:391–399. doi: 10.1016/0304-3959(94)00209-W. [DOI] [PubMed] [Google Scholar]
- 18.Berde CB, Sethna NF, Conrad LS, et al. Subarachnoid bupivacaine analgesia for seven months for a patient with a spinal cord tumor. Anesthesiology. 1990;72:1094–1096. doi: 10.1097/00000542-199006000-00025. [DOI] [PubMed] [Google Scholar]
- 19.Lau WC, Green CR, Faerber GJ, et al. Determination of the effective therapeutic dose of intrathecal sufentanil for extracorporeal shock wave lithotripsy. Anesth Analg. 1999;89:889–892. doi: 10.1097/00000539-199910000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Nitescu P, Dahm P, Appelgren L, Curelaru I. Continuous infusion of opioid and bupivacaine by externalized intrathecal catheters in long-term treatment of “refractory” nonmalignant pain. Clin J Pain. 1998;14:17–28. doi: 10.1097/00002508-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Budd K. High dose buprenorphine for postoperative analgesia. Anaesthesia. 1981;36:900–903. doi: 10.1111/j.1365-2044.1981.tb08867.x. [DOI] [PubMed] [Google Scholar]
- 22.Canavero S, Bonicalzi V, Clemente M. No neurotoxicity from long-term (>5 years) intrathecal infusion of midazolam in humans. J Pain Symptom Manage. 2006;32:1–3. doi: 10.1016/j.jpainsymman.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Yaksh TL, Allen JW. The use of intrathecal midazolam in humans: a case study of process. Anesth Analg. 2006;98:1536–1545. doi: 10.1213/01.ANE.0000122638.41130.BF. [DOI] [PubMed] [Google Scholar]
- 24.Kroin JS, Ling ZD, Buvanendran A, Tuman KJ. Upregulation of spinal cyclooxygenase-2 in rats after surgical incision. Anesthesiology. 2004;100:364–369. doi: 10.1097/00000542-200402000-00027. [DOI] [PubMed] [Google Scholar]
- 25.Eisenach JC, Curry R, Hood DD, Yaksh TL. Phase I safety assessment of intrathecal ketorolac. Pain. 2002;99:599–604. doi: 10.1016/S0304-3959(02)00208-7. [DOI] [PubMed] [Google Scholar]
- 26.Yaksh TL, Horais KA, Tozier N, et al. Intrathecal ketorolac in dogs and rats. Toxicol Sci. 2004;80:322–334. doi: 10.1093/toxsci/kfh168. [DOI] [PubMed] [Google Scholar]
- 27.Chaudhari LS, Kane DG, Shivkumar B, Kamath SK. Comparative study of intrathecal pethidine versus lignocaine as an anaesthetic and a postoperative analgesic for perianal surgery. J Postgrad Med. 1996;42:43–45. [PubMed] [Google Scholar]
- 28.Coderre TJ, Kumar N, Lefebvre CD, Yu JS. Evidence that gabapentin reduces neuropathic pain by inhibiting the spinal release of glutamate. J Neurochem. 2005;94:1131–1139. doi: 10.1111/j.1471-4159.2005.03263.x. [DOI] [PubMed] [Google Scholar]
- 29.Cheng JK, Chen CC, Yang JR, Chiou LC. The antiallodynic action target of intrathecal gabapentin: Ca2+ channels, KATP channels or N-methyl-d-aspartic acid receptors? Anesth Analg. 2006;102:182–187. doi: 10.1213/01.ane.0000189550.97536.83. [DOI] [PubMed] [Google Scholar]
- 30.Luer MS, Hamani C, Dujovny M, et al. Saturable transport of gabapentin at the blood-brain barrier. Neurol Res. 1999;21:559–562. doi: 10.1080/01616412.1999.11740975. [DOI] [PubMed] [Google Scholar]
- 31.Deer T, Kim CK, Bowman RG, et al. The use of continuous intrathecal infusion of octreotide in patients with chronic pain of noncancer origin: an evaluation of side-effects and toxicity in a prospective double-blind fashion. Neuromodulation. 2005;8:171–175. doi: 10.1111/j.1525-1403.2005.05235.x. [DOI] [PubMed] [Google Scholar]
- 32.Tsai YC, So EC, Chen HH, et al. Effect of intrathecal octreotide on thermal hyperalgesia and evoked spinal c-Fos expression in rats with sciatic constriction injury. Pain. 2002;99:407–413. doi: 10.1016/S0304-3959(02)00107-0. [DOI] [PubMed] [Google Scholar]
- 33.Paice JA, Penn RD, Shott S. Intraspinal morphine for chronic pain: a retrospective, multicenter study. J Pain Symptom Manage. 1996;11:71–80. doi: 10.1016/0885-3924(95)00099-2. [DOI] [PubMed] [Google Scholar]
- 34.Yoon MH, Bae HB, Choi JI, et al. Roles of adenosine receptor subtypes in the antinociceptive effect of intrathecal adenosine in a rat formalin test. Pharmacology. 2006;78:21–26. doi: 10.1159/000094762. [DOI] [PubMed] [Google Scholar]
- 35.Eisenach JC, Hood DD, Curry R, et al. Intrathecal but not intravenous opioids release adenosine from the spinal cord. J Pain. 2004;5:64–68. doi: 10.1016/j.jpain.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Eisenach JC. Adenosine reduces glutamate release in rat spinal synaptosomes. Anesthesiology. 2005;103:1060–1065. doi: 10.1097/00000542-200511000-00021. [DOI] [PubMed] [Google Scholar]
- 37.Sharma M, Mohta M, Chawla R. Efficacy of intrathecal adenosine for postoperative pain relief. Eur J Anaesthesiol. 2006;23:449–453. doi: 10.1017/S0265021506000342. [DOI] [PubMed] [Google Scholar]
- 38.Hayashida M, Fukuda K, Fukunaga A. Clinical application of adenosine and ATP for pain control. J Anesth. 2005;19:225–235. doi: 10.1007/s00540-005-0310-8. [DOI] [PubMed] [Google Scholar]
- 39.Belfrage M, Segerdahl M, Arnér S, Sollevi A. The safety and efficacy of intrathecal adenosine in patients with chronic neuropathic pain. Anesth Analg. 1999;89:136–142. doi: 10.1097/00000539-199907000-00023. [DOI] [PubMed] [Google Scholar]
- 40.Karlsten R, Gordh T., Jr An A1-selective adenosine agonist abolishes allodynia elicited by vibration and touch after intrathecal injection. Anesth Analg. 1995;80:844–847. doi: 10.1097/00000539-199504000-00037. [DOI] [PubMed] [Google Scholar]
- 41.Rane K, Segerdahl M, Goiny M, Sollevi A: Intrathecal adenosine administration: a phase 1 clinical safety study in healthy volunteers, with additional evaluation of its influence on sensory thresholds and experimental pain. Anesthesiology 1998, 89:1108–1115; discussion 1109A. [DOI] [PubMed]
- 42.Deer T, Krames ES, Hassenbusch S, et al. Future directions for intrathecal pain management: a review and update from the Interdisciplinary Polyanalgesic Consensus Conference 2007. Neuromodulation. 2008;11:92–97. doi: 10.1111/j.1525-1403.2008.00148.x. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen CK, Lewis RJ, Alewood D, et al. Anti-allodynic efficacy of the chi-conopeptide, Xen2174, in rats with neuropathic pain. Pain. 2005;118:112–124. doi: 10.1016/j.pain.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Szabo T, Olah Z, Iadarola MJ, Blumberg PM. Epidural resiniferatoxin induced prolonged regional analgesia to pain. Brain Res. 1999;840:92–98. doi: 10.1016/S0006-8993(99)01763-1. [DOI] [PubMed] [Google Scholar]
- 45.Clinical Trials: resiniferatoxin to treat severe pain associated with advanced cancer. Available at http://clinicaltrials.gov/ct2/show/NCT00804154. Accessed December 2009.
- 46.Allen JW, Mantyh PW, Horais K, et al. Safety evaluation of intrathecal substance P-saporin, a targeted neurotoxin, in dogs. Toxicol Sci. 2006;91:286–298. doi: 10.1093/toxsci/kfj143. [DOI] [PubMed] [Google Scholar]
- 47.Wiley RG. Substance P receptor-expressing dorsal horn neurons: lessons from the targeted cytotoxin, substance P-saporin. Pain. 2008;136:7–10. doi: 10.1016/j.pain.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Smith TJ, Staats PS, Deer T, et al. Randomized clinical trial of an implantable drug delivery system compared with comprehensive medical management for refractory cancer pain: impact on pain, drug-related toxicity, and survival. J Clin Oncol. 2002;20:4040–4049. doi: 10.1200/JCO.2002.02.118. [DOI] [PubMed] [Google Scholar]
- 49.Yaksh TL, Reddy SV. Studies in the primate on the analgetic effects associated with intrathecal actions of opiates, alpha-adrenergic agonists and baclofen. Anesthesiology. 1981;54:451–467. doi: 10.1097/00000542-198106000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Wallace MS, Yaksh TL. Long-term intraspinal drug therapy: a review. Reg Anesth Pain Med. 2000;25:117–157. doi: 10.1053/rapm.2000.0250117. [DOI] [PubMed] [Google Scholar]
- 51.Yaksh TL, Malmberg, AB: Interaction of spinal modulatory receptor systems. In Progress in Pain Research and Management, vol 1. Edited by Fields HL, Liebeskind JC. Seattle: IASP Press; 1994:151–171.
- 52.Yaksh TL, Rathbun M, Provencher JC: Preclinical drug safety evaluation. In Spinal Drug Delivery. Edited by Yaksh TL. Elsevier; 1999.
- 53.Kan J, Yang Z, Tang K, Cheng G: Pumping performance of a new piezoelectric pump for drug delivery. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2004, 21:297–301. [PubMed]
- 54.InSet Technologies, Inc.: Information on file with the company. Available at http://www.insetinc.com. Accessed December 2009.
- 55.Rosen S, Deer T, Rauck R, et al. Multi-center evaluation of drug delivery accuracy with the Prometra intrathecal infusion pump. Pain Med. 2009;10:124. doi: 10.1111/j.1526-4637.2009.00672.x. [DOI] [Google Scholar]
- 56.Clinical Trials: Prometra implantable programmable pump (PUMP). Available at http://clinicaltrials.gov/ct2/show/NCT00817596. Accessed December 2009.
- 57.Clinical Trials: Prometra’s utilization in mitigating pain II (PUMP2). Available at http://clinicaltrials.gov/ct2/show/NCT00866164. Accessed December 2009.
- 58.Berg A, Barsa T, Deer T, et al.: Efficacy of morphine sulfate infusion via the Prometra® intrathecal infusion pump. A prospective multicenter evaluation. Presented at the 5th World Congress Institute of Pain. New York, NY; March 15, 2009.
- 59.Codman: Information on file with the company. Available at http://www.codman.com/DePuy/home/index.html. Accessed December 2009.
- 60.Smith Medical: Infusion systems. Available at http://www.smiths-medical.com/products/infusion/. Accessed December 2009.
- 61.US Food and Drug Administration: PMA: premarket approval. Available at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/PMA.cfm?ID=4499. Accessed December 2009.
- 62.Ilias W, le Polain B, Buchser E. Demartini L; oPTiMa study group: Patient-controlled analgesia in chronic pain patients: experience with a new device designed to be used with implanted programmable pumps. Pain Pract. 2008;8:164–170. doi: 10.1111/j.1533-2500.2008.00187.x. [DOI] [PubMed] [Google Scholar]
- 63.Rainov NG, Buchser E. Making a case for programmable pumps over fixed rate pumps for the management of fluctuations in chronic pain and spasticity: a literature review. Neuromodulation. 2002;5:88–99. doi: 10.1046/j.1525-1403.2002.02013.x. [DOI] [PubMed] [Google Scholar]
- 64.Coffey RJ, Owens ML, Broste SK, et al. Mortality associated with implantation and management of intrathecal opioid drug infusion systems to treat noncancer pain. Anesthesia. 2009;111:881–892. doi: 10.1097/ALN.0b013e3181b64ab8. [DOI] [PubMed] [Google Scholar]
- 65.Ghafoor VL, Epshteyn M, Carlson GH, et al. Intrathecal drug therapy for long-term pain management. Am J Health Syst Pharm. 2007;64:2447–2461. doi: 10.2146/ajhp060204. [DOI] [PubMed] [Google Scholar]
- 66.Hassenbusch S, Burchiel K, Coffey RJ, et al. Management of intrathecal catheter-tip inflammatory masses: a consensus statement. Pain Med. 2002;3:313–323. doi: 10.1046/j.1526-4637.2002.02055.x. [DOI] [PubMed] [Google Scholar]