

Abstract
The phosphoinositide 3-kinase (PI3K) family has multiple vascular functions, but the specific regulatory isoform supporting lymphangiogenesis remains unidentified. Here we report that deletion of the Pik3r1 gene, encoding the regulatory subunits p85α, p55α, and p50α impairs lymphatic sprouting and maturation, and causes abnormal lymphatic morphology, without major impact on blood vessels. Pik3r1 deletion had the most severe consequences among gut and diaphragm lymphatics, which share the retroperitoneal anlage, initially suggesting that the Pik3r1 role in this vasculature is anlage-dependent. However, whereas lymphatic sprouting toward the diaphragm was arrested, lymphatics invaded the gut, where remodeling and valve formation were impaired. Thus, cell-origin fails to explain the phenotype. Only the gut showed lymphangiectasia, lymphatic up-regulation of the TGFβ co-receptor endoglin, and reduced levels of mature VEGF-C protein. Our data suggest that Pik3r1 isoforms are required for distinct steps of embryonic lymphangiogenesis in different organ microenvironments, whereas they are largely dispensable for hemangiogenesis.
Keywords: Lymphangiogenesis, Phosphatidyl inositol 3-kinase
INTRODUCTION
PI3Ks control cell size, metabolism, differentiation, survival, migration, and proliferation (Engelman et al., 2006; Fruman and Bismuth, 2009). PI3K-mediated pathways are being pursued as pharmacological targets for halting tumor growth, metastasis, and angiogenesis (Stephens et al., 2005; Garcia-Echeverria and Sellers, 2008), but drug-specificity remains challenging due to widespread expression of PI3K proteins. Identifying and targeting specific subunits may help address those concerns.
There are four subgroups of PI3K enzyme (class Ia, Ib II and III) that differ in their regulation and substrate selectivity. Growth factor receptors activate primarily the class Ia subgroup, which are dimeric proteins containing a catalytic subunit (p110α, p110β, or p110δ) and a regulatory subunit (p85α, p55α, p50α, p85β, or p55γ). The dimers are targeted to membrane-associated signaling complexes through several protein interaction domains in the regulatory subunits, and through a Ras-binding domain in the catalytic subunit. Vascular cells express multiple class Ia catalytic and regulatory isoforms. A reasonable hypothesis is that different regulatory subunits afford specificity to the multiple (lymph)angiogenic signaling receptors linked to PI3Ks, including Tie1/Tie2 (Peters et al., 2004), and VEGFR-1, -2 and -3 (Saharinen et al., 2004). However, no step in lymph vessel development has been ascribed to a specific PI3K regulatory subunit.
Our previous studies prompted us to investigate the hypothesis that regulatory isoforms encoded by the Pik3r1gene (p85α/p55α/p50α) have more critical functions in lymphangiogenesis than in hemangiogenesis. Specifically, we previously showed that Pik3r1 null mice survive to birth but develop chylous ascites, a hallmark of lymphatic insufficiency (Fruman et al., 2000). Mice lacking only p85α (p55α/p50α isoforms intact) (Fruman et al., 1999), p55α/p50α (p85α isoform intact) (Chen et al., 2004), or p85β encoded by Pik3r2 (Ueki et al., 2002) showed no lymphatic defects. However, chylous ascites was also reported in newborn mice with a point mutation abrogating the interaction between p110α and Ras (Gupta et al., 2007), implicating p110α as an important Ras effector in perinatal lymphatic development. Here we report that the class Ia regulatory isoforms encoded by Pik3r1 are required for organ-specific steps of lymphangiogenesis.
RESULTS AND DISCUSSION
We sought to determine if abnormal lymphatic development underlies chylous ascites (Fruman et al., 2000) and intestinal edema (Figures 1A,B) in Pik3r1 null mice as follows: Using LYVE1 (Schledzewski et al., 2006), VEGFR-3 (Lymboussaki et al., 1998) and Prox1 (Rodriguez-Niedenfuhr et al., 2001) as lymphatic endothelial cell markers (Kim et al., 2007), we analyzed lymphatic density and morphology in Pik3r1-targeted newborns, all of which presented with chylous ascites (Supplemental Figure s.1). To visualize the complete vascular tree, we used the pan-endothelial markers VEGFR-2 and PECAM1, whose expression did not change upon Pik3r1 deletion (data not shown). As shown by LYVE1 whole mount immunohistochemistry (IHC), there was a paucity of serosal lymphatics at birth in Pik3r1 null mice (Figure 1C,D), as determined by the mean number of vessel branch points/area (n=5/group; wild-type 11.6 versus null 3.8, p<1×10−5). In contrast, the lymphatic capillaries (lacteals, (Papp et al., 1962)) were normal in number and size (Figure 1H, (Kim et al., 2007)). The morphology of the remaining Pik3r1 null mesenteric and serosal lymphatics was abnormal (Figures 1E–G). Confocal microscopy indicated that segments of the mesenteric collectors of Pik3r1 null mice were more variable than wild-type; i.e., some were twice as wide as those of wild-type littermates (Figures 1I,J) and others were less than half the normal size (Figures 1E,F). Wider distribution of diameter classes in null versus wild-type lymphatics resulted in a failure to achieve statistical significance for mean diameter (data not shown). Hemangiogenesis appeared normal except that the duodenal arcades exhibited additional branching as visualized by alpha smooth muscle actin (SMA) staining (Figures 1K,L).
Figure 1. Intestinal lymphatic defects in Pik3r1 null newborns.
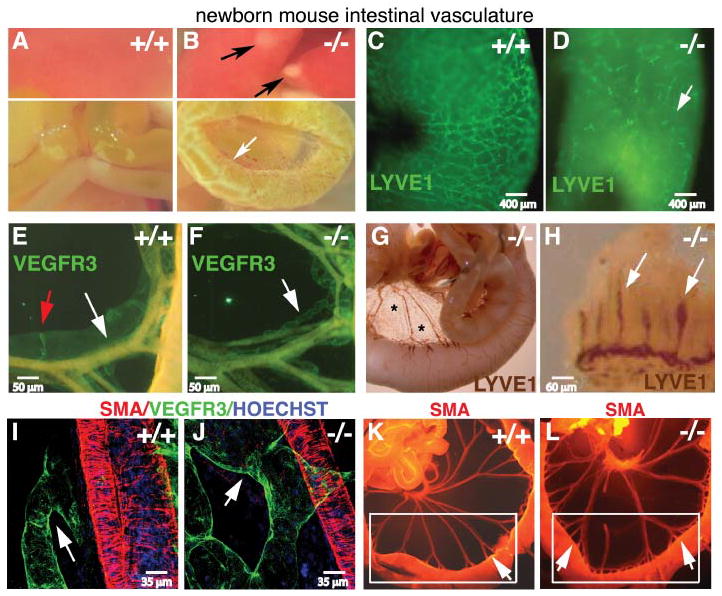
(A,B): Gross anatomy of a wild-type (+/+) and Pik3r1 (−/−) null newborn littermates. Chylous ascites was evident in the intact peritoneum of Pik3r1 null mice (Supplemental Data). Intestinal edema (white arrow), and liver necrosis (black arrows) are indicated in the Pik3r1 null organs. (C–F): Intestinal lymphatics stained by whole mount immunofluorescence for LYVE1 (C,D), VEGFR3 (green, E,F), and SMA (orange, that bleeds through the green channel (E,F)). The white arrow in panel D highlights the lack of intestinal lymphatics. (E,F): The white arrows indicate segments of the mesenteric collectors highlighting the difference in lymph vessel diameter and morphology; the red arrow in panel E highlights a lymphatic valve of the sort lacking in the null mouse specimens.
(G,H): Intestinal lymphatics LYVE1-stained by whole mount immunoperoxidase method. LYVE1+ presumptive macrophages in the mesentery (Kim et al., 2007; Kubota et al., 2009) are marked with asterisks. (H): Brown-stained lymphatic vessels (lacteals) inside the villi are indicated with white arrows. (I,J): Confocal immunofluorescence for VEGFR3 (green) and SMA (red). The white arrows highlight the increased diameter of the lymphatic collectors of Pik3r1 null (J) compared to wild-type mice (I). (K,L): Whole mount immunofluorescence of newborn small intestine stained for SMA. Blood vessels are highlighted by this method because they have higher numbers of smooth muscle cells (arteries>veins⋙lymphatics). Increased blood vessel branching (arrows, panel K versus L) at the level of the arcades is visible within the white boxes. Scale bars are as indicated in the panels.
Overall, the phenotype of the Pik3r1-targeted newborns was reminiscent of intestinal lymphangiectasia, a clinical condition in which inflammation or obstructing tumors (Kolbjornsen et al., 1994) results in poor lymphatic drainage. Our data suggest that in the absence of Pik3r1, lymphangiectasia resulted from abnormal lymphatic development. Liver and heart necrosis (Fruman et al., 2000) may also contribute to impaired lymphatic function in Pik3r1 null mice by impairing venous function, though widespread venous insufficiency is unlikely because: (i) the newborns lacked pleural effusions (Figures 2A–F) and (ii), despite significantly decreased dermal lymphatic branching and increased diameter (Figure 2G), subcutaneous edema was barely detectable. Interestingly, Angiopoietin2 null newborn mice have chylous ascites, but also show pleural effusions, subcutaneous edema, with fewer and smaller lacteals than control littermates, thus their lymphatic defects differ significantly from Pik3r1 null mice (Gale et al., 2002). Because angiopoietins signal via PI3K (Peters et al., 2004), it is still possible that aspects of the Pik3r1 null phenotype reflect a defect in this pathway. Post-natal blood vessel remodeling is also defective in Angiopoietin2 null mice (Gale et al., 2002), but this could not be studied in Pik3r1 null mice because they died by post natal day one. However, Pik3r1 null newborn mice had normal blood vessel density and morphology, exemplified by whole mount-IHC for SMA in ventral skin (Figures 2H,I), with the exception of the additional branching of the arcades noted already (Figures 1K,L).
Figure 2. Dermal lymphatic defects in Pik3r1 null newborns.
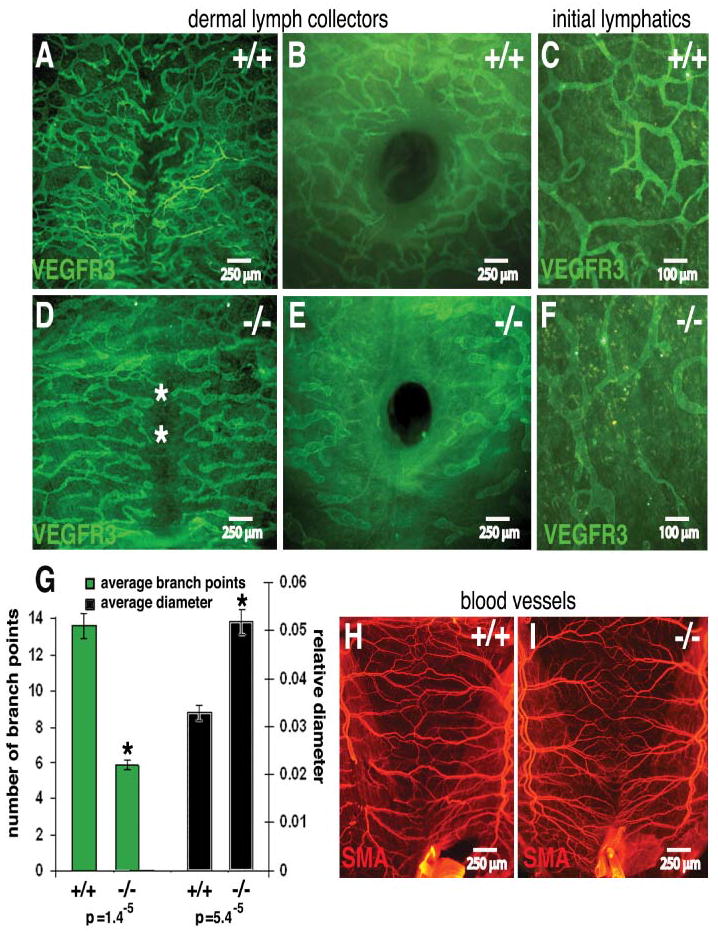
Lymphatics (A–F) and blood vessels (H,I) from the ventral skin of a wild-type and a Pik3r1 null newborn littermate, stained whole mount for VEGFR3 (green) and SMA (red). The central hole (B,E) is the umbilicus. The asterisks mark the ventral midline (D). (G): Quantitative image analysis of wild-type and Pik3r null lymphatic mean branch point count and diameter. Asterisks mark significantly different values with the associated Student’s T-test p values indicated below the figure.
Interestingly, lymphatics from the abdominal surface were significantly reduced (Figures 3A–D, and 3G), but were present in the thoracic surface (Figures 3E,F and 3G), appearing capable of extending from the body wall but not from the retroperitoneal sac (Oliver, 2004). These results are consistent with Pik3r1 having different roles on lymph and blood vessel development, suggesting that Pik3r1 isoforms are required for proper lymphatic patterning but are dispensable for, or to some extent inhibitory of, blood vessel branching.
Figure 3. Abdominal lymphatic defects in Pik3r1 null newborns.
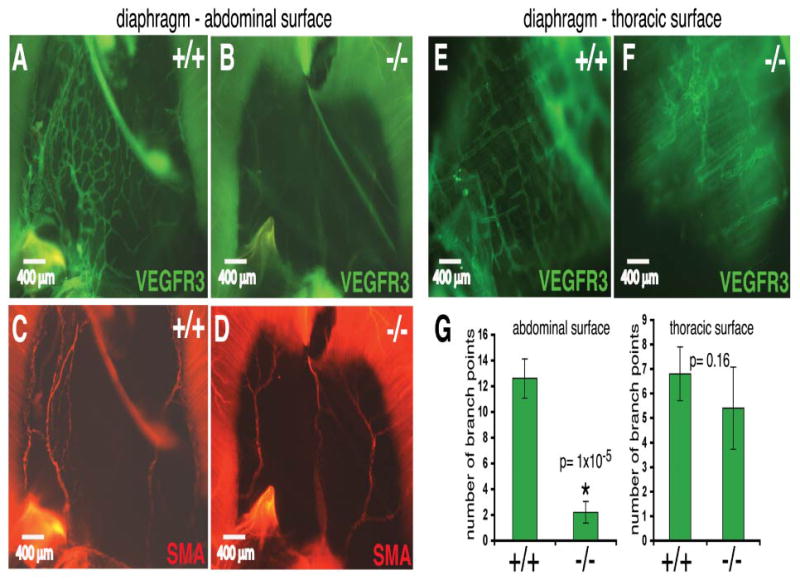
(A–F): Diaphragm lymphatics and blood vessels of a representative Pik3r1 null newborn and a wild-type littermate, stained whole mount for VEGFR3 (green) and SMA (red), respectively. In contrast to the abdominal surface of the diaphragm (A–D), the thoracic surface contained a lymphatic network in both wild-type and Pik3r1 null mice (E,F). (G): Quantitative image analysis of wild-type and Pik3r1 null mouse lymphatic vessel mean branch point count per constant area.
To gain insight into the cellular basis for the organ-specific dependency of lymphangiogenesis, we first questioned whether Pik3r1 loss causes vessel regression, or arrests vessel growth during development. Seminal work by Sabin ((Sabin, 1913), for a review see Oliver (Oliver, 2004)) indicates that lymphatics from the gut and abdominal diaphragm surface originate from the retroperitoneal/mesenteric lymph sac after E14. In turn, dermal lymphatics extend from paired jugular lymph sacs (after E10.5), a process halted in Prox1 (Oliver, 2004) and Vegfc (Karkkainen et al., 2004) null mice. By staining E10.5 to E19.5 embryos, we disproved the hypothesis that impaired anlagen development explains the organ-specific phenotype because we observed no early differences in timing, position, and expansion of lymph sacs between Pik3r1 null mice and wild-type littermates (data not shown). Instead, we found defects at later stages. Specifically, diaphragm invasion by lymphatics sprouting from the retroperitoneal lymph sac stopped at the central tendon (Figures 4E–H). In the gut, sprouts reached some microenvironments (mesentery; Figures 4A–D, Figure 5) but were reduced in others (serosa; Figures 5A–D). Moreover, our data suggest that lymphatic remodeling was dysregulated by Pik3r1 deletion: E16 Pik3r1 null mice established normal primitive mesenteric vascular plexi (Figures 5A–D) that failed to mature into the normal network of wild-type littermates in later stage E18.5 embryos. Indeed, even before suckling, which stimulates lymphatic transport, mesenteric lymphatics acquired abnormal constrictions and dilations (Figures 5E–J).
Figure 4. Arrested diaphragm invasion underlies lymphangiogenesis abnormalities in Pik3r1 null embryos.
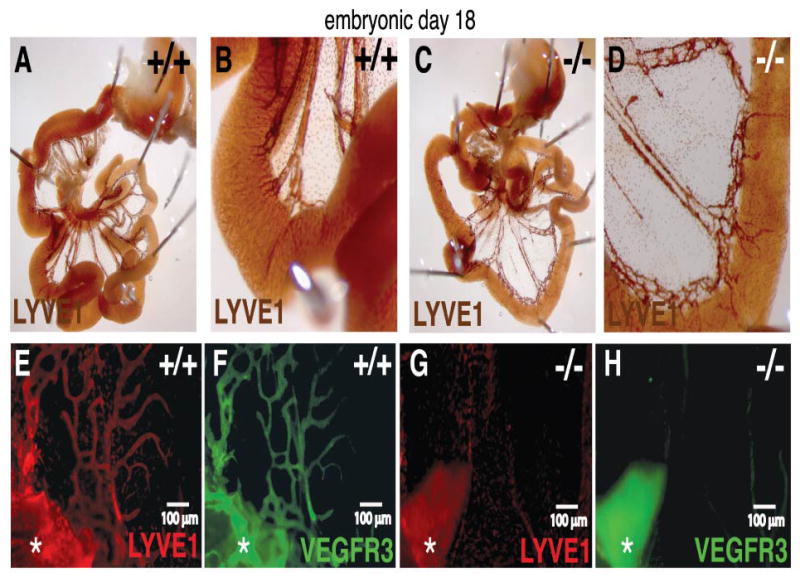
(A–H): E18 embryos stained whole mount with antibodies to detect LYVE1, VEGFR3, and SMA. (A–D): LYVE1 whole mount mesenterics stained using HRP substrate DAB. (E–H): Diaphragm lymphatics expanding past the central tendon into the pleuro-peritoneal membrane at E18 in wild-type but not in a null Pik3r1 littermate embryo, as visualized by whole mount double-immunofluorescence for LYVE1 and VEGFR3. The asterisk indicates a segment of attached liver.
Figure 5. Inappropriate gut remodeling contributes to the lymphangiogenesis abnormalities in Pik3r1 null embryos.
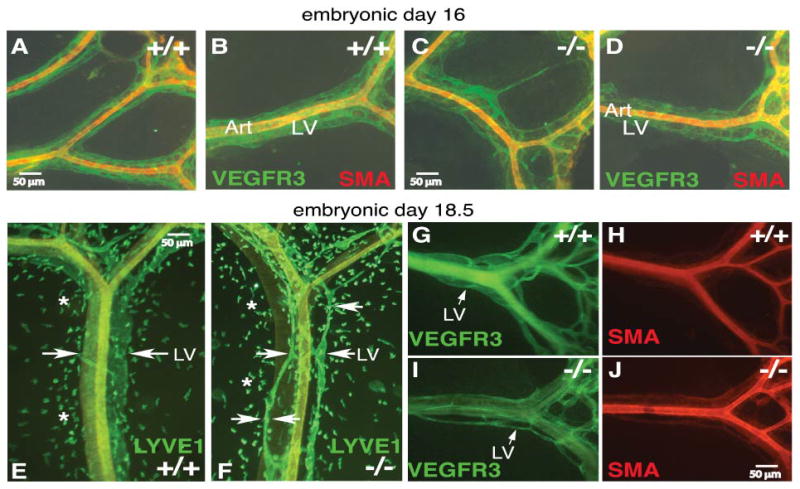
Mesenteric lymphatics of wild-type and Pik3r1 null littermate mice at E16 (A–D), E18.5 (E–J). (E–G, I): Lymphatic vessels are indicated by white arrows; white asterisks (E,F) mark LYVE1+ macrophages (Kim et al., 2007). Art, artery; LV, lymphatic vessel.
Pharmacological inhibitors of PI3K activity and PI3K mutants decrease endothelial cell proliferation and survival in vitro (Makinen et al., 2001; Wang et al., 2004). However, loss of Pik3r1 alone is not always sufficient to impair proliferation; for example, in T cells, Pik3r2 (p85β) compensates (Deane et al., 2007). Indeed, mesenteric lymphatics of Pik3r1 null mice and wild-type embryos did not differ in proliferation or cell death, as assessed by BrdU-incorporation and phosphohistone-H3 analyses, or by TUNEL staining, respectively (data not shown). Instead, our results are consistent with mouse lymphatic development studies (Kim et al., 2007) that support a vessel-branching mechanism driving mesenteric plexi remodeling without changes in cell number (Patan, 2004). Alternatively, Pik3r1 null mice may lack a subpopulation of lymphatic progenitors, like those detected in quail-mouse chimeras (Pudliszewski and Pardanaud, 2005). Although descriptions of (lymph)angiogenic mechanisms in the diaphragm are lacking, decreased invasion was the most visible defect in Pik3r1 null diaphragms. Collectively, these results suggest that Pik3r1 is required for distinct steps of lymphangiogenesis in distinct organ microenvironments, despite common vessel origins.
VEGF-C is needed for embryonic lymphangiogenesis (Karkkainen et al., 2004), and its tumor expression is PI3K-dependent (Tang et al., 2003). Interestingly, we found only decreased levels of mature ΔNΔC-VEGF-C in the Pik3r1 null gut, without obvious differences in the levels of precursor pro-VEGF-C, which is the substrate for N-terminal and C-terminal cleavages by plasmin, furin, and proconvertases (Figure 6A, (Jeltsch et al., 2003)). Interpreting these results will require a better understanding of post-translational processing of VEGF-C in vivo, and of the role of the different products, which signal through different VEGFRs in lymph vessel growth, remodeling, and maturation (Jeltsch et al., 2003).
Figure 6. Decreased VEGF-C levels are associated with altered lymphatic wall and valve structure.
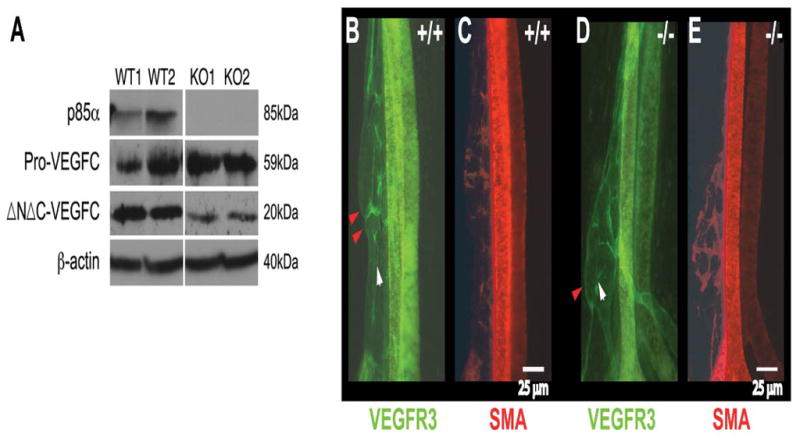
(A): p85α and VEGF-C protein levels in western blots of homogenates prepared from newborn mesenteries and diaphragm crura (three mice were pooled per lane). β-actin was used as a total protein loading control. (B–E): Whole mount staining for lymphatic endothelial cell VEGFR3 (green) and mural cell SMA (red). Red arrowheads point to lymph valves in mesenteric collectors of a wild-type newborn and the corresponding region in a Pik3r1-null, which display an abnormal wall structure instead of a valve. The white arrowhead indicates the normal direction of lymph flow.
To better understand the impact of Pik3r1 deletion on maturation, we assessed smooth muscle/pericyte investment and valve formation. Mesenteric lymphatics of Pik3r1 null mice lacked valves (Figures 6B–E); instead of endothelial lumenal flaps, we found abnormal vessel-wall constrictions (Figures 1–5, 6B,D). Only two other mutants show valve-less phenotypes: the knockout of Foxc2 (a member of the forkhead family of transcription factors, (Petrova et al., 2004)), and the knock-in of a PDZ-less ephrinB2 mutant (Makinen et al., 2005). Although Eph-receptors may signal via PI3K (Makinen et al., 2005), it is not understood how ephrinB2 affects valve formation.
Previous data indicate that Foxc2 ablation causes lymph valve agenesis and abnormal pericyte/SMC investment, due to up-regulation of Pdgfb and endoglin transcription in lymphatics (Petrova et al., 2004). Therefore, to test the hypothesis that PI3K regulates endoglin transcription, luciferase reporter assays were conducted on cultured human primary lymphatic endothelial cells transfected using human-derived endoglin promoter-luciferase constructs (Rius et al., 1998; Botella et al., 2002). First, a series of endoglin promoter deletion mutations comprising sequential human 5′ non-coding promoter regions extending from the minimal promoter (Figure 7A, base pairs -148–281, relative to the endoglin transcription start site) to a position 782 base pairs upstream of the transcription start site (Figure 7A, 782–281) were cotransfected with either the wild-type or dominant-negative p85α expression construct. These constructs were characterized elsewhere (Rius et al., 1998; Botella et al., 2002). Analysis of the endoglin promoter deletion mutations indicated that PI3K-mediated repression of endoglin expression is governed at least in part by elements contained in the proximal promoter DNA segment 5′ to position -281, relative to the transcription initiation site in the minimal endoglin promoter (Figure 7A). Moreover, cotransfection of the wild-type p85α subunit expression inhibited the shortest maximally-responsive endoglin luciferase reporter construct (−397/+281, Figure 7B) in a dose-dependent fashion. In contrast, the dominant-negative construct, p85αdn, was significantly less effective in terms of inhibition of endoglin promoter reporter expression (Figure 7B).
Figure 7. Increased lymphatic endothelial endoglin expression in the mesentery of Pik3r1 null mice.
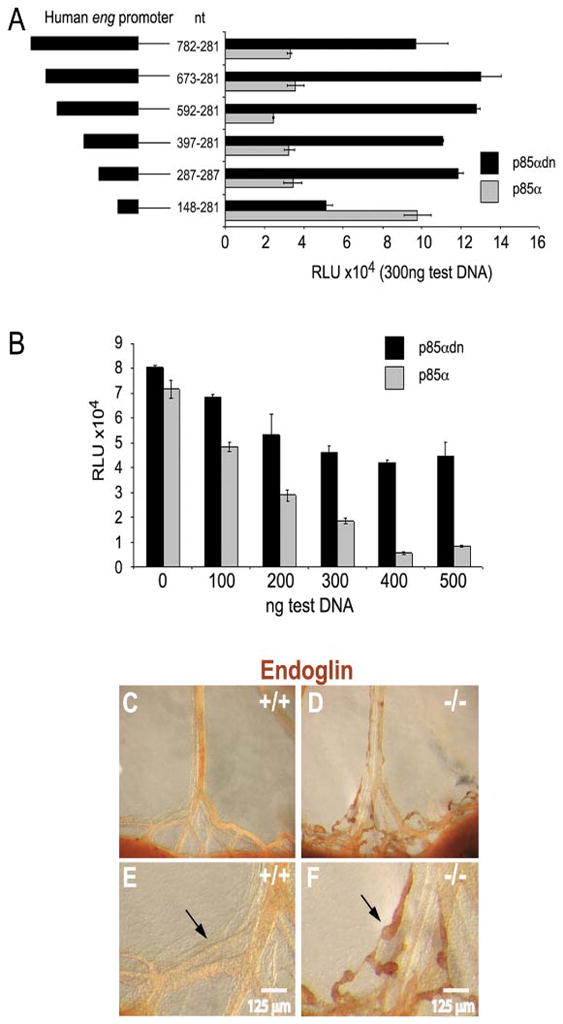
(A,B): Endoglin promoter luciferase constructs were tested in primary human lymphatic endothelial cell culture for regulation by p85α. (A): Successive endoglin promoter 5′ deletion constructs were transfected along with p85α protein expression construct corresponding to maximal endoglin promoter inhibition (300 ng/ml). The endoglin promoter segment numbers represent base pairs numbered from the 5′ terminus to the transcriptional start site. (B): Lymphatic endothelial cell cultures were transfected with the 397/281 endoglin promoter luciferase construct and either wild-type or dominant-negative p85αdn protein expression constructs. Histogram black bars, wild- type p85α gray bars, dominant negative p85α, p85αdn. For expression plasmid dosage studies, empty pcDNA was used to maintain constant total DNA levels. (C–F): Anti-endoglin whole mount staining of E19 mesenteries from a Pik3r1 null mouse and a wild-type littermate. Black arrows highlight the increased endoglin protein expression within the abnormal lymphatic collectors of Pik3r1 null mice.
A limitation of this approach is that the efficiency of transfection of the primary human lymphatic endothelial cells with p85 constructs, while sufficient for the reporter assays, was not sufficient to produce a statistically significant change in endogenous endoglin protein levels overall, as determined by western blotting. However, consistent with the reporter data shown, luciferase reporter experiments using constitutively active forms of the PI3K p110 catalytic subunit and constitutively active Akt, but not dominant-negative forms of these proteins, showed increasing inhibition of the endoglin promoter with plasmid dosage (data not shown). Overexpression of wild-type p85α may exhibit a dominant-negative effect due to overproduction of p85α monomers, which could compete with endogenous p85/p110 dimers. Thus, in in vitro systems, p85α overexpression could produce a similar effect as the p85α dominant negative mutant that lacks any ability to bind p110. Our results suggest that this does not occur in this system, but are consistent with loss of repression of endoglin expression in Pik3r1 null lymphatics, providing support for the view that endoglin expression is inhibited by PI3K at the transcriptional level, and suggesting that the regulation of endoglin by PI3K in mice is recapitulated in human lymphatic endothelial cells. To further test the hypothesis that PI3K regulates endoglin expression in developing lymphatics, we prepared whole mount sections, which were immunostained with anti-endoglin antibody. Consistent with the preceding results, we found increased endoglin expression in gut lymphatics of Pik3r1 null mice (Figures 7C-7F).
Valve formation is either independently controlled by Foxc2 and Pik3r1, or Pik3r1 acts downstream of Foxc2. However, in contrast to Foxc2 null mice, Pik3r1 ablation was not accompanied by increased mural cell recruitment, as assessed with IHC for SMA (Figures 1–5) and NG2 (data not shown). Foxc2 expression in adipocytes is p85/PI3K-dependent (Gronning et al., 2002), and so is the subcellular localization and phosphorylation status of other forkhead superfamily members (Abid et al., 2004). However, we found no difference in Foxc2 between Pik3r1 null mice and wild-type littermates by immunohistochemistry and western blot analyses (data not shown). Our data suggest that Pik3r1 isoforms are dispensable for proper mural cell investment, but are necessary for lymph valve formation.
Endoglin has been implicated in blood vessel maturation and stability. Mutations in endoglin are responsible for hereditary hemorrhagic telangiectasia (McAllister et al., 1994), which is caused by reduced levels of endoglin protein expression ((Pece-Barbara et al., 1999), reviewed in (Bernabeu et al., 2007)). Recent studies using endoglin transgenic mice indicate that endoglin overexpression in vascular precursor cells promotes vascular smooth muscle cell investment of major vessels (Mancini et al., 2007). The finding that endoglin, a prohemangiogenic endothelial cell marker, is upregulated in Pik3r1 null mouse lymphatic tissues and human lymphatic endothelial cells suggests that PI3K-dependent endoglin repression opposes angiogenesis and plays a role in lymphangiogenic vessel identity.
These data not only suggest a mechanism whereby abnormal endoglin expression in Pik3r1 null lymphatic vessels contributes to the pathology observed in Pik3r1 null mice, but also provide the novel insight that PI3K-dependent regulation of endoglin may play a role in lymph vessel homeostasis. Endoglin expression is thought to repress TGFβ signaling by inhibiting the TGFβ receptor ALK5 (Lastres et al., 1996; Blanco et al., 2005). Pharmacologic inhibition of ALK5-dependent TGFβ signaling in lymphatic cells accelerates lymphangiogenesis in a mouse model of chronic peritonitis (Oka et al., 2008). Thus, our study suggests that endoglin may play a role in regulating lymphangiogenic homeostasis. This view is supported by the observation that endoglin is absent in normal blood vascular smooth muscle, but is increased following vessel injury (Ma et al., 2000) and in atherosclerotic vascular smooth muscle (Conley et al., 2000).
The present study, combined with the absence of lymphatic defects in p85α null (p55α/p50α isoforms intact) and p55α/p50α null (p85α isoform intact) embryos, indicates that p85α/p55α/p50α isoforms have redundant but required functions in lymphatic development. These functions are most likely to involve phosphotyrosine-based associations with Tie1/Tie2, PDGFRs, VEGFRs, or Eph receptors, because what is preserved among the Pik3r1 isoforms are the SH2 domains. Based on the lymphatic defects of p110α-mutant mice (Gupta et al., 2007), an attractive model is that effective activation of class Ia PI3K requires at least two interactions downstream of receptor tyrosine kinases: SH2 domains of a regulatory isoform binding to phosphotyrosine, and binding of p110α to activated Ras. Further in vitro and in vivo studies to determine the lymphangiogenic signaling pathway(s) dependent on each isoform, and to elucidate the extent to which their function in vascular development is endothelial cell-autonomous will provide new insight into the mechanism of lymphangiogenesis.
METHODS
Protocols were pre-approved by our Institutional Animal Care and Use Committee. We followed published methods for generation of the Pik3r-targeted mice (Fruman et al., 2000), western blot analysis (Fruman et al., 2000), whole mount immunohistochemistry (IHC) (Karkkainen et al., 2004; Petrova et al., 2004), lymphatic endothelial cell culture (Petrova et al., 2004), and luciferase reporter analyses (Rius et al., 1998; Botella et al., 2002). At least eight surviving mice of each genotype were analyzed per data point.
Image analysis of lymphatic vessel branching and diameter was estimated using a morphometric approach (Kumar et al., 1997). For vessel branching, the number of branch points was determined for a minimum of five independent samples each from wild-type and null mouse specimens. For vessel diameters, the width of the vessel lumen was obtained by delineating the lumenal diameter, in triplicate, using Photoshop CS2 (Adobe Systems). Statistical significance was assessed using a two-tailed Student’s t-Test and assuming unequal variances.
Supplementary Material
Acknowledgments
Grant Information: This study was supported in part by grants from the NIH: AI050831 (DAF), P20 RR 15555 from the NIH COBRE program of the National Center for Research Resources (LL, IP, CPHV), and HL083151 from the National Heart, Lung, and Blood Institute (CPHV).
The authors thank Dr. William B. Stallcup for the gift of anti NG2 antibody, Dr. F. L. Lucas, Center for Outcomes Research and Evaluation, Maine Medical Center Research Institute, for review of statistical analyses, and the DNA and Protein Analysis and Cell Imaging Core Facility for confocal immunofluorescence analysis.
References
- Abid MR, Guo S, Minami T, Spokes KC, Ueki K, Skurk C, Walsh K, Aird WC. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:294–300. doi: 10.1161/01.ATV.0000110502.10593.06. [DOI] [PubMed] [Google Scholar]
- Bernabeu C, Conley BA, Vary C. Novel Biochemical Pathways of Endoglin in Vascular Cell Physiology. J Cell Bioch. 2007;102:1375–1388. doi: 10.1002/jcb.21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CP, Bernabeu C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol. 2005;204:574–584. doi: 10.1002/jcp.20311. [DOI] [PubMed] [Google Scholar]
- Botella LM, Sanchez-Elsner T, Sanz-Rodriguez F, Kojima S, Shimada J, Guerrero-Esteo M, Cooreman MP, Ratziu V, Langa C, Vary CP, Ramirez JR, Friedman S, Bernabeu C. Transcriptional activation of endoglin and transforming growth factor-beta signaling components by cooperative interaction between Sp1 and KLF6: their potential role in the response to vascular injury. Blood. 2002;100:4001–4010. doi: 10.1182/blood.V100.12.4001. [DOI] [PubMed] [Google Scholar]
- Chen D, Mauvais-Jarvis F, Bluher M, Fisher SJ, Jozsi A, Goodyear LJ, Ueki K, Kahn CR. p50alpha/p55alpha phosphoinositide 3-kinase knockout mice exhibit enhanced insulin sensitivity. Mol Cell Biol. 2004;24:320–329. doi: 10.1128/MCB.24.1.320-329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley BA, Smith JD, Guerrero-Esteo M, Bernabeu C, Vary CP. Endoglin, a TGF-beta receptor-associated protein, is expressed by smooth muscle cells in human atherosclerotic plaques. Atherosclerosis. 2000;153:323–335. doi: 10.1016/s0021-9150(00)00422-6. [DOI] [PubMed] [Google Scholar]
- Deane JA, Kharas MG, Oak JS, Stiles LN, Luo J, Moore TI, Ji H, Rommel C, Cantley LC, Lane TE, Fruman DA. T-cell function is partially maintained in the absence of class IA phosphoinositide 3-kinase signaling. Blood. 2007;109:2894–2902. doi: 10.1182/blood-2006-07-038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunological Reviews. 2009 doi: 10.1111/j.1600-065X.2008.00750.x. In Press. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Mauvais-Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT, Kahn CR, Cantley LC. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, Cantley LC. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999;283:393–397. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- Gronning LM, Cederberg A, Miura N, Enerback S, Tasken K. Insulin and TNF alpha induce expression of the forkhead transcription factor gene Foxc2 in 3T3-L1 adipocytes via PI3K and ERK 1/2-dependent pathways. Mol Endocrinol. 2002;16:873–883. doi: 10.1210/mend.16.4.0803. [DOI] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Jeltsch M, Tammela T, Alitalo K, Wilting J. Genesis and pathogenesis of lymphatic vessels. Cell Tissue Res. 2003;314:69–84. doi: 10.1007/s00441-003-0777-2. [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Kim KE, Sung HK, Koh GY. Lymphatic development in mouse small intestine. Dev Dyn. 2007;236:2020–2025. doi: 10.1002/dvdy.21200. [DOI] [PubMed] [Google Scholar]
- Kolbjornsen O, Press CM, Landsverk T. Gastropathies in the Lundehund. I. Gastritis and gastric neoplasia associated with intestinal lymphangiectasia. Apmis. 1994;102:647–661. [PubMed] [Google Scholar]
- Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, Saya H, Suda T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009 doi: 10.1084/jem.20081605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hoover JL, Simmons CA, Lindner V, Shebuski RJ. Remodeling and neointimal formation in the carotid artery of normal and P-selectin-deficient mice. Circulation. 1997;96:4333–4342. doi: 10.1161/01.cir.96.12.4333. [DOI] [PubMed] [Google Scholar]
- Lastres P, Letamendia A, Zhang H, Rius C, Almendro N, Raab U, Lopez LA, Langa C, Fabra A, Letarte M, Bernabeu C. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol. 1996;133:1109–1121. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymboussaki A, Partanen TA, Olofsson B, Thomas-Crusells J, Fletcher CD, de Waal RM, Kaipainen A, Alitalo K. Expression of the vascular endothelial growth factor C receptor VEGFR-3 in lymphatic endothelium of the skin and in vascular tumors. Am J Pathol. 1998;153:395–403. doi: 10.1016/S0002-9440(10)65583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Labinaz M, Goldstein J, Miller H, Keon WJ, Letarte M, O’Brien E. Endoglin Is overexpressed after arterial injury and is required for transforming growth factor-ss-induced inhibition of smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2000;20:2546–2552. doi: 10.1161/01.atv.20.12.2546. [DOI] [PubMed] [Google Scholar]
- Makinen T, Adams RH, Bailey J, Lu Q, Ziemiecki A, Alitalo K, Klein R, Wilkinson GA. PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev. 2005;19:397–410. doi: 10.1101/gad.330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG, Alitalo K. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. Embo J. 2001;20:4762–4773. doi: 10.1093/emboj/20.17.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini ML, Verdi JM, Conley BA, Nicola T, Spicer DB, Oxburgh LH, Vary CP. Endoglin is required for myogenic differentiation potential of neural crest stem cells. Dev Biol. 2007;308:520–533. doi: 10.1016/j.ydbio.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- Oka M, Iwata C, Suzuki HI, Kiyono K, Morishita Y, Watabe T, Komuro A, Kano MR, Miyazono K. Inhibition of endogenous TGF-beta signaling enhances lymphangiogenesis. Blood. 2008;111:4571–4579. doi: 10.1182/blood-2007-10-120337. [DOI] [PubMed] [Google Scholar]
- Oliver G. Lymphatic vasculature development. Nat Rev Immunol. 2004;4:35–45. doi: 10.1038/nri1258. [DOI] [PubMed] [Google Scholar]
- Papp M, Rohlich P, Rusznyak I, Toro I. An electron microscopic study of the central lacteal in the intestinal villus of the cat. Z Zellforsch Mikrosk Anat. 1962;57:475–486. [PubMed] [Google Scholar]
- Patan S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004;117:3–32. doi: 10.1007/978-1-4419-8871-3_1. [DOI] [PubMed] [Google Scholar]
- Pece-Barbara N, Cymerman U, Vera S, Marchuk DA, Letarte M. Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum Mol Genet. 1999;8:2171–2181. doi: 10.1093/hmg/8.12.2171. [DOI] [PubMed] [Google Scholar]
- Peters KG, Kontos CD, Lin PC, Wong AL, Rao P, Huang L, Dewhirst MW, Sankar S. Functional significance of Tie2 signaling in the adult vasculature. Recent Prog Horm Res. 2004;59:51–71. doi: 10.1210/rp.59.1.51. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Karpanen T, Norrmen C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Yla-Herttuala S, Miura N, Alitalo K. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974–981. doi: 10.1038/nm1094. [DOI] [PubMed] [Google Scholar]
- Pudliszewski M, Pardanaud L. Vasculogenesis and angiogenesis in the mouse embryo studied using quail/mouse chimeras. Int J Dev Biol. 2005;49:355–361. doi: 10.1387/ijdb.041956mp. [DOI] [PubMed] [Google Scholar]
- Rius C, Smith JD, Almendro N, Langa C, Botella LM, Marchuk DA, Vary CP, Bernabeu C. Cloning of the promoter region of human endoglin, the target gene for hereditary hemorrhagic telangiectasia type 1. Blood. 1998;92:4677–4690. [PubMed] [Google Scholar]
- Rodriguez-Niedenfuhr M, Papoutsi M, Christ B, Nicolaides KH, von Kaisenberg CS, Tomarev SI, Wilting J. Prox1 is a marker of ectodermal placodes, endodermal compartments, lymphatic endothelium and lymphangioblasts. Anat Embryol (Berl) 2001;204:399–406. doi: 10.1007/s00429-001-0214-9. [DOI] [PubMed] [Google Scholar]
- Sabin FR. The Origin and Development of the Lymphatic System. Baltimore, MD: The Johns Hopkins Press; 1913. [Google Scholar]
- Saharinen P, Tammela T, Karkkainen MJ, Alitalo K. Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. Trends Immunol. 2004;25:387–395. doi: 10.1016/j.it.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Schledzewski K, Falkowski M, Moldenhauer G, Metharom P, Kzhyshkowska J, Ganss R, Demory A, Falkowska-Hansen B, Kurzen H, Ugurel S, Geginat G, Arnold B, Goerdt S. Lymphatic endothelium-specific hyaluronan receptor LYVE-1 is expressed by stabilin-1+, F4/80+, CD11b+ macrophages in malignant tumours and wound healing tissue in vivo and in bone marrow cultures in vitro: implications for the assessment of lymphangiogenesis. J Pathol. 2006;209:67–77. doi: 10.1002/path.1942. [DOI] [PubMed] [Google Scholar]
- Stephens L, Williams R, Hawkins P. Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol. 2005;5:357–365. doi: 10.1016/j.coph.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhang D, Fallavollita L, Brodt P. Vascular endothelial growth factor C expression and lymph node metastasis are regulated by the type I insulin-like growth factor receptor. Cancer Res. 2003;63:1166–1171. [PubMed] [Google Scholar]
- Ueki K, Yballe CM, Brachmann SM, Vicent D, Watt JM, Kahn CR, Cantley LC. Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2002;99:419–424. doi: 10.1073/pnas.012581799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JF, Zhang X, Groopman JE. Activation of vascular endothelial growth factor receptor-3 and its downstream signaling promote cell survival under oxidative stress. J Biol Chem. 2004;279:27088–27097. doi: 10.1074/jbc.M314015200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.