

Abstract
The serine/threonine protein kinase B (PKB, also known as Akt) constitutes an important node in diverse signaling cascades downstream of growth factor receptor tyrosine kinases. Akt plays an essential role in cell survival, growth, migration, proliferation, polarity, and metabolism (lipid and glucose); cell cycle progression; muscle and cardiomyocyte contractility; angiogenesis; and self-renewal of stem cells. Altered Akt activity has been associated with cancer and other disease conditions, such as diabetes mellitus, neurodegenerative diseases, and muscle hypotrophy. In the past decade, the upstream signals that lead to Akt activation, the downstream substrates that exert the effects of Akt, and the secondary binding proteins that regulate Akt activation have been well documented. Recent reports from our group and others have revealed how the stability of Akt protein is regulated through phosphorylation on its Thr-Pro motifs. This literature review details findings of those reports and others relevant to the regulation of Akt activation by its upstream kinases, with a focus on mammalian target of rapamycin complexes (mTORCs) and inactivation by PHLDA3 and the protein phosphatases PP2A and pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP). Reports on ubiquitin-dependent Akt degradation, caspase-dependent cleavage, and the roles of molecular chaperone heat shock protein 90 (Hsp90) in the regulation of Akt stability are summarized. The highlight will be on the role of “turn motif” phosphorylation and an isomerase, Pin1, in the regulation of Akt stability. We also discuss issues related to the intricate mTORC2-AktmTORC1 loop and the contradictory regulation of Akt phosphorylation and stabilization of Akt by mTORC2. Finally, we offer perspective on potential future directions for investigation, particularly on translating the knowledge we learned on the regulation of Akt stability into therapeutic intervention on human cancer with Akt alteration.
Keywords: Serine/threonine protein kinase B, PKB, Akt, growth factor receptor, tyrosine kinases, physiological activity regulation, stability, mammalian target of rapamycin complexes, mTORCs, Pin1, caspase, DEPTOR, PP2A, pleckstrin homology domain, PH domain, PHLPPs, PHLDAs, heat shock protein
Background
Akt was originally identified as the human homologue of the viral oncogene v-akt from the transforming retrovirus AKT8, which was isolated from an AKR mouse T-cell lymphoma [1, 2]. Akt is now classified as a family of kinases that bears significant homology to both protein kinase A (PKA) and protein kinase C (PKC). In mammalian cells, Akt has three closely related and highly conserved (>80% sequence identity) cellular homologues, designated as Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ (Figure 1). The three isoforms of human (mouse) Akt are located at chromosomes 14q32 (12F1-2), 19q13 (7B1), and 1q44 (1H4-6), respectively [2, 3]. Each Akt family member contains an N-terminal pleckstrin homology (PH) domain, a central kinase domain, and a carboxyl-terminal regulatory domain that contains the hydrophobic motif (HM). The HM is a characteristic of AGC kinases (for PKA, protein kinase G [PKG], and PKC), which in mammalian cells include Akt, p70 ribosomal S6 kinase (S6K1), and serumand glucocorticoid-inducible kinase (SGK) [2, 4]. Analysis of mice lacking either individual Akt isoforms or various combinations of Akt isoforms has indicated that the Akt1 isoform has a dominant role in embryonic development, fetal growth, and fetal survival, whereas Akt2 and Akt3 have non-redundant functions in glucose homoeostasis and postnatal brain development, respectively [4-6].
Figure 1.

Akt domains and comparison of Akt isoforms (% of homology). Chromosome location of each Akt isoform in human as well as reported phosphorylation sites in Akt1 are also depicted. Color-embedded sites are the major focus of the current review.
Aberrant activation of the Akt pathway has been widely implicated in many cancers [7-17]. Elevated Akt activation in human cancers can result from enhanced activation phosphorylation of Akt on the Ser473 site, owing to enhanced phosphatidyliositoal-3 kinase (PI3K) activation resulting from: (1) the mutation or amplification/overexpression of growth factor receptors (such as Her-2/neu and EGFR), somatic mutations of Ras oncogenes, somatic mutations as well as amplification/overexpression of PI3K (either PI3CA or the p85 subunit); (2) loss of function mutations (somatic or germ line) and decreased expression (loss of heterozygosity or methylation) of a 3'-phosphatase with tensin homology or mutated in multiple advanced cancers (PTEN/MMAC1), which converts the lipid second messenger phosphatidylinositol-3,4,5- triphosphate (PIP3) to phosphatidylinositol- 4,5-biphosphate (PIP2) and thus shuts off PI3K signaling [8, 10, 11, 18-23]. Amplification, overexpression, and somatic mutation (at a very low frequency, ∼2%) of Akt itself also contribute to the elevated expression of Akt in human cancers [8, 14, 16, 22, 24-26]. Additionally, altered expression of PHLPP, a Ser473-specific protein phosphatase, may also affect Akt activity as reduced PHLPP in certain cancer cell lines correlates with Akt activity [27-31] (discussed in the next section). Gene amplifications and mutations of Akt and other molecules in the PI3K-Akt pathway were discussed in a recent review by Brugge et al [16].
Activation phosphorylation sites of Akt
Akt is activated through receptor tyrosine kinase pathways, such as those of plateletderived growth factor receptor (PDGF-R), insulin, epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), and insulin-like growth factor I (IGF-I) (Figure 2) [2, 32-34]. In the absence of growth factor stimulation in quiescent cells, all three isoforms of the Akt kinase are catalytically inactive. Growth factor stimulation activates Akt through a PI3Kdependent process [2, 32]. PI3Ks comprise a family of intracellular lipid kinases that phosphorylate the 3'-hydroxyl group of phosphatidylinositols (PIs) and phosphoinositides. PI3Ks are classified into three groups (classes I, II, and III) based on structure and substrate specificity. Class I PI3Ks are the primary lipid kinases that generate PIP3 from PIP2 [35-37]. Class I PI3Ks are further subdivided according to signaling receptors into class Ia (PIK3s that are activated by growth factor receptor tyrosine kinases) and class Ib (PIK3s that are activated by G protein-coupled receptors) [35-37]. Class II and class III PI3Ks use PI to generate PI-3-P. Mammals have one class III PI3K, VPS34, which is also conserved in yeast and acts as a sensor of available amino acids and signals mTOR to regulate cell growth and autophagy in response to low nutrient pools. Class II PI3Ks bind clathrin in coated pits, suggesting class II PI3Ks function in membrane trafficking and receptor internalization. Whereas, class Ia PI3Ks transduce signals from insulin and growth factors to regulate proliferation, survival, growth, and glucose homeostasis (reviewed in [36, 37]).
Figure 2.
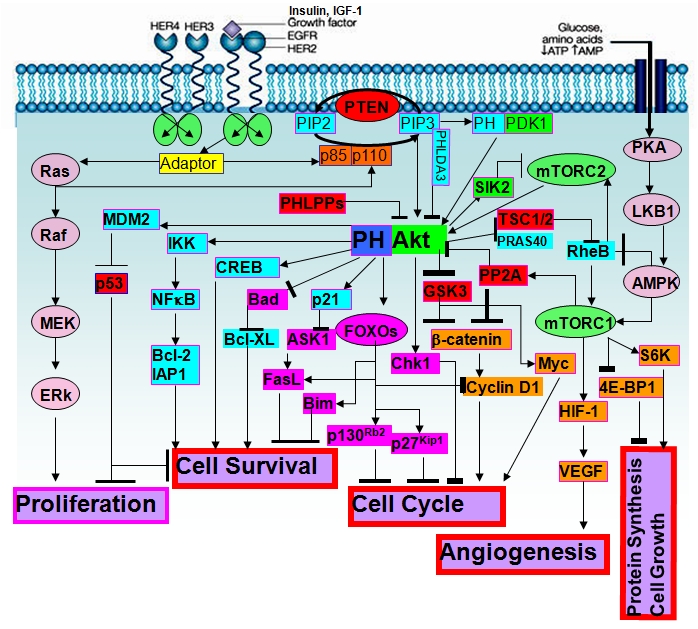
Cellular signaling around Akt and Akt substrates regulated major cellular processes.
Class Ia PI3K, which is engaged by the growth factor receptor tyrosine kinases through adaptor proteins such as Gab1, is a heterodimer comprising the p85 regulatory and p110 catalytic subunits (PI3CA). When class Ia PI3K is activated, it phosphorylates PIP2 at the 3' position on its inositol ring and converts PIP2 to PIP3 at the plasma membrane [2, 32, 36]. Subsequently, PIP3 recruits PI3K-downstream molecules such as Akt and 3-phosphoinositide- dependent kinase 1 (PDK1) by binding to their PH domain [2, 23, 32, 38-40]. Full activation of Akt is a multistep process, and the final step is to phosphorylate Akt on two sites, Thr308 (for human Akt1) and Ser473 (for Akt1), via PDK1 and PDK2, respectively [41]. Point mutants at these two sites with Alanine (T308A and S473A for Akt1) show little activity, even after stimulation with insulin or IGF-I, while the phosphorylation-mimicking mutant (T308D/S473D) shows constitutive kinase activation, indicating that Thr308 and Ser473 are necessary and sufficient for the full activation of Akt [2, 32, 38, 41-46]. Interestingly, in serum-starved cells, Akt is constitutively phosphorylated at Ser124 and Thr450, which are independent of PI3K, and neither serum starvation nor treatment of cells with the PI3K inhibitor wortmannin interferes with phosphorylation at Ser124 and Thr450 [41]. In addition, inactive mutation of Ser124 and Thr450 into Ala (Ser124A and T450A) only marginally inhibits the activation of Akt by growth factors. Therefore, it has been proposed by Tsichlis's group that phosphorylation of Akt on these sites is the first step for full activation of Akt [2, 41, 42, 47, 48].
The PH domain containing protein PDK1 is the only known mammalian isoform of the Thr308- Akt kinase [40, 49]. The C-terminal PH domain of PDK1 binds phospholipids, keeping it constitutively localized at the plasma membrane. Upon growth factor stimulation and PIP3 production by PI3Ks, Akt interacts with these phospholipids, causing Akt to translocate to the inner membrane, where PDK1 is located. The interaction of the Akt PH domain with 3'- phosphoinositides is thought to impose conformational changes in Akt, exposing its two main phosphorylation sites at the kinase domain (T308 for Akt1) and the HM of the Cterminal (S473 for Akt1). The direct homodimerization of the two PH domains between Akt and PDK1 might also mediate protein proximity and subsequently phosphorylate Thr-308 in Akt, which stabilizes the activation loop in an active conformation and renders Ser473 phosphorylation by the rapamycin-insensitive mTORC2, resulting in full activation of Akt kinase [39, 40, 48]. In certain contexts, kinases including PDK1, ataxia telangectasia mutated (ATM), DNA-dependent protein kinase (DNA-PK), integrin-linked kinase (ILK), PKCς/ι, PKC-related kinase-2 (PRK2), mitogen-activated protein kinase-associated protein kinase-2 (MAPKAP-K2), mTOR, and Akt itself were implicated in the phosphorylation of Akt at Ser473 before mTORC2 was identified as a Ser473 kinase [46-48, 50-63]. In a later phase, activated Akt is translocated to the nucleus, where several of its substrates are located (Figure 2).
Mechanistically, activation of Akt will profoundly affect cellular processes by phosphorylating numerous Akt substrates (listed and reviewed in [64]). Consensus motif analysis indicates that there are potentially thousands of cellular substrates for Akt; about 50 of them have been characterized so far [64]. Through phosphorylation, Akt may either positively or negatively affect the functions of these substrates, alter their subcellular localization, or modify their protein stabilities [64]. A major outcome of Akt activation is toward cell survival and cell growth; many Akt substrates play important roles. Among those substrates are: (1) regulators of cell survival or cell death, such as Bad, caspase-9, ASK1, apoptosis signal-regulating kinase 1 (ASK1), forkhead box O transcription factors (FoxOs), Bim1, FasL, inhibitor of nuclear factor-κB kinase (IKK-NFκB), and p53; (2) regulators of cells cycle progression; such as p21, p27, cyclin D1, and glycogen synthase kinase-3β (GSK-3β); (3) regulators of protein synthesis or cell growth, such as tuberous sclerosis complexes 1 and 2 (TSC1/2), mTOR, elongation-initiation factor 4E binding protein-1 (4E-BP1), and S6K; (4) regulators of angiogenesis, such as mTOR and hypoxia-inducible factor-1 (HIF-1); and (5) regulators of cell metabolism, such as glucose transporter 1 (Glut1), GSK3, and a Ras homologue enriched in brain (RheB) (reviewed in [64] and summarized in Figure 2). Aktmediated regulation of hormone receptors such as estrogen receptor (ER) and androgen receptor (AR) are not listed in Figure 2. Positive and negative regulators of Aktinteracting proteins have also been identified. These include oncogenes identified in human T-cell leukemia (Tcl1 and Tcl1 family members); JNK interacting protein 1 (JIP1); growth factor receptor-binding protein-10 (Grb10); Ras GTPase-activation protein (RasGAP); Hsp90/Cdc37 molecular chaperone or co-chaperone complex; a tribbles homologue 3 (TRB3); adaptor protein containing PH domain, PTB domain, and Leucine- zipper motif (APPL); C-terminal modulator protein (CTMP); Akt phosphorylation enhancer or hook-related protein-1 (APE); SH3 domaincontaining protein, Src and Arg-binding protein 2γ (ArgBP2γ); breast tumor kinase (Btk); prohibin 2 (PHB2); and pleckstrin homology-like domain, family A, member 3 (PHLDA3) [65-69] (Figure 2).
mTORC1 and mTORC2
The mTOR serine/threonine kinase is a member of the PI3K-like kinase family (PIKK), which includes ATM, ataxia-telangiectasis and Rad3- related (ATR), and DNA-PK. mTOR integrates many cellular signals that coordinate cell growth and division in response to growth factors, nutrients, and the energy status of the cell [14, 18, 23, 70]. Dysregulation of mTOR is implicated in various human diseases, including cancer, diabetes, and cardiovascular disease [58, 70, 71]. mTOR is the catalytic component of mTORC1 and mTORC2, which are evolutionarily conserved signaling complexes [48, 53, 54, 57, 58, 62, 63, 72, 73]. The rapamycin-sensitive mTORC1 is a key regulator of insulin, growth factor, and nutrient signaling and has three components: the mTOR catalytic subunit; regulatory-associated protein of mTOR (Raptor); and mammalian ortholog of yeast LST8, lethal with SEC13 protein 8 (mLST8) or small WD-repeat (i.e., tryptophan- aspartate repeat) protein Gβ-like protein (GβL). S6 kinase 1 and 2 (S6K1 and S6K2) and the elF-4E binding protein 1 (4E-BP1) are the best-characterized downstream effectors of mTORC1; they regulate ribosome biogenesis, mRNA translation, cell growth, and autophagy [63, 71, 74, 75].
The rapamycin-resistant mTORC2 contains mTOR, mLST8, rapamycin-insensitive companion of mTOR (Rictor), and mammalian stress activated protein kinase-interacting protein (mSin1) variants. Both Rictor and mSin1 are necessary for mTORC2 to phosphorylate Akt on its C-terminal HM, and this function is conserved evolutionarily [53, 54, 57]. mSin1 is also necessary for the assembly of mTORC2. Alternative splicing of mSin1 generates at least five isoforms of the mSin1 protein, three of which assemble into three distinct mTORC2s [48, 54, 58, 62, 63]. It is proposed that Rictor dictates the specificity of mTORC2 toward Akt and away from S6K and 4E-BP1 [51, 58]. The adaptor protein mLST8 is common to both mTORC1 and mTORC2. mLST8 may increase the activity of mTORC1 toward S6K and 4E-BP1. It may also play a positive role in mTORC2 function [72]. Recently, proteins observed with Rictor-1 (Protor-1) and its isoform Protor-2 have also been proposed to be components of mTORC2 but not mTORC1 [59]. Protor-1 is not required for the assembly of other mTORC2 subunits, and its expression is regulated by the expression of Rictor [59]. However, the roles of Protor-1 and -2 in mTORC2 and Akt S473 phosphorylation require further elucidation. By far, the best-characterized function of mTORC2 is the phosphorylation of Akt on Ser473 at the HM, but mTORC2 is also involved in the phosphorylation of a similar motif of PKCα, which along with S6K belongs to the AGC kinase family [48, 76-78]. Recent reports indicate that mTORC2 also phosphorylates a second conserved site on Akt and PKCα referred to as the “turn motif” (Thr450 on Akt1) [48, 77, 78]. As discussed earlier, Thr450 is constitutively phosphorylated on Akt and is independent of PI3K, and neither serum starvation nor treatment of cells with the PI3K inhibitor wortmannin interferes with phosphorylation of this site. Therefore, the question of how mTORC2 differentially phosphorylates Akt on Thr450 but not on Ser473 is quite intriguing (discussed further in section “Ser/Thr-Pro motif” phosphorylation and Akt stability. The conditions under which mTORC2 phosphorylates Akt on Ser473 but not on Thr450 are not clear. Also unclear is whether Thr450 phosphorylation is required for mTORC2-mediated phosphorylation of Ser473.
The drug rapamycin strongly and acutely inhibits mTORC1, but it affects mTORC2 assembly and activity only after prolonged exposure in certain cell types [56, 58]. A recent report from Hunter's group demonstrated that mTOR is phosphorylated differentially when it is associated with mTORC1 and mTORC2 and that intact complexes are required for these mTORC-specific mTOR phosphorylations [79]. The report also showed that mTORC1 contains mTOR phosphorylated predominantly on Ser2448, while mTORC2 contains mTOR phosphorylated predominantly on Ser2481. Using Ser2481 phosphorylation as a marker for mTORC2 sensitivity to rapamycin, the investigators found that mTORC2 formation is rapamycin sensitive in several cancer cell lines; those cell lines had previously been reported to have rapamycin–insensitive mTORC2 assembly and function, as deduced from phosphorylation of Akt Ser473. Therefore, the authors proposed that Ser2481 phosphorylation in mTOR is a better biomarker than Akt Ser473 phosphorylation for intact mTORC2 and its sensitivity to rapamycin [79]. Although phosphorylation of mTOR may have an important role in mTOR regulation and the formation of mTOR complexes, the identities of the kinases that phosphorylate mTOR on Ser2448 and Ser2481 are not known. Since mTOR is the only known target of rapamycin identified in mammalian cells, it is not clear how treatment with rapamycin mechanistically blocked mTOR phosphorylation on Ser2448 and Ser2481. It is also unclear whether rapamycin- mediated dissociation of mTOR from the complexes comes first or whether dephosphorylation of mTOR is a prerequisite for mTOR dissociation from the complexes. Along the same line, so far the upstream signals that lead to mTORC2 formation and activation by growth factors and insulin are not known. What is clear is that mTOR lies both upstream (in the form of mTORC2) and downstream (in the form of mTORC1) of Akt and negatively regulates Akt activation through the S6K insulin receptor substrate (S6K-IRS) negative feedback loop (reviewed in [63, 80]). Akt also regulates mTORC1 through direct phosphorylation of tuberous sclerosis complex-2 (TSC2) and proline-rich Akt substrate 40 kDa (PRAS40), proteins that suppress mTORC1 kinase activity and thereby activate mTORC1 [63, 75, 80]. Phosphorylation of TSC2 by Akt impairs the ability of the TSC1-TSC2 complex to act as a GTPase-activating protein toward small GTPase Rheb, which allows Rheb-GTP to accumulate and potently activate mTORC1 [63, 80]. It has been proposed that Aktmediated cell proliferation and oncogenic transformation may occur exclusively via mTORC1 [80, 81]. Additionally, negative feedback regulation between Akt and mTORC2 may also exist. Dentin et al [82] reported that Akt2-mediated phosphorylation and activation of salt-inducible kinase 2 (SIK2, a Ser/Thr kinase) may in turn stimulate SIK2-induced phosphorylation of mTOR at Ser171 and subsequent cytoplasmic translocation and ubiquitin proteasome–dependent degradation of mTORC2. Intriguingly, recent identification of a new mTOR-interacting protein, DEP domain– containing mTOR-interacting protein (DEPTOR), demonstrates another feedback regulation loop among Akt, mTORC1, and mTORC2 [83]. Immunoprecipitation indicates that DEPTOR associates with mTOR, Rictor, and Raptor; thus, DEPTOR is a component of both mTORC1 and mTORC2. Levels of DEPTOR are negatively regulated by mTORC1 and mTORC2 through mTOR-mediated phosphorylation and subsequent degradation of DEPTOR. While overexpression of DEPTOR suppresses S6K and mTORC1, it relieves feedback inhibition from mTORC1 to PI3K-Akt signaling; thus, overexpression of DEPTOR activates mTORC2 and Akt. Consistently, depletion or loss of DEPTOR activates S6K1, Akt, SGK1, mTORC1, and mTORC2. Therefore, mTORC1, mTORC2, and DEPTOR form a feed-forward loop in which overexpression of DEPTOR represses mTOR, as observed in multiple myeloma, and is required for the maintenance of PI3K activation, Akt activation, and cell survival [83].
A newly identified oncoprotein, GOLPH3 (Golgi phosphoprotein 3), which is amplified in several solid tumor types, has also been shown to modulate mTOR signaling by upregulating phosphorylation of both mTORC1 (eg, S6K) and mTORC2 (eg, Akt-S473) substrates. However, unlike the link between DEPTOR and mTOR signaling, the link between GOLPH3 and mTOR signaling is weak, and it is not clear how GOLPH3 regulates mTOR signaling mechanistically [84]. Although GOLPH3 directly interacts with vacuolar protein sorting 35 (VPS35) in yeast (a component of the large multimeric retromer complex, which is involved in retrograde transport of proteins from endosomes to the trans-Golgi network), there is no evidence yet to validate whether GOLPH3 regulates mTOR signaling indirectly through its interaction with VPS35 or directly through interactions with mTORC components.
Inactivation of Akt––PP2A and the HM phosphatases
PP2A and Akt dephosphorylation
The core enzyme of PP2A is a dimer consisting of a catalytic subunit (PP2A/Cα or β) and a regulatory or structural A subunit (PP2A/Aα or β). A third regulatory B subunit (PP2A/B) that determines substrate specificity can be associated with this core structure [85-89]. The A and C subunits each exist as two isoforms, whereas the 16 B subunits fall into four families. PP2A A subunits are composed of 15 nonidentical tandem repeats of a 39–amino acid sequence, termed a HEAT motif (named after the proteins that contain them: huntingtin, elongation factor, A subunit, and TOR kinase). The B subunits bind to repeats 1-10 of the A subunit, and the C subunits bind to repeats 11-15 of the A subunit. In the cell, the C subunit of PP2A forms a complex with an array of regulatory B subunits that modulate its catalytic activity, substrate specificity, and subcellular localization. The B subunits comprise four unrelated families: B, B', B”, and B'”. Each of these families has several members, all of which are able to bind to the A subunit in a mutually exclusive manner to form distinct ABC holoenzyme complexes. In various combinations, the subunits can produce more than 75 different trimeric holoenzymes. It is believed that PP2A exercises regulatory flexibility and differential substrate specificity through the preferential association of the core dimer with one of the regulatory B subunits (ie, the B subunit determines the substrate specificity of PP2A). The B subunits of PP2A are expressed differentially by tissue and temporally during development; therefore, it is the B subunits that specify substrate specificity and likely target the PP2A catalytic complex to intracellular sites, i.e., the B subunits define the individual PP2A isoforms and their physiological roles.
Recent evidence indicates that PP2A forms stable complexes with protein kinase signaling molecules, indicating that it plays a central regulatory role in signal transduction mediated by reversible protein phosphorylation [90-92]. Certain viruses have chosen to target this enzyme system to manage the host cell machinery to their own benefit and to program cells into a malignant state [93-95]. The small t antigen (ST) of the DNA tumor virus simian virus 40 (SV40) may directly interact with a pre-existing dimer, or it may displace the third regulatory B subunit from a trimeric holoenzyme [93-95]. Similarly, suppression of PR61/B'γ, a specific B subunit of PP2A that might be replaced by ST, can substitute for the viral SV40 ST and cause tumorigenic transformation [88, 93, 96]. Based on the observations that okadaic acid is a strong inhibitor of PP2A, yet at the same time a potent tumor promoter, it was suggested that the PP2A holoenzyme might, to some extent, be a tumor suppressor [88, 96, 97]. Indeed, both the α and β isoforms of the A subunit are genetically altered at low frequency in a variety of primary human tumors [93, 94, 98-105]. Ruediger et al discovered that some of these cancer-associated isoform mutations were defective in their binding of other PP2A subunits [100-102]. Ito et al reported that a truncated isoform of the B56γ subunit of PP2A promotes genetic instability and causes tumor progression [106].
There is evidence that PP2A targets Akt to cause cellular transformation. The PI3K-Akt signaling cascade is activated by ST of SV40 [107], and by repressing PP2A, ST promotes phosphorylation of Akt on both Thr308 and Ser473 in the absence of growth factors [108]. Further, studies from our group and others have demonstrated that Akt is a direct substrate of PP2A [68, 109-111]. Pim et al reported that the human papilloma virus E7 protein also binds to the C and A subunits of PP2A, sequesters these subunits, and inhibits their interaction with Akt, thereby maintaining Akt signaling by inhibiting its dephosphorylation [112].Our group and others have shown that either exposing cells to the PP2A-specific inhibitor okadaic acid or knocking down PP2A catalytic (or specific regulatory B) subunits resulted in elevated Akt phosphorylation and activation [68, 109-111, 113, 114]. Ceramide, an intermediate in sphingomyelin biosynthesis and a powerful second-signal effector molecule that enhances PP2A activity, has also been shown to repress Akt by maintaining Akt in an inactive, dephosphorylated state [115]. More recently, particular B subunits of the PP2A holoenzyme that preferentially bind Akt to PP2A and inactive Akt by dephosphorylation have been confirmed in mammalian cells and C. elegans [114, 116, 117]. Although PP2A may preferentially dephosphorylate Akt on the Thr-308 site, it can also dephosphorylate Akt on the Ser473 site under certain conditions. Using in vitro dephosphorylation assays, we demonstrated that PP2A dephosphorylates Akt on both Thr-308 and Ser473 sites [110]. In cell culture under in vivo conditions, we were also able to show that both Thr-308 and Ser473 phosphorylation were affected by PP2A [110]. In both mammalian cells and C. elegans, knocking down the B56 regulatory subunit of PP2A induces enhanced Akt phosphorylation (on both Thr308 and Ser473 in mammalian cells and Thr-350 and Ser-527 in C. elegans), though to a lesser extent [112, 114, 117].
PHLPPs and isoform-specific HM phosphatases
The lipid protein phosphatase PTEN acts as a potent negative regulator of Akt activation phosphorylation by converting PI3K-generated PIP3 into PIP2, thereby blocking Akt phosphorylation on both Thr308 and Ser473 by PDK1 and PDK2, respectively [111, 118]. However, PTEN does not directly dephosphorylate Akt on either the Thr308 site or the Ser473 site. As discussed earlier, PP2A preferentially dephosphorylates Akt on the Thr308 site [108, 113, 116, 119]. In a query of the National Center for Biotechnology Information database for genes that encode a PH domain and a phosphatase domain, Newton's group identified the PHLPP family as HM phosphatases that specifically dephosphorylate Akt on Ser473 but not Thr308 residue [27, 28, 120]. The PHLPP family comprises three members, PHLPP1α, PHLPP1β, and PHLPP2. PHLPP1α and β are splice variants of the same gene, located at chromosome 18q21.33, while the geneencoding PHLPP2 resides at chromosome 16q22.3 [29]. The PHLPP proteins contain an identical domain structure, with a PH domain followed by region of leucine-rich repeats (LRR), a PP2C phosphatase domain, and a Cterminal PDZ ligand. Both PHLPP1β and PHLPP2 also contain a Ras-associated domain that precedes the PH domain. PHLPPs are expressed in a majority of human tissues and numerous cancer cell lines, including cancers of the brain, breast, lung, prostate, and ovary. PHLPP proteins are present in the cytosolic, nuclear, and membrane fractions of cells, which may reflect their potential substrate localizations [27, 29, 30, 121]. So far, only Akt and PKC have been identified as direct substrates of PHLPPs. AGC kinase family p70S6K, SGK, and p90RSK, all of which also have phosphorylation HMs but not necessarily the PH domain, have not been confirmed as a reliable direct substrate of PHLPPs. For PHLPPmediated dephosphorylation of Akt, both the PP2C domain and the C-terminal PDZ-binding motif are required; intriguingly, the PH domain of PHLPPs is dispensable for PHLPP-mediated dephosphorylation of Akt on Ser473 [27, 28]. However, the PH domain, but not the PDZbinding motif, is indispensable for PHLPPmediated dephosphorylation of PKCβII, which does not contain a PH domain but relies on its lipid second messenger and membrane targeting to be active in the cell. Unlike that mediated by PKC, dephosphorylation of Akt on Ser473 mediated by PHLPP neither destabilizes the enzyme nor promotes its degradation [27-30, 120]. A common polymorphism that results in an amino acid change from a Leu to Ser at codon 1016 in the phosphatase domain of PHLPP2 demonstrates reduced regulation of Akt and PKC and may likely contribute to elevated Akt phosphorylation and increased PKC levels in human cancer [30].
Further studies of each member of the PHLPP family by Newton's group revealed that specific PHLPP isoforms regulate the phosphorylation state of specific Akt isoforms as well as specific downstream substrates of these Akt isoforms [28]. First, the group discovered that PHLPP1 co-immunoprecipitated with only Akt2 and Akt3, while PHLPP2 co-immunoprecipitated with only Akt1 and Akt3. Accordingly, knocking down PHLPP1 increased HM phosphorylation of only Akt2 and Akt3, while knocking down PHLPP2 increased hydrophobic motif phosphorylation of only Akt1 and Akt3. Second, depletion of endogenous PHLPP2 resulted in increased phosphorylation of Akt1 substrates GSK3β, TSC2, FoxO, and p27. Third, depletion of endogenous PHLPP1 increased the phosphorylation state of a unique set of Akt substrates, including HDM2 (human homologue to murine double minute 2) and GSK3α, in addition to the same penal of Akt1 substrates (i.e., GSK3β, TSC2, and FoxO) regulated by PHLPP2. These results showed that only PHLPP2 regulates Akt1 Ser473 phosphorylation on the HM, only PHLPP1 regulates Akt2 Ser 474 phosphorylation on the HM, and both PHLPP1 and PHLPP2 regulate Akt3 Ser472 phosphorylation on the HM [28, 120]. It is not clear whether the PH domain of PHLPPs can bind with 3'-phosphorylated phosphoinositides and compete with Akt for binding to PIP3 as do some other PH domain–containing proteins. It is also not clear whether the PH domain of PHLPPs is required for their physical interaction with Akt. So far, there is no evidence to document how PHLPPs themselves are regulated and under what physiological or pathological conditions PHLPPs differentially dephosphorylate Akt isoforms on the HM.
PHLDA3 and Akt-MDM2-p53-PHLDA3 feedback regulation
A PH domain–only protein named PH domain, family A, member 3 (PHLDA3) also inhibits Akt activity, but its inhibition is dependent on its PH domain [66]. The PHLDA3 gene resides in chromosome 1q31.1, which is transcribed as a 127–amino acid protein (∼14 kDa). PHLDA3 was originally identified as a p53-inducible protein involved in DNA damage-mediated stress response [122, 123]. It contains two consensus p53 binding motifs in its promoter region, and its expression is regulated directly by p53. PHLDA3 also participates in p53- mediated apoptosis. Overexpression of PHLDA3 itself induces apoptosis, and an intact PH domain is required for PHLDA3-induced apoptosis [66]. Through the PH domain, which in most cases is known to mediate specific binding with phosphoinositides and to drive membrane recruitment in response to phosphatidylinositol 3-kinase activation, PHLDA3 binds with each single PI(3)P, PI(4)P, or PI(5)P and any combination of PIP2s [PI(3,4)P2; PI(4,5)P2; and PI(3,5)P2] or PI(3,4,5)P3. Thus, PHLDA3 competes with Akt for binding to both PI(3,4)P2 and PI(3,4,5)P3, the only two lipids to which Akt binds [66]. Interestingly, Kawase et al [66] showed via an immunoprecipitation assay that PHLDA3 does not physically interact with Akt. Therefore, the PHLDA3-mediated repression of Akt activation phosphorylation on both Thr308 and Ser473 is achieved through PHLDA3's superior binding affinity with PI(3,4)P2 and PI(3,4,5)P3, which eventually blocks Akt from binding with PIPs and disrupts Akt membrane localization and subsequent activation by PDKs.
Earlier reports from our group and others demonstrated that Akt represses p53 protein stability via Akt-mediated phosphorylation of MDM2 [124-133]. Phosphorylation of MDM2 by Akt enhances nuclear accumulation of MDM2 and consequently augments the destabilization of p53 by MDM2. Therefore, Akt- MDM2-p53-PHLDA3 may form a negative feedback loop [129] (Figure 3). Recruitment of PI3K components (p85 adaptor and p110 catalytic subunit) and non-receptor tyrosine kinases, such as Src kinase, occurs in the presence of growth/survival factors, growth factor–induced receptor dimerization, and autophosphorylation of tyrosine residues within the intracellular domain. Activated PI3K generates PIP3, which directly and indirectly (through PDKs) leads to Akt activation and subsequent phosphorylation of MDM2 and degradation of p53. Activated PI3K thereby blocks p53-mediated transcriptional activation of its target genes, including PHLDA3 and PTEN. In addition, when Src kinase is phos-phorylated and activated by receptor tyrosine kinases, it phosphorylates PTEN on tyrosine residues within the PTEN C2 domain and prevents PTEN from translocating to the plasma membrane and dephosphorylating PI3K-generated PIP3; this leads to cell growth and cell proliferation [134-136]. Under stress conditions, stress signal–mediated activation of p53 counteracts the inhibitory effects of this PI3K-Akt survival pathway by multiple mechanisms. One mechanism may be the p53-mediated transcriptional activation of PTEN and PHLDA3, both of which block PI3Kgenerated PIP3 and subsequent Akt activation (Figure 3). In this model, survival is achieved by the inhibition of p53 by Akt, and apoptosis is achieved by the counteracting of Akt by p53.
Figure 3.
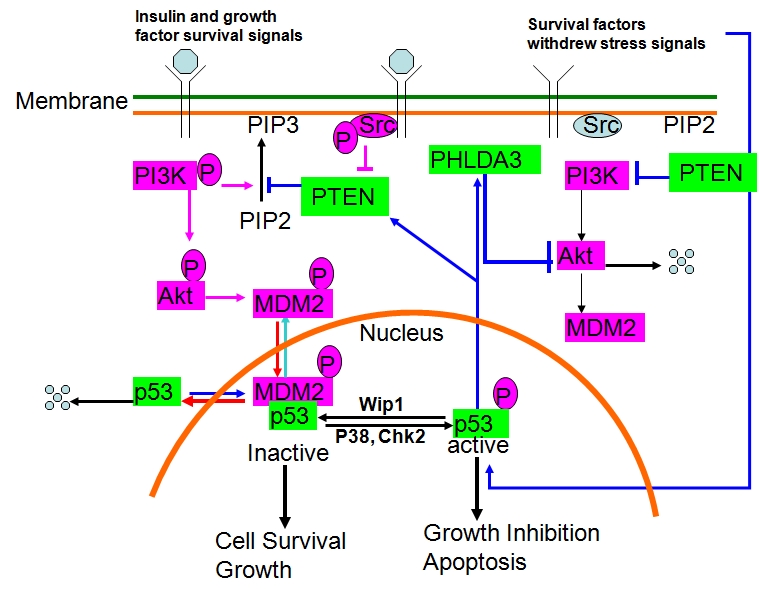
The PTEN-PI3K-Akt-p53-PHLDA3 feedback loop.
Degradation of Akt protein
As discussed earlier, proteins that negatively regulate Akt activation may not necessarily lead to Akt degradation [68]. In fact, under certain physiological or pathophysiological conditions, Akt protein can be degraded by the ubiquitin proteasome-dependent pathway, caspase-mediated cleavage, and caspase-dependent ubiquitination, as summarized below.
Ubiquitin-dependent Akt degradation
Degradation of proteins by the ubiquitin-proteasome system (UPS) is important for the regulation of many cellular functions, including cell cycle, growth, and cell polarity. The UPS involves the sequential activation of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), which determine substrates' specificity; thus, activation of E1, E2, and E3 results in the conjugation of ubiquitin to the lysine residues of target proteins. Those proteins, tagged with poly-ubiquitin, are then degraded by the proteasome complex [137-140]. Few reports have described how the Akt protein is degraded by the ubiquitin proteasome pathway, and it is not clear which specific E3 ligases are involved in Akt degradation by the UPS. Kim et al [141] reported that mannito-linduced degradation of Akt by the UPS can be blocked by IGF-1, thereby dissociating Akt from binding and subsequent degradation by the UPS, yet no published mechanistic studies have elucidated which E3 ligase links to Akt or how IGF-1 regulates this E3 ligase. Riesterer et al [142] reported that deprivation of vascular endothelial growth factor (VEGF) or blockade of the VEGF signal transduction cascade with the VEGF tyrosine kinase inhibitor PTK787/ZK222584 resulted in a specific decrease of Akt protein level; subsequent cellular stimulation with VEGF rescued Akt stability in endothelial cells. VEGF-controlled Akt proteolysis is blocked by a broad range of inhibitors of caspases and the proteasome complex (with proteasome inhibitor MG-262 showing particular efficacy). In addition, the mTOR inhibitor rapamycin also neutralized the VEGF-protective effect in proteolysis-dependent reduction of Akt protein, suggesting that mTOR signaling is involved in VEGF-protected Akt degradation [142]. As discussed earlier, mTORC2 is generally rapamycin insensitive, while the rapamycin-sensitive mTORC1 lies downstream of Akt; thus, it is not clear how mTOR signaling by itself regulates Akt stability in endothelial cells. It is unlikely that the regulation of Akt stability by rapamycin observed in endothelial cells will be shown to be a general phenomenon. Yan et al [143] reported that selective Akt degradation by the UPS in dendrites is required for generating neuronal polarity under physiological conditions and that suppressing the UPS with proteasomal inhibitor MG-132 leads to the symmetric distribution of Akt and the formation of multiple axons. Those findings indicate that local protein degradation of Akt mediated by the UPS is important in determining neuronal polarity. Mechanistically, why Akt was degraded in the dendrites and which E3 ligase was involved in the degradation of Akt is not known.
Caspase-mediated Akt cleavage
Unlike that of the UPS mediated Akt degradation, the mechanism by which Akt protein is cleaved by caspases is relatively clear. As a critical survival factor downstream of the receptor protein tyrosine kinases, Akt-transmitted survival signals have protective effects against apoptosis induced by a variety of stimuli. It thus follows that the Akt protein is degraded when cells undergo apoptosis [144-147]. By incubating Akt protein with active caspases (namely caspase-3, -6, and -7) in vitro, Rokudai et al [148] found that Akt was cleaved at three sites to produce 40- and 44-kDa fragments. Two cleavage sites were between the N-terminal PH domain and the kinase domain (TVAD108↓G and EEMD119↓ F) and a third site was in the C-terminal regulatory domain (SETD 434↓T). As can be expected, the loss of the C-terminal domain of the Akt protein reduced its kinase activity, owing to the loss of the HM (where Ser473 resides). Over-expression of Akt fragment with either deletion of the N-terminal domain or deletion of the Cterminal domain increased cell sensitivity to apoptosis-inducing stimuli, indicating that caspase-dependent cleavage of anti-apoptotic Akt turns off survival signals and augments apoptotic cell death [148]. In interleukin-3 (IL-3)-dependent 32D cells, Xu et al discovered that cytokine withdrawal resulted in Akt degradation by caspases as well [146]. The authors found the Asp462 residue of Akt1 (ECVD462↓) in the upstream of HM to be the primary cleavage site for caspase-3. Mutation of this site (Akt1-D462N) prevented caspase cleavage. Similar to the earlier-described C-terminal deletion fragment, the Akt truncation mutant mimicking the caspase cleavage product lost its kinase activity, functioned as a dominant negative mutant, and promoted cell death [146]. These results indicate that the balance between Akt and caspase activity controls cell survival in 32D cells, and upon survival factor withdrawal, caspases are able to render Akt inactive and dominantly inhibit the Akt pathway to allow apoptosis to occur. In an artificial in vitro caspase-3 cleavage assay, Jahani-Asl et al [149] recently found that both the caspase-3 consensus site (DQDD456↓ SM) as well as non-consensus caspase-3 cleavage sites (QEEE116↓E117↓MD; EEMD119↓; TPPD453↓QD; and DAKE398↓IM) of Akt1 can be cleaved by caspase-3. In addition, the authors demonstrated that phosphorylation of Akt1 modulates its cleavage in a site-specific manner (i.e., there is resistance to cleavage at the site DAKE398 within the kinase domain in response to phosphorylation), suggesting a possible mechanism by which the anti-apoptotic role of Akt1 is regulated [149]. However, it is not yet clear whether similar events might occur with Akt under physiological or pathological conditions in vivo. In Jurkat cells treated with 4-hydroxynonenal (HNE) to induce caspase-dependent apoptosis, Liu et al observed a decrease in Akt activity owing to dephosphorylation at Ser473 by PP2A. The effect could be prevented by either okadaic acid, a potent PP2A inhibitor, or DEVD-CHO, a caspase-3 specific inhibitor [150]. HNE treatment resulted in an increase in the total level of PP2A and PP2A-Akt association, both of which were depended on caspase-3 activation. The activity of Src, a representative caspase-sensitive kinase that phosphorylates and inactivates PP2A at tyrosine residues, was also downregulated by HNE treatment [150]. The authors of this report also showed that activated caspase-3 partially cleaves Akt at a late stage of apoptosis; however, they did not identify which specific cleavage sites on Akt were involved in HNE-induced apoptosis. In 3T3-lL adipocytes, Medina et al reported that the Akt protein was degraded via a caspase-dependent ubiquitination during tumor necrosis factor (TNF)-α-induced apoptosis [151]. Although no specific E3 ligase was identified, the authors demonstrated that TNF-α enhances E3 ligase activity associated with Akt and that both caspase and proteasome inhibition rescued Akt stability as well as insulin-stimulated Akt phosphorylation in adipocytes pre-exposed to TNF-α [151].
Heat shock proteins and Akt stability
Among molecule chaperones, Hsp90 is of prime importance to the survival of cancer cells [152-160]. Hsp90 contains a unique nucleotide binding domain at its animo terminal pocket. It binds with ATP and contains ATP hydrolyzing activity. Hsp90 forms the basis of a super-chaperone machine that promotes the proper folding of client proteins so that they can respond to a stimulus or bind ligand. ATP hydrolysis and ADP/ATP nucleotide exchange drive the cycling of the Hsp-90–based super-chaperone machine. A client protein's half-life may be stochastically determined by the period of time it resides in association with the Hsp-90-Hsp-70 form of the chaperone machine, because during this period, the client protein is susceptible to ubiquitination and delivery to the proteasome where it is degraded [155-159, 161, 162]. The list of Hsp90 client proteins includes key components of multiple signaling pathways utilized by cancer cells for growth and/or survival (reviewed in [152, 155-157, 163, 164]). Hsp90 also stabilizes Akt and prevents Akt from PP2A-mediated inactivation [165-169]. When cells are exposed to heat shock, Akt is activated. The heat shock-induced Akt activation is PI3K dependent, as blocking PI3K leads to rapid dephosphorylation of Akt and detachment of Akt from Hsp90 [165]. Detachment of Akt from the Hsp90 complex increases Akt sensitivity to PP2A-mediated dephosphorylation and subsequent degradation. Blockade of the Akt-HSP-90 complex formation by the Hsp90–specific inhibitor 17-allyamino 17- demethoxy-geldanamycin (17-AAG), a geldanamycin analogue that is undergoing clinical testing in malignant melanoma and breast and prostate cancers, induces Akt dephosphorylation and degradation [164, 168, 169]. As a nucleotide mimetic inhibitor of Hsp90, 17-AAG can block the intrinsic ATPase activity of Hsp90 and thereby block the formation of a multi-chaperone complex, including the Akt- Hsp90 complex. It is not yet clear under what patho-physiological conditions will the ubiquitin proteasome pathway recycle or degrade Hsp90-bound Akt.
“Ser/Thr-Pro motif” phosphorylation and Akt stability
Although Akt can be degraded by either the UPS or caspases, very few studies have directly addressed whether Akt phosphorylation status affects Akt's stability and which phosphorylation sites are responsible for the regulation of that stability. Phosphorylation of Akt on either Thr 308 or Ser473 affects Akt activity, but it may not directly, physiologically affect Akt stability. Two groups recently reported that phosphorylation of Akt at the Thr450-Pro site of the turn motif by mTORC2 controls Akt protein folding and maturation, similar to results of phosphorylation of the conserved turn motifs in the PKC family [29-31, 48, 77, 78, 170-172]. Both groups demonstrated that genetic deletion of mTORC2 components, including mSin1, Rictor, and mTOR, affected Akt phosphorylation not only at Ser473 on the HMs but also at Thr450- Pro on the turn motif. Since Thr450 is followed by a proline residue, screening of Erk, JNK, p38, CDK, GSK3, CK2 and other proline-directed protein kinases indicated that such kinases had no significant effect on Akt phosphorylation at the Thr450-Pro motif [77]. As discussed earlier, phosphorylation of Akt at the Thr450-Pro was identified more than 10 years ago and has since been demonstrated to be a constitutively phosphorylated residue that is not dependent on either growth factors or PI3K kinase [41, 42]. One report claimed that mTORC-mediated phosphorylation of Akt at the Thr450-Pro site on the turn motif also regulates Akt stability, similar to the effect of PKC phosphorylation on the turn motifs; the authors further demonstrated that the Hsp90 inhibitor 17-AAG facilitates degradation of Akt in mSin−/− or Rictor−/− cells but not in wildtype cells [78]. They also showed that in the absence of Thr450 phosphorylation, there was an increase in the level of Akt that was bound to Hsp90; thus, exposing cells to 17-AAG abolished Thr450 phosphorylation by mSin1−/− or Rictor−/− and enhanced Akt degradation by the UPS. They further showed that this turn motif phosphorylation of Akt may mediate proper carboxyl-terminal folding and stabilization of the Akt structure by Akt's interacting with three conserved basic residues (K163, K182, and R222) at the catalytic domain. Mutations of the conserved basic residues (K163M, K182M, and R222M) in Akt1 were predicted to interact with the turn motif site phosphate and led to increased Akt binding with Hsp90 and subsequent destabilization of Akt upon 17-AAG-mediated inhibition of Hsp90. Although Facchinetti et al [78] tried to further show that mTORC2 directly phosphorylates Thr450 on Akt and that this action requires the mTOR kinase, the data was not very stringently controlled and may support a different mechanism; they were able to detect Akt phosphorylation at both the turn motif and HM in the mSin1−/− mouse embryo fibroblast (MEF) cells after infection with HA-tagged Akt1 [78]. In addition, a kinase-dead mutant form of HA-mTOR (which is supposed to be the major kinase of the mTOR complexes) sufficiently phosphorylated GST-Akt on both the turn motif and the HM in the in vitro kinase assay. This result is likely derived from an incomplete dephosphorylation of the GST-Akt prepared as the substrate for mTORC2; however, the opposite (i.e., mTORC2 may not be directly responsible for Akt phosphorylation at the turn motif) may also be possible. Indeed, data from Ikenoue et al clearly demonstrate that mTORC2 phosphorylates the HM but not the turn motifs in Akt and PKCα; thus, mTORC2 may not be the kinase directly responsible for the turn motif phosphorylation [48, 77]. Also intriguing is how mTORC2 independently controls Akt phosphorylation at Thr450-Pro but not Ser473 at the same time and how it directly phosphorylates Akt on Ser473 but not Thr450-Pro at another time under physiological conditions. Given the constitutively phosphorylated nature of the Thr450-Pro site of Akt, the claim that the stress-responsive kinase JNK mediates Akt phosphorylation on Thr450 under hypoxic injury in cardiomyocytes cannot be generalized or reproduced in other cell types [173]. Indeed, the report from Ikenoue et al on screening proline-directed kinases excluded JNK as a kinase for Thr450-Pro phosphorylation [77]. Our unpublished data also demonstrate that JNK is unlikely to be the kinase that directly phosphorylates Akt on the Thr450-Pro motif. Therefore, the identity of the “turn motif kinase” is still elusive [48].
The regulation of PKCβII activity and stability by turn motif phosphorylation involves the deletion mutation of either mSin1 or Rictor, which drastically affects the expression levels of endogenous and exogenous PKCs (mainly members of the conventional PKC family, including PKC-α, -βII, -γ, and -βI but not atypical or novel PKC-ζ, -η, and -δ). In contrast, data from two groups [77, 78] as well as previously published papers on genetic disruption or siRNA-mediated knock-down of the mTORC2 components essential for the formation of mTORC2 (such as mSin1, Rictor, and mTOR), have demonstrated that disruption of mTORC2 does not dramatically affect the level of Akt protein expression [50, 53-57, 59, 60, 62, 77, 78]. As discussed earlier, Bellacosa et al [41, 42] demonstrated that the inactivating mutation of Thr450 into alanine (T450A) alone did not affect Akt kinase activity or protein stability. In fact, data from both Ikenoue et al [77] and Facchinetti et al [78] also demonstrated that disruption of mTORC2 affected Akt protein migration (or phosphorylation), but the effect on the level of Akt protein expression or Akt stability was unconvincing [77, 78].
The Thr450-Pro motif is conserved not only within the Akt family and among different species but also in other AGC kinases, including PRK2, p70S6K, PKA, and the PKC family [29, 31, 48, 174]. The Thr450 residue is located in the turn motif, which anchors the C-terminus HM at the top of the upper lobe of the kinase domain, with the phosphorylated threonine residues at the apex of a tight turn [29, 31, 174]. Although mutation of this residue (T450) to alanine (T450A) does not dramatically affect Akt stability, activation phosphorylation, or kinase activity as demonstrated in an earlier report by Bellacosa et al [41, 42], mutation of the conserved site in PKA and an adjacent compensating site in PKC βII does destabilize the kinase domain and abolish the kinase activity of PKA and PKC βII [29, 31, 174]. Facchinetti et al [78] showed that a combined mutation of Thr450-Pro and the adjacent Thr443 to alanine in Akt does not affect the expression levels of HA-tagged single (T450A) or double (T450A/T443A) mutant Akt protein compared with the expression level of wildtype Akt. Even though the binding of Akt mutants with Hsp90 or myc-tagged ubiquitin was enhanced, the levels of HA-tagged Akt mutant proteins were comparable with the wild-type HA-Akt in mSin1 MEFs (wild-type and null) and in the presence or absence of proteasome inhibitor MG132. Although Facchinetti et al [78] claimed that the turn motif phosphorylation on Akt controls folding and stability of Akt similar to that observed in PKC, it is difficult to explain why the kinase activity (as demonstrated by Bellacosa et al [41]) and the mutant Akt levels in mSin1 wild-type and null MEFs were comparable with the wild-type Akt. Since PKA and family members of PKC do not have the PH domain, which plays an inhibitory effect on activation phosphorylation on Akt, we speculated that mutation of T450 alone in Akt is not sufficient to destabilize the kinase domain as it is for PKA or PKC (i.e., the PKA and PKC turn motif phosphorylation, protein stability, and activation model may not necessarily fit the Akt family of proteins). Indeed, when we performed an alanine screening of all five Ser/Thr-Pro motifs in human Akt1, we discovered that double mutation of Thr450-Pro in the turn motif and Thr92- Pro in the PH domain, but not Ser124-Pro, Thr312-Pro, Ser422-Pro or their combination, generates an inactive, labile Akt mutant protein with undetectable phosphorylation on both Thr308 and Ser473 [175]. Further elucidation of Akt stability and activation phosphorylation regulated by the Thr-Pro motifs led us to discover that the peptidylprolyl isomerase Pin1 plays an important role in the regulation of Akt stability and activation phosphorylation (discussed later).
The role of a peptidyl-prolyl isomerase Pin1 in the regulation of Akt stability
Interaction between Pin1 and Akt
Pin1, a peptidyl-prolyl cis/trans isomerase (PPIase), has recently been reported to modulate the balance between protein phosphorylation and dephosphorylation at the cellular level [176-183] and to regulate the stability of its substrates, including cyclin D1, Myc, p53, p73, beta-catenin, cyclin E, and p65/RelA [181-189]. Given the fact that both Pin1 and Akt are direct upstream regulators of cyclin D1, we sought to determine whether Pin1 and Akt interact with each other and whether Pin1 regulates Akt stability and activation phosphorylation. Interestingly, we observed that elevated expression of Akt-S473 strongly correlated with Pin1 expression in a human breast cancer samples (P < 0.0001). The expression levels of Pin1 and Akt-pS473 were also associated with tumor stage in human breast cancer (P < 0.05 for both). In addition, the combination of Akt-pS473 with Pin1 expression predicted a poorer prognosis than either one alone in patients with breast cancer (P = 0.0052) [175].
We demonstrated that Akt associated with Pin1 by co-immunoprecipitation of endogenous Akt and vice versa. Although the Akt that is associated with Pin1 is phosphorylated on both T308 and S473, we excluded the involvement of these residues in the regulation of Akt stability and association with Pin1 since neither stimulation with IGF-1 nor inhibition with LY294002 affected the levels of Pin1- associated Akt protein. When we examined phosphorylation of Akt on the Ser/Thr-Pro motifs using the monoclonal antibody MPM2, which specifically recognizes the phosphorylated Ser/Thr-Pro (pSer/Thr-Pro) motifs [190, 191], we observed that the immunoprecipitated Akt protein was recognized by the MPM2 antibody, while a protein immunoprecipitated with the MPM2 antibody was also recognized by a specific antibody against Akt. In addition, neither IGF-1 nor LY294002 affected Akt immunoreactivity against the MPM2 antibody, suggesting that the Akt and Pin1 interaction is independent of activation phosphorylation of Akt at T308 or S473 but may depend on Akt phosphorylation at the Ser/Thr-Pro motifs.
Akt phosphorylation on Ser/Thr-Pro motifs and Pin1-Akt interaction
In order to determine which Ser/Thr-Pro sites are required for the Akt and Pin1 interaction, we mutated Ser/Thr to Ala in the five Ser/Thr- Pro sites of human Akt1 (T92A, S124A, T312A, S422A, and T450A) (Table 1). We found that single mutations of T312A, S124A, and S422A did not attenuate Akt immunoreactivity with the MPM2 antibody, while a point mutation of either T92A or T450A reduced Akt immunoreactivity with MPM2 and a double mutation of T92-Pro and T450-Pro (T92A/T450A) completely abrogated Akt immunoreactivity against MPM2. Those findings suggested that the T92 and T450 sites may both be important for the Akt-Pin1 interaction. Sequence alignment analysis revealed that the Thr92-Pro and Thr450-Pro motifs are highly conserved in different Akt isoforms and species, including C. elegans and humans (Table 2). Consistently, a GST-Pin1 pull-down assay demonstrated that all three isoforms of human Akt (Akt1, 2, and 3) were pulled down by GST-Pin1. In addition, a double mutation of Akt on T92 and T450 (T92A/T450A) resulted in a labile Akt protein that lacked activation phosphorylation on either S473 or T308, while single mutation of either T92 or T450 only slightly affected Akt expression and activation phosphorylation. A cycloheximide chase experiment demonstrated that the half-life of Akt1-T92A/T450A mutant protein was much shorter than the wild-type Akt1 [175]. A kinase assay also showed that the Akt1-T92A/T450A double mutant transfected into 293 cells expressed no kinase activity toward its substrate GST-GSK3 β in vitro (Liao et al, unpublished observation). These results suggest that the phosphorylated Thr-Pro motifs of Akt on T92 and T450 are critical for the maintenance of Akt stability and activity.
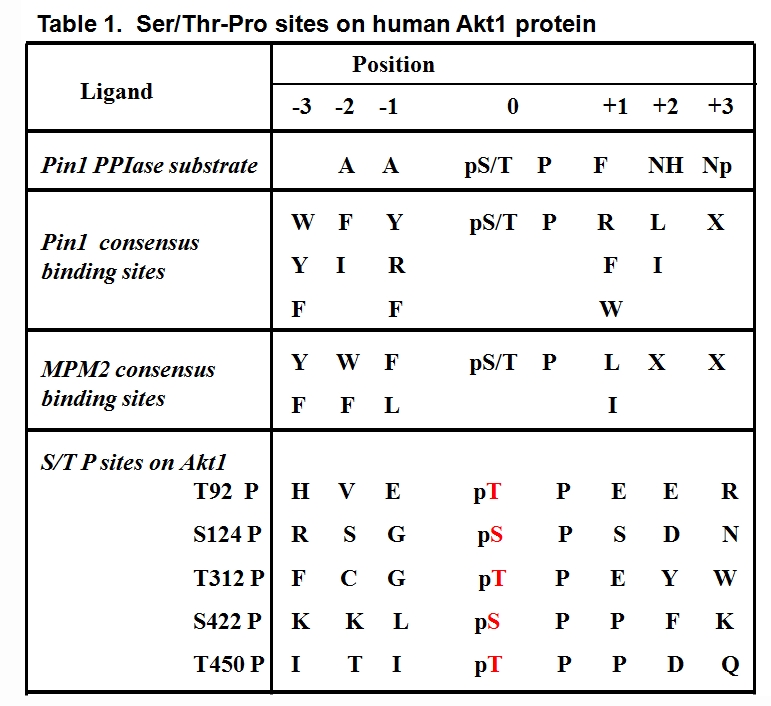 |
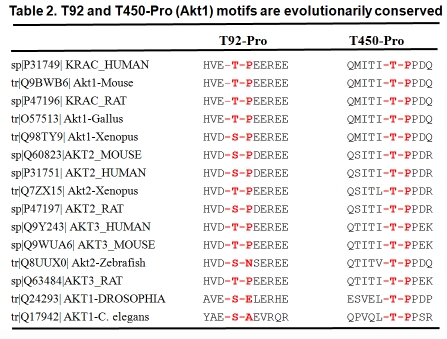 |
Pin1 is required for the maintenance of Akt stability and activation phosphorylation
To test whether Pin1 affects Akt stability, we examined the expression levels of total Akt and Akt activation phosphorylations on both T308 and S473 sites in Pin1 wild-type and Pin1 (−/−) MEFs in the presence or absence of IGF-1 stimulation. As expected, reduced levels of total Akt protein were detected in Pin1-null MEF cell lysates with or without stimulation of IGF-1, and activation phosphorylations on T308 and S473 were reduced. The effective stimulation of Akt activation phosphorylation on T308 and S473 by IGF-1 in Pin1 (−/−) cells indicates that Pin1 did not affect upstream PI3K signalling toward Akt phosphorylation, which suggests that Pin1 may be required for the maintaining Akt phosphorylation through a post-phosphorylation regulatory mechanism. Similar results have been obtained when Pin1 expression was knocked down by specific small interfering RNAs (siRNA). In addition, we showed that the re-introduction of Pin1 enhanced Akt protein stability both in vitro and in vivo in Pin1-null MEFs. However, Pin1 mutants (Pin1-W34A or Pin1-R68,69A, which lost its substrate binding capability or isomerase activity, respectively) did not restore Akt protein stability or activation phosphorylation [175]. Thus, our results demonstrate that Pin1 regulates Akt protein stability and activation phosphorylation.
Based on our results and previous reports, we propose a model to document the role of Pin1- mediated conformational modifications during multiple-step Akt activation (Figure 4). Briefly, a nascent Akt protein is unstable and inactive; after it has been phosphorylated on the T92- Pro and T450-Pro motifs, the phosphorylated Akt protein (Akt-pT92/T450) may remain unstable (Figure 4A) until Pin1-mediated conformational modification, the Akt-pT92/T450 protein becomes stable, but remains inactive (Figure 4B) unless it has been further phosphorylated by PDK1/2 (or mTORC2) (Figure 4C). In the absence of Pin1 (e.g., in Pin1-null MEFs), the Akt-pT92/T450 protein may still be able to translocate to the inner plasma membrane and be further activated by PDK1/2 (or mTORC2) (Figure 4D); however, the protein will be unstable unless Pin1 is reintroduced, after which Akt will become stable and fully active (Figure 4E). Under certain conditions, the fully active and stable Akt protein will also become inactive and unstable as a result of protein phosphatase-mediated dephosphorylation (such as that mediated by PHLPPs and PP2A) (Figure 4F). The balance between an active and stable Akt versus an inactive and unstable Akt at the cellular level largely depends on the kinetics of the upstream kinases and protein phosphatases as well as the expression level of Pin1. Therefore, identification of the kinases that phosphorylate Akt on the Thr92-Pro and Thr450-Pro motifs and further elucidation of the mechanisms of Akt phosphorylation at the Thr-Pro motifs under physiological conditions will be important directions for future research.
Figure 4.

A plausible model on Pin1-mediated regulation of Akt stability and activation phosphorylation.
Perspective
In recent years, the mechanisms for Akt activation by PI3K-mTORC2 and inactivation by protein phosphatase PTEN, PP2A, and PHLPPs have been well characterized. A negative feedback regulation loop between the major oncogenic survival factor Akt and the critical pro-apoptotic factor p53 has also been established. Components of the different mTORCs and their functional roles in Akt activation phosphorylation have been revealed. Another intricate negative feedback loop among mTORC2-Akt-mTORC1-S6K-IRS1-PI3K has emerged, and its regulation under physiological and pathological conditions needs further elucidation. It is not yet clear how the different mTORCs are assembled physiologically and what upstream signals activate the mTORC2 kinase. mTORC2 has been shown to be involved in the regulation of Akt stability on Ser473 at the HM and phosphorylation on Thr450-Pro at the turn motif. However, the spatial-temporal dynamics of mTORC2 that control Akt phosphorylation at Thr450-Pro (which may be required for a proper folding of newly synthesized Akt protein) and Ser473 (which is required for a full activation) under physiological conditions still must be elucidated. It is debatable whether mTORC2 kinase is directly responsible for Thr450-Pro motif phosphorylation as well as regulation of Akt folding and stability. The E3 ligases that mediate Akt degradation by the UPS and kinases that directly phosphorylate Akt on the Thr92- and Thr450-Pro motifs are still elusive and need to be revealed so that the physiological regulation of Akt stability may be better understood. We believe that further elucidation of the cellular mechanisms regulating Akt stability and the identification of specific E3 ligases that mediate Akt degradation will not only show how Akt stability is regulated physiologically but also open a new avenue for targeted degradation of Akt protein in human cancer.
Acknowledgments
This work has been supported by the U.S. Department of Defense Breast Cancer Research Program (DAMD17-01-1-0300), the Susan G. Komen Breast Cancer Foundation (BCTR0504146), and a Research Development Award from the Specialized Program of Research Excellence (SPORE) in ovarian cancer (P50-CA83639-A01) (to L.Y.); and grant funding from the U.S. National Institutes of Health P01 099031, P50 CA116199 (breast cancer SPORE at The University of Texas M. D. Anderson Cancer Center [MDACC]), P50 CA83639 (ovarian cancer SPORE at MDACC), National Breast Cancer Foundation, and Kadoorie Charitable Foundations (to M.-C. H.).
The authors would also like to acknowledge Dr. Walter N. Hittelman at the Department of Experimental Therapeutics, The University of Texas M.D. Anderson Cancer Center, for the stimulating discussions on Akt signaling and its feedback regulation during the preparation of this review. The authors also greatly appreciate the editing and language improvement of this manuscript by Dr. Stephanie A. Miller at the Department of Molecular & Cellular Oncology and John C. LeBas at the Department of Scientific Publications, The University of Texas M.D. Anderson Cancer Center.
References
- 1.Bos JL. A target for phosphoinositide 3-kinase: Akt/PKB. Trends Biochem Sci. 1995;20:441–442. doi: 10.1016/s0968-0004(00)89097-0. [DOI] [PubMed] [Google Scholar]
- 2.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 3.Murthy S, Tosolini A, Taguchi T, Testa JR. Mapping of AKT3, encoding a member of the Akt/protein kinase B family, to human and rodent chromosomes by fluorescence in situ hybridization. Cytogenet Cell Genet. 2000;88:38–40. doi: 10.1159/000015481. [DOI] [PubMed] [Google Scholar]
- 4.Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT–a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007;35:231–235. doi: 10.1042/BST0350231. [DOI] [PubMed] [Google Scholar]
- 6.Yang ZZ, Tschopp O, Baudry A, Dummler B, Hynx D, Hemmings BA. Physiological functions of protein kinase B/Akt. Biochem Soc Trans. 2004;32:350–354. doi: 10.1042/bst0320350. [DOI] [PubMed] [Google Scholar]
- 7.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 8.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 9.Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene. 2005;24:7391–7393. doi: 10.1038/sj.onc.1209100. [DOI] [PubMed] [Google Scholar]
- 10.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 11.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Vestey SB, Sen C, Calder CJ, Perks CM, Pignatelli M, Winters ZE. Activated Akt expression in breast cancer: correlation with p53, Hdm2 and patient outcome. Eur J Cancer. 2005;41:1017–1025. doi: 10.1016/j.ejca.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Dillon RL, White D.E., Muller W.J. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–1345. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- 14.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 15.Tokunaga E, Oki E, Egashira A, Sadanaga N, Morita M, Kakeji Y, Maehara Y. Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets. 2008;8:27–36. doi: 10.2174/156800908783497140. [DOI] [PubMed] [Google Scholar]
- 16.Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12:104–107. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Huang WC, Hung MC. Induction of Akt activity by chemotherapy confers acquired resistance. J Formos Med Assoc. 2009;108:180–194. doi: 10.1016/S0929-6646(09)60051-6. [DOI] [PubMed] [Google Scholar]
- 18.Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr Opin Cell Biol. 2009 doi: 10.1016/j.ceb.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 20.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–361. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 23.Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Curr Opin Genet Dev. 2009;19:12–17. doi: 10.1016/j.gde.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 25.Soung YH, Lee JW, Nam SW, Lee JY, Yoo NJ, Lee SH. Mutational analysis of AKT1, AKT2 and AKT3 genes in common human carcinomas. Oncology. 2006;70:285–289. doi: 10.1159/000096289. [DOI] [PubMed] [Google Scholar]
- 26.Carpten JD, Faber A.L., Horn C., Donoho G.P., Briggs S.L., Robbins C.M., Hostetter G., Boguslawski S., Moses T.Y., Savage S., Uhlik M., Lin A., Du J., Qian Y.W., Zeckner D.J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M.H., Blanchard K.L., Thomas J.E. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 27.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Brognard J, Newton AC. PHLPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab. 2008 doi: 10.1016/j.tem.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brognard J, Niederst M, Reyes G, Warfel N, Newton AC. Common polymorphism in the phosphatase PHLPP2 results in reduced regulation of Akt and protein kinase C. J Biol Chem. 2009;284:15215–15223. doi: 10.1074/jbc.M901468200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newton AC. Lipid activation of protein kinases. J Lipid Res. 2009;50(Suppl):S266–271. doi: 10.1194/jlr.R800064-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. (Pt 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 34.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 35.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 36.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 38.Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 39.Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. ChemMedChem. 2008;3:1810–1838. doi: 10.1002/cmdc.200800195. [DOI] [PubMed] [Google Scholar]
- 40.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 41.Bellacosa A, Chan T.O., Ahmed N.N., Datta K., Malstrom S., Stokoe D., McCormick F., Feng J., Tsichile P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 42.Alessi DR, Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen, P, Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 43.Kandel E, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKA. Exp. Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Zhang S, Yamane H, Wahl R, Ali A, Lofgren JA, Kendall RL. Kinetic mechanism of AKT/PKB enzyme family. J Biol Chem. 2006;281:13949–13956. doi: 10.1074/jbc.M601384200. [DOI] [PubMed] [Google Scholar]
- 45.Yang J, Cron P., Thompson V., Good V.M., Hess D., Hemmings B.A., Bardford D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002;9:1227–1240. doi: 10.1016/s1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- 46.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 47.Chan TO, Tsichlis PN. PDK2: a complex tail in one Akt. Sci STKE. 2001;2001:PE1. doi: 10.1126/stke.2001.66.pe1. [DOI] [PubMed] [Google Scholar]
- 48.Alessi DR, Pearce LR, Garcia-Martinez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal. 2009;2:pe27. doi: 10.1126/scisignal.267pe27. [DOI] [PubMed] [Google Scholar]
- 49.Bayascas JR. Dissecting the role of the 3- phosphoinositide-dependent protein kinase-1 (PDK1) signalling pathways. Cell Cycle. 2008;7:2978–2982. doi: 10.4161/cc.7.19.6810. [DOI] [PubMed] [Google Scholar]
- 50.Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/Protein Kinase B in 3T3- L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 51.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 52.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 53.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 55.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 56.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 57.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 58.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 59.Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, Ibrahim A, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR-complex-2. Biochem J. 2007 doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 61.Hietakangas V, Cohen SM. Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev. 2007;21:632–637. doi: 10.1101/gad.416307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 63.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang P, Ostrander JH, Faivre EJ, Olsen A, Fitzsimmons D, Lange CA. Regulated association of protein kinase B/Akt with breast tumor kinase. J Biol Chem. 2005;280:1982–1991. doi: 10.1074/jbc.M412038200. [DOI] [PubMed] [Google Scholar]
- 66.Kawase T, Ohki R, Shibata T, Tsutsumi S, Kamimura N, Inazawa J, Ohta T, Ichikawa H, Aburatani H, Tashiro F, Taya Y. PH domainonly protein PHLDA3 is a p53-regulated repressor of Akt. Cell. 2009;136:535–550. doi: 10.1016/j.cell.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 67.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 68.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24:7401–7409. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
- 70.Strimpakos AS, Karapanagiotou EM, Saif MW, Syrigos KN. The role of mTOR in the management of solid tumors: an overview. Cancer Treat Rev. 2009;35:148–159. doi: 10.1016/j.ctrv.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 71.Goberdhan DC, Boyd CA. mTOR: dissecting regulation and mechanism of action to understand human disease. Biochem Soc Trans. 2009;37:213–216. doi: 10.1042/BST0370213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25:6347–6360. doi: 10.1038/sj.onc.1209885. [DOI] [PubMed] [Google Scholar]
- 73.Polak P, Hall MN. mTORC2 Caught in a SINful Akt. Dev Cell. 2006;11:433–434. doi: 10.1016/j.devcel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 74.Porstmann T, Santos CR, Lewis C, Griffiths B, Schulze A. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem Soc Trans. 2009;37:278–283. doi: 10.1042/BST0370278. [DOI] [PubMed] [Google Scholar]
- 75.Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal. 2009;21:827–835. doi: 10.1016/j.cellsig.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 76.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serumand glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 77.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821–1827. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009;21:656–664. doi: 10.1016/j.cellsig.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 82.Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 83.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scott KL, Kabbarah O., Liang M.-C., Ivanova E., Anagnostou V., Wu J., Dahkal S., Wu M., Chen S., Feinberg T., Huang J., Saci A., Widlund H.R., Fisher D.E., Xiao Y., Protopopov A., Wong K.-K., Chin L. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature. 2009;459:1085–1090. doi: 10.1038/nature08109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang B, Zhang P, Wei Q. Recent progress on the structure of Ser/Thr protein phosphatases. Sci China C Life Sci. 2008;51:487–494. doi: 10.1007/s11427-008-0068-y. [DOI] [PubMed] [Google Scholar]
- 86.Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends Biochem Sci. 2008;33:113–121. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 87.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 88.Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009;1795:1–15. doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 89.Shi Y. Assembly and structure of protein phosphatase 2A. Sci China C Life Sci. 2009;52:135–146. doi: 10.1007/s11427-009-0018-3. [DOI] [PubMed] [Google Scholar]
- 90.Petersen P, Chou DM, You Z, Hunter T, Walter JC, Walter G. Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7- independent DNA damage checkpoint. Mol Cell Biol. 2006;26:1997–2011. doi: 10.1128/MCB.26.5.1997-2011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chiang CW, Yan L, Yang E. Phosphatases and regulation of cell death. Methods Enzymol. 2008;446:237–257. doi: 10.1016/S0076-6879(08)01614-5. [DOI] [PubMed] [Google Scholar]
- 92.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 2008;14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 93.Sablina AA, Hahn WC. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev. 2008;27:137–146. doi: 10.1007/s10555-008-9116-0. [DOI] [PubMed] [Google Scholar]
- 94.Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- 95.Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–7745. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 96.Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr Opin Genet Dev. 2005;15:34–41. doi: 10.1016/j.gde.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 97.Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130:21–24. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 98.Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, Evans G. Alteration of the PPP2R1B gene in human lung and colon cancer. Science. 1998;282:284–287. doi: 10.1126/science.282.5387.284. [DOI] [PubMed] [Google Scholar]
- 99.Calin GA, di Iasio MG, Caprini E, Vorechovsky I, Natali PG, Sozzi G, Grocec CM, Barbanti-Brodano G, Russo G, Negrini M. Low frequency of alterations of the a (PPP2R1A) and b (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene. 2000;19:1191–1195. doi: 10.1038/sj.onc.1203389. [DOI] [PubMed] [Google Scholar]
- 100.Ruediger R, Pham H.T., Walter G. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the Aa subunit gene. Oncogene. 2001;20:10–15. doi: 10.1038/sj.onc.1204059. [DOI] [PubMed] [Google Scholar]
- 101.Ruediger R, Pham H.T., Walter G. Alterations in protein phosphatase 2A subunit interaction in human cancinomas of the lung and colon with mutations in the beta subunit gene. Oncogene. 2001;20:1892–1899. doi: 10.1038/sj.onc.1204279. [DOI] [PubMed] [Google Scholar]
- 102.Zhu D, Tate RI, Ruediger R, Meigs TE, Denker BM. Domains Necessary for G{alpha}12 Binding and Stimulation of PP2A: is G{alpha} 12 a Novel Regulatory Subunit of PP2A? Mol Pharmacol. 2007 doi: 10.1124/mol.106.033555. [DOI] [PubMed] [Google Scholar]
- 103.Schonthal AH. Role of serine/threonine protein phosphatase 2A in cancer. Cancer Lett. 2001;170:1–13. doi: 10.1016/s0304-3835(01)00561-4. [DOI] [PubMed] [Google Scholar]
- 104.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005;65:8183–8192. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 105.Yeh LS, Hsieh YY, Chang JG, Chang WW, Chang CC, Tsai FJ. Mutation analysis of the tumor suppressor gene PPP2R1B in human cervical cancer. Int J Gynecol Cancer. 2007 doi: 10.1111/j.1525-1438.2007.00880.x. [DOI] [PubMed] [Google Scholar]
- 106.Ito A, Kataoka TR, Watanabe M, Nishiyama K, Mazaki Y, Sabe H, Kitamura Y, Nojima H. A truncated isoform of the PP2A B56 subunit promotes cell motility through paxillin phosphorylation. Embo J. 2000;19:562–571. doi: 10.1093/emboj/19.4.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 108.Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc Natl Acad Sci U S A. 2007;104:19011–19016. doi: 10.1073/pnas.0706696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM. Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol. 2004;24:8778–8789. doi: 10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liao Y, Hung MC. A new role of protein phosphatase 2A in adenoviral E1A proteinmediated sensitization to anticancer drug-induced apoptosis in human breast cancer cells. Cancer Res. 2004;64:5938–5942. doi: 10.1158/0008-5472.CAN-04-1533. [DOI] [PubMed] [Google Scholar]
- 111.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pim D, Massimi P, Dilworth SM, Banks L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene. 2005;24:7830–7838. doi: 10.1038/sj.onc.1208935. [DOI] [PubMed] [Google Scholar]
- 113.Vereshchagina N, Ramel MC, Bitoun E, Wilson C. The protein phosphatase PP2A-B' subunit Widerborst is a negative regulator of cytoplasmic activated Akt and lipid metabolism in Drosophila. J Cell Sci. 2008;121:3383–3392. doi: 10.1242/jcs.035220. [DOI] [PubMed] [Google Scholar]
- 114.Padmanabhan S, Mukhopadhyay A, Narasimhan SD, Tesz G, Czech MP, Tissenbaum HA. A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell. 2009;136:939–951. doi: 10.1016/j.cell.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ruvolo PP. Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia. 2001;15:1153–1160. doi: 10.1038/sj.leu.2402197. [DOI] [PubMed] [Google Scholar]
- 116.Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. 2008;283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- 117.Rocher G, Letourneux C, Lenormand P, Porteu F. Inhibition of B56-containing protein phosphatase 2As by the early response gene IEX-1 leads to control of Akt activity. J Biol Chem. 2007;282:5468–5477. doi: 10.1074/jbc.M609712200. [DOI] [PubMed] [Google Scholar]
- 118.Petersen Shay K, Hagen TM. Ageassociated impairment of Akt phosphorylation in primary rat hepatocytes is remediated by alpha-lipoic acid through PI3 kinase, PTEN, and PP2A. Biogerontology. 2008 doi: 10.1007/s10522-008-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tremblay ML, Giguere V. Phosphatases at the heart of FoxO metabolic control. Cell Metab. 2008;7:101–103. doi: 10.1016/j.cmet.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 120.Mendoza MC, Blenis J. PHLPPing it off: phosphatases get in the Akt. Mol Cell. 2007;25:798–800. doi: 10.1016/j.molcel.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 121.Cozzone D, Frojdo S, Disse E, Debard C, Laville M, Pirola L, Vidal H. Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia. 2008;51:512–521. doi: 10.1007/s00125-007-0913-8. [DOI] [PubMed] [Google Scholar]
- 122.Day KR, Jagadeeswaran P. Microarray analysis of prothrombin knockdown in zebrafish. Blood Cells Mol Dis. 2009 doi: 10.1016/j.bcmd.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kerley-Hamilton JS, Pike AM, Li N, DiRenzo J, Spinella MJ. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embryonal carcinoma. Oncogene. 2005;24:6090–6100. doi: 10.1038/sj.onc.1208755. [DOI] [PubMed] [Google Scholar]
- 124.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, MacRae EJ, Vousden KH. Phosphorylation of HDM2 by Akt. Oncogene. 2002;21:1955–1962. doi: 10.1038/sj.onc.1205276. [DOI] [PubMed] [Google Scholar]
- 126.Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–1303. doi: 10.1038/sj.onc.1205181. [DOI] [PubMed] [Google Scholar]
- 127.Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002;277:5484–5489. doi: 10.1074/jbc.M108302200. [DOI] [PubMed] [Google Scholar]
- 128.Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- 129.Oren M, Damalas A, Gottlieb T, Michael D, Taplick J, Leal JF, Maya R, Moas M, Seger R, Taya Y, Ben-Ze'ev A. Regulation of p53: intricate loops and delicate balances. Biochem Pharmacol. 2002;64:865–871. doi: 10.1016/s0006-2952(02)01149-8. [DOI] [PubMed] [Google Scholar]
- 130.Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. 2004;279:35510–35517. doi: 10.1074/jbc.M404936200. [DOI] [PubMed] [Google Scholar]
- 131.Milne D, Kampanis P, Nicol S, Dias S, Campbell DG, Fuller-Pace F, Meek D. A novel site of AKT-mediated phosphorylation in the human MDM2 onco-protein. FEBS Lett. 2004;577:270–276. doi: 10.1016/j.febslet.2004.09.081. [DOI] [PubMed] [Google Scholar]
- 132.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 133.Zhou BP, Hung MC. Novel targets of Akt, p21(Cipl/WAF1), and MDM2. Semin Oncol. 2002;29:62–70. doi: 10.1053/sonc.2002.34057. [DOI] [PubMed] [Google Scholar]
- 134.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 135.Cheng GZ, Park S, Shu S, He L, Kong W, Zhang W, Yuan Z, Wang LH, Cheng JQ. Advances of AKT pathway in human oncogenesis and as a target for anti-cancer drug discovery. Curr Cancer Drug Targets. 2008;8:2–6. [PubMed] [Google Scholar]
- 136.Laurent-Puig P, Lievre A, Blons H. Mutations and response to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2009;15:1133–1139. doi: 10.1158/1078-0432.CCR-08-0905. [DOI] [PubMed] [Google Scholar]
- 137.Herrmann J, Lerman LO, Lerman A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ Res. 2007;100:1276–1291. doi: 10.1161/01.RES.0000264500.11888.f0. [DOI] [PubMed] [Google Scholar]
- 138.Thompson SJ, Loftus LT, Ashley MD, Meller R. Ubiquitin-proteasome system as a modulator of cell fate. Curr Opin Pharmacol. 2008;8:90–95. doi: 10.1016/j.coph.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Konstantinova IM, Tsimokha AS, Mittenberg AG. Role of proteasomes in cellular regulation. Int Rev Cell Mol Biol. 2008;267:59–124. doi: 10.1016/S1937-6448(08)00602-3. [DOI] [PubMed] [Google Scholar]
- 140.Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009 doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 141.Kim B, Feldman EL. Insulin-like growth factor I prevents mannitol-induced degradation of focal adhesion kinase and Akt. J Biol Chem. 2002;277:27393–27400. doi: 10.1074/jbc.M201963200. [DOI] [PubMed] [Google Scholar]
- 142.Riesterer O, Zingg D, Hummerjohann J, Bodis S, Pruschy M. Degradation of PKB/Akt protein by inhibition of the VEGF receptor/mTOR pathway in endothelial cells. Oncogene. 2004;23:4624–4635. doi: 10.1038/sj.onc.1207596. [DOI] [PubMed] [Google Scholar]
- 143.Yan D, Guo L, Wang Y. Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J Cell Biol. 2006;174:415–424. doi: 10.1083/jcb.200511028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Widmann C, Gibson S, Johnson GL. Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. J Biol Chem. 1998;273:7141–7147. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]
- 145.Bachelder RE, Wendt MA, Fujita N, Tsuruo T, Mercurio AM. The cleavage of Akt/protein kinase B by death receptor signaling is an important event in detachment-induced apoptosis. J Biol Chem. 2001;276:34702–34707. doi: 10.1074/jbc.M102806200. [DOI] [PubMed] [Google Scholar]
- 146.Xu J, Liu D, Songyang Z. The role of Asp- 462 in regulating Akt activity. J Biol Chem. 2002;277:35561–35566. doi: 10.1074/jbc.M203805200. [DOI] [PubMed] [Google Scholar]
- 147.Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 2001;61:1862–1868. [PubMed] [Google Scholar]
- 148.Rokudai S, Fujita N, Hashimoto Y, Tsuruo T. Cleavage and inactivation of antiapoptotic Akt/PKB by caspases during apoptosis. J Cell Physiol. 2000;182:290–296. doi: 10.1002/(SICI)1097-4652(200002)182:2<290::AID-JCP18>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 149.Jahani-Asl A, Basak A, Tsang BK. Caspase- 3-mediated cleavage of Akt: involvement of non-consensus sites and influence of phosphorylation. FEBS Lett. 2007;581:2883–2888. doi: 10.1016/j.febslet.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 150.Liu W, Akhand AA, Takeda K, Kawamoto Y, Itoigawa M, Kato M, Suzuki H, Ishikawa N, Nakashima I. Protein phosphatase 2A-linked and -unlinked caspase-dependent pathways for downregulation of Akt kinase triggered by 4-hydroxynonenal. Cell Death Differ. 2003;10:772–781. doi: 10.1038/sj.cdd.4401238. [DOI] [PubMed] [Google Scholar]
- 151.Medina EA, Afsari RR, Ravid T, Castillo SS, Erickson KL, Goldkorn T. Tumor necrosis factor-{alpha} decreases Akt protein levels in 3T3-L1 adipocytes via the caspase-dependent ubiquitination of Akt. Endocrinology. 2005;146:2726–2735. doi: 10.1210/en.2004-1074. [DOI] [PubMed] [Google Scholar]
- 152.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 153.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 154.Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Invest New Drugs. 1999;17:361–373. doi: 10.1023/a:1006382320697. [DOI] [PubMed] [Google Scholar]
- 155.Neckers L. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb Exp Pharmacol. 2006:259–277. doi: 10.1007/3-540-29717-0_11. [DOI] [PubMed] [Google Scholar]
- 156.Neckers L, Neckers K. Heat-shock protein 90 inhibitors as novel cancer chemotherapeutics - an update. Expert Opin Emerg Drugs. 2005;10:137–149. doi: 10.1517/14728214.10.1.137. [DOI] [PubMed] [Google Scholar]
- 157.Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15:419–424. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 158.Li Y, Zhang T, Schwartz SJ, Sun D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updat. 2009;12:17–27. doi: 10.1016/j.drup.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Niu G, Chen X. From protein-protein interaction to therapy response: molecular imaging of heat shock proteins. Eur J Radiol. 2009;70:294–304. doi: 10.1016/j.ejrad.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 160.Mahalingam D, Swords R, Carew JS, Nawrocki ST, Bhalla K, Giles FJ. Targeting HSP90 for cancer therapy. Br J Cancer. 2009;100:1523–1529. doi: 10.1038/sj.bjc.6605066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Amolins MW, Blagg BS. Natural product inhibitors of Hsp90: potential leads for drug discovery. Mini Rev Med Chem. 2009;9:140–152. doi: 10.2174/138955709787316056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Karapanagiotou EM, Syrigos K, Saif MW. Heat shock protein inhibitors and vaccines as new agents in cancer treatment. Expert Opin Investig Drugs. 2009;18:161–174. doi: 10.1517/13543780802715792. [DOI] [PubMed] [Google Scholar]
- 163.Davenport EL, Morgan GJ, Davies FE. Untangling the unfolded protein response. Cell Cycle. 2008;7:865–869. doi: 10.4161/cc.7.7.5615. [DOI] [PubMed] [Google Scholar]
- 164.Messaoudi S, Peyrat JF, Brion JD, Alami M. Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agents Med Chem. 2008;8:761–782. doi: 10.2174/187152008785914824. [DOI] [PubMed] [Google Scholar]
- 165.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Theodoraki MA, Kunjappu M, Sternberg DW, Caplan AJ. Akt shows variable sensitivity to an Hsp90 inhibitor depending on cell context. Exp Cell Res. 2007;313:3851–3858. doi: 10.1016/j.yexcr.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Georgakis GV, Li Y, Younes A. The heat shock protein 90 inhibitor 17-AAG induces cell cycle arrest and apoptosis in mantle cell lymphoma cell lines by depleting clcin D1, Akt, bid, and activating caspase-9. Br J Haematology. 2006;135:68–71. doi: 10.1111/j.1365-2141.2006.06247.x. [DOI] [PubMed] [Google Scholar]
- 168.Georgakis GV, Younes A. Heat-shock protein 90 inhibitors in cancer therapy: 17AAG and beyond. Future Med. 2005;1:273–281. doi: 10.1517/14796694.1.2.273. [DOI] [PubMed] [Google Scholar]
- 169.Power MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocrine-Related Cancer. 2006;13:S125–135. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 170.Gallegos LL, Newton AC. Spatiotemporal dynamics of lipid signaling: protein kinase C as a paradigm. IUBMB Life. 2008;60:782–789. doi: 10.1002/iub.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gould CM, Newton AC. The life and death of protein kinase C. Curr Drug Targets. 2008;9:614–625. doi: 10.2174/138945008785132411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283:6300–6311. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 173.Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, Wang YM, Kyriakis J, Mohanlal R, Kuida K, Namchuk M, Salituro F, Yao YM, Hou WM, Chen X, Aronovitz M, Tsichlis PN, Bhattacharya S, Force T, Kilter H. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ Res. 2006;98:111–118. doi: 10.1161/01.RES.0000197781.20524.b9. [DOI] [PubMed] [Google Scholar]
- 174.Newton AC. Regulation of ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liao Y, Wei Y, Zhou X, Yang JY, Dai C, Chen YJ, Agarwal NK, Sarbassov D, Shi D, Yu D, Hung MC. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene. 2009 doi: 10.1038/onc.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liou YC, Ryo A, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchida T, Hunter T, Lu KP. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci U S A. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell. 2003;4:175–180. doi: 10.1016/s1535-6108(03)00218-6. [DOI] [PubMed] [Google Scholar]
- 178.Ryo A, Liou YC, Lu KP, Wulf G. Prolyl isomerase Pin1: a catalyst for oncogenesis and a potential therapeutic target in cancer. J Cell Sci. 2003;116:773–783. doi: 10.1242/jcs.00276. [DOI] [PubMed] [Google Scholar]
- 179.Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wulf G, Finn G, Suizu F, Lu KP. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat Cell Biol. 2005;7:435–441. doi: 10.1038/ncb0505-435. [DOI] [PubMed] [Google Scholar]
- 181.Yeh ES, Means AR. PIN1, the cell cycle and cancer. Nat Rev Cancer. 2007;7:381–388. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- 182.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 183.Takahashi K, Uchida C, Shin RW, Shimazaki K, Uchida T. Prolyl isomerase, Pin1: new findings of post-translational modifications and physiological substrates in cancer, asthma and Alzheimer's disease. Cell Mol Life Sci. 2008;65:359–375. doi: 10.1007/s00018-007-7270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang H, Zhang J, Feng W, Zhang S, Liang H, Wang Y, Zheng Q, Li Z. PIN1 gene overexpression and beta-catenin gene mutation/expression in hepatocellular carcinoma and their significance. J Huazhong Univ Sci Technolog Med Sci. 2007;27:54–57. doi: 10.1007/s11596-007-0116-z. [DOI] [PubMed] [Google Scholar]
- 185.Lu KP, Suizu F, Zhou XZ, Finn G, Lam P, Wulf G. Targeting carcinogenesis: A role for the prolyl isomerase Pin1? Mol Carcinog. 2006;45:397–402. doi: 10.1002/mc.20216. [DOI] [PubMed] [Google Scholar]
- 186.Oberst A, Rossi M, Salomoni P, Pandolfi PP, Oren M, Melino G, Bernassola F. Regulation of the p73 protein stability and degradation. Biochem Biophys Res Commun. 2005;331:707–712. doi: 10.1016/j.bbrc.2005.03.158. [DOI] [PubMed] [Google Scholar]
- 187.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 188.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1- dependent prolyl isomerization and ubiquitinmediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 189.Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853–857. doi: 10.1038/nature01120. [DOI] [PubMed] [Google Scholar]
- 190.Lu KP. Pinning down cell signaling, cancer and Alzheimer's disease. Trends Biochem Sci. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 191.Lu PJ, Zhou XZ, Shen M, Lu KP. Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]