

Abstract
Arrest-defect-1 protein (ARD1), an acetyltransferase, catalyzes N—α-acetylation in yeast. In mammalian cells, both N-α-acetylation and ε-acetylation induced by ARD1 have been reported. Emerging evidence has revealed that ARD1 is involved in a variety of cellular functions, including proliferation, apoptosis, autophagy, and differentiation and that dysregulation of ARD1 is associated with tumorigenesis and neurodegenerative disorder. This review will discuss recent discoveries regarding variations among the different ARD1 isoforms, the associated biological functions of ARD1, and ARD1 localization in different cells. We will also discuss the potential upstream regulators and downstream targets of ARD1 to provide new avenues for resolving its controversial roles in cancer development.
Keywords: ARD1, acetyltransferase, acetylation, tumorigenesis
Introduction
Arrest-defect-1 protein (ARD1) was first identified in yeast and is responsible for the N-terminal α-acetylation. N-α-acetylation is an enzymatic process in which an acetyl group is transferred from acetyl coenzyme A to the very N-terminal of nascent polypeptides. It neutralizes positive charges and thereby may affect protein function, stability, association with other molecules, or subsequent modifications. In eu-karyotes, N-α-acetylation is one of the most common modifications. Studies have shown that around 50% of yeast proteins and 30% of mammalian proteins are N-α-acetylated [1, 2]. Given that protein turnover mediated by a ubiq-uitin-dependent degradation system depends on the presence of a free α-NH2 group at the N-terminal [3, 4], it is believed that N-α-acetylation may play a critical role in preventing proteolytic degradation of proteins.
N-a-acetylation can occur on the first Met or the newly exposed residue when the Met is cleaved. Although the most frequently acetylated residues are Ser, Ala, and Met, other residues may also be substrates for this type of modification [5]. Based on substrate specificity and subunit composition, N-acetyltransferases (Nats) are classified into three major groups, NatA, NatB, and NatC. Proteins with Ser, Ala, Gly, or Thr termini are NatA substrates, while proteins with Met-Glu, Met-Asp, Met-Asn, or Met-Met are NatB substrates, and proteins with Met-lle, Met-Leu, Met-Trp, or Met-Phe are NatC substrates [1, 6]. Nevertheless, researchers have not excluded the possibility of a new Nat being identified, especially for proteins with unusual N-terminal sequences that are not substrates for any of these three Nats.
Current data suggest that ARD1 is the catalytic subunit of NatA acetyltransferase. It associates with NAT1 and NAT5 and further cross-links to nascent polypeptides to exert its acetylation function [7, 8]. Of great interest, ARD1 represents a novel type of enzyme with both N-terminal α-protein and ε-protein acetylation activities in mammalian cells [9-11]. It has been shown that mouse ARD1 acetylation of HIF-1α at Lys532 enhances its interaction with pVHL and degradation of HIF-1α. In addition, ARD1 induced cyclin Dl transcription through s-acetylation of β-catenin. On the other hand, our group recently demonstrated that ARD1 does in fact contribute to the a-acetylation of tuberous sclerosis complex 2 (TSC2) [11] and that acetylation occurred on the first Met. It will be important to determine whether ARD1 mediates different types of acetylation when acting on different substrates and recruiting different complex partners.
Characterization of ARD1 isoforms
Various isoforms of ARD1 have been identified, including mouse variants (mARD1198, mARD1225, and mARD1235) and human variants (hARD1131 and hARD1235). These ARD1 isoforms share a conserved N-acetyltransferase domain but contain different sequences and lengths in their C-terminal region [12], which contribute to differential hydrophobicity among ARD1 isoforms. Whereas the N-terminal domain forms a globular structure, the C-terminal region is unstructured and flexible [13]. It has been reported that mouse ARD1225 induces HIF-1α acetylation and degradation, but other forms of ARD1 have no such effect [14-17], suggesting that the C-terminal region of ARD1 may have important functions independent of its acetyl-transferase activity.
Although ARD1 isoforms share a high sequence identity, their differential regulation and subcellular localization have been demonstrated previously [18]. We have observed that siRNAs used in previous studies for hARD1235 depletion might also target other isoforms of ARD1, for example, hARD1131. Given the variance among ARD1 isoforms, an isoform-specific experimental tool should be used in future studies to help decipher the isoforms’ respective roles. In addition, caspase-dependent cleavage of ARD1 has been previously reported in HeLa cells treated with daunorubicin [10], but whether the truncated ARD1 loses its activity or has a gain-of-function still remains to be determined. Future studies should focus on comparison of these isoforms and their specific roles in different cells.
Subcellular localization of ARD1
Growing evidence has suggested that proteins may be associated with different biological functions according to their subcellular localization. For example, AKT induces the cytoplasmic localization of p21Cip1/WAF1 through phosphorylation, thereby promoting cell growth [19]. In addition, phosphorylation by AKT and IkB kinase β (IKKβ) leads to the release of forkhead box O3a (FOXO3a) from DNA and translocation of FOXO3a into cytoplasm, thereby suppressing its activity [20-24]. A detailed analysis of the ARD1 sequence illustrates a possible nuclear localization signal (NLS) among amino acids 78–83 (KRSHRR), indicating that ARD1 might be imported into the nucleus. Indeed, an early study by Arnesen et al. reported that ARD1 is expressed in both the nucleus and cytoplasm in HeLa, GaMg, HEK-293, MCF-7, and NB4 cells [10]. In agreement with these data, the nuclear and cytoplasmic localizations of ARD1 in HEK-293 cells were also shown by Suzuki's group [25]. However, a discrepant localization of ARD1 in HeLa cells has also been reported, showing that the majority of ARD1 expression is in the cytoplasm [15]. Consistent with this observation, Ren et al. identified a predominantly cytoplasmic localization of ARD1 in the colorectal carcinoma cell line LoVo [26].
According to our preliminary results, both nuclear and cytoplasmic localizations of ARD1 were observed in SKOV-ip1 and MCF-7 cells; however, the ARD1 expression patterns in these two cell lines were distinct. While ARD1 was predominantly located in the nucleus in SKOV-ipl cells, in MCF-7 cells the majority of ARD1 was located in the cytoplasm (Figures 1A, B). Whether the discrepancy in ARD1 localization comes from the diversity of the cells or from the cross-reactivity of antibodies may require further investigation. On the other hand, Chun et al. reported differential localization of ARD1 isoforms. Human ARD1235 is distributed in both the cytoplasm and nucleus, whereas mouse ARD1225 and mouse ARD1235 are present in cytoplasm and nucleus, respectively [18]. It is conceivable that ARD1 may be associated with different functions based on its localization, and this would be an interesting topic for future research.
Figure 1.

Distinct localization of ARD1 in different cell types. The localization of ARD1 was determined by immunoblotting (A) and immunostaining (B).
Biological functions of ARD1
In yeast, ARD1 is involved in the switch control between mitosis and alternative development [27]. According to mutation studies, ARD1 plays an essential role in regulating entry into the stationary phase and sporulation during nitrogen deprivation, which is critical for survival with limited nutrients. In addition, ARD1 is required for a-specific gene expression and mating process in response to pheromone α-factor [28].
In mammalian cells, controversial roles of ARD1 in cancer development have been reported (Figure 2). For instance, Fisher et al. concluded that ARD1 is required for cell proliferation maintenance [16] and depletion of ARD1 in HepG2 cells caused impaired cell division. Consistent with these findings, Lim et al. suggested that ARD1 participates in the proliferation process of lung cancer cells through activation of β-catenin [29]. However, our group recently identified a tumor suppression activity of ARD1 in breast cancer, in which ARD1 reduced cell growth and induced autophagy by inhibiting mTOR signaling [11, please see more later]. Discrepant results have also been shown regarding the regulation of apoptosis by ARD1. Arnesen et al. demonstrated that knockdown of ARD1 triggers apoptosis [30], while Yi et al. reported an essential role of ARD1 in DNA damage-induced caspase activation and apoptosis in a genome-wide RNAi screening study [31]. Thus, whether ARD1 possesses opposing functions in different cells or under distinct conditions may require further investigation.
Figure 2.
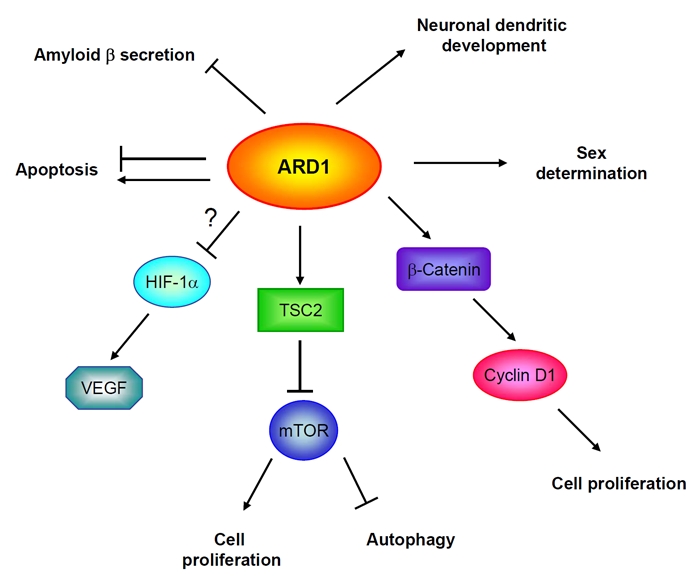
The biological functions of ARD1 in mammalian cells.
In addition to participating in tumorigenesis, ARD1 is also associated with brain development and neurodegenerative disorders. ARD1 plays an essential role in neuronal dendritic development, and downregulation of ARD1 has been observed during differentiation of neurons [32, 33]. Through interaction with amyloid β-protein (Aβ), ARD1 suppresses the secretion of Aβ which is the first step in the development of Alzheimer's disease [25]. We also observed increase in lipid droplets in MDA-MB-435 ARD1 stable transfectants (data not shown), which may suggest a role for ARD1 in the differentiation of breast cancer cells [34-37]. It is interesting that the human ARD1 gene is involved in sex determination [38], which is a conserved function of ARD1 in yeast for which mutation of the ARD1 gene causes a defect in mating. Collectively, ARD1 functions on an unknown set of proteins and to affect diverse cellular activities including cell growth, apoptosis, autophagy, and differentiation. Thus, ARD1 may have pleiotropic effects on many biological systems and understanding its regulation could be important for these systems.
Upstream regulators of ARD1 signaling
Although ARD1 plays an essential role in control of a variety of cellular functions, the upstream stimuli responsible for ARD1 regulation in mammalian cells remain largely unknown (Figure 3). Reduced ARD1 expression under hypoxic conditions has been shown by Kim and Li groups [9, 16]; however, conflicting data raise questions about the foundation of this mechanism [15, 17]. Moreover, ARD1 has been reported to be cleaved during the apoptosis process, thereby decreasing its acetyltransferase activity [10, 18].
Figure 3.
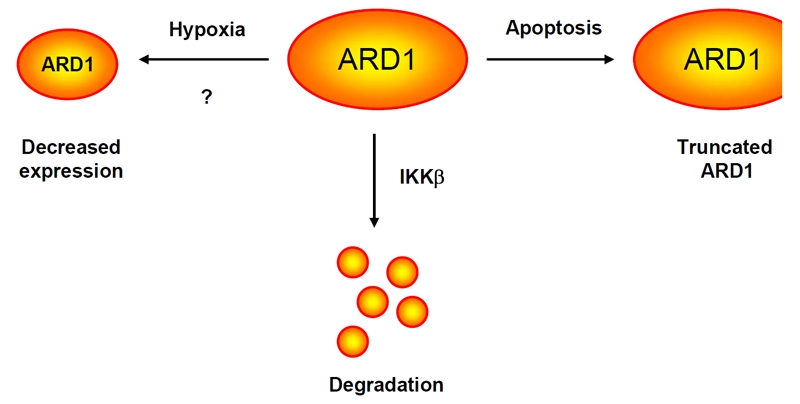
The mechanisms that regulate ARD1 protein level.
Our group recently identified IkB kinase β (IKKβ) as a kinase of ARD1. IKKβ associates with, phosphorylates, and induces proteasome-mediated degradation of ARD1 [39]. Based on previous reports, IKKα and IKKβ, in addition to forming a complex with IKKy and exerting kinase activities, have their own substrates when functioning individually [40, 41]. Thus, it would be interesting to further ask whether the whole IKK complex may be required for ARD1 regulation. Moreover, two additional oncogenic kinases, AKT and extracellular signal-regulated kinase (ERK), also frequently phosphorylate common substrates and are involved in some of the same signaling pathways as IKK. For example, AKT, IKK, and ERK all phosphorylate FOXO3a and TSC1/2, resulting in the inactivation of these two tumor suppressors [20, 40, 42-46]. Therefore, it raises the possibility that AKT and ERK might also be associated with ARD1 regulation.
Since IKKβ is the major kinase activated by TNFα stimulation, an intriguing question remains: whether TNFα or various inflammation signals have any effect on ARD1 phosphoryla-tion and biological functions? Investigating different kinds of stimuli, for example, amino acids, glucose, growth factors, inflammatory cyto-kines, and heat shock on ARD1 transcription, translation, stability, enzyme activity, and functions may advance our understanding of the physiological importance of ARD1.
Downstream targets mediate ARD1 functions
In yeast, a defect in the response to pheromone α-factor caused by ARD1 mutation implies an involvement of ARD1 in the expression of a-specific genes which are required for the mating process [27]. In addition, ARD1 has been shown to be essential for cell survival during nutrient deprivation and resistance to various stimuli like heat shock [47]. Because autophagy is a process that maintains cell survival under various stress conditions, our recent finding that ARD1 increases autophagy in breast cancer cell lines suggests that autophagy induction may be an evolutionary conserved function of ARD1 [11].
Early work has shown that ARD1 increases the RNA levels of Beclin 1 [16], a mammalian autophagy gene. In our recent report, we identified ARD1 as a suppressor of the mTOR signaling pathway [11]. Based on the understanding that suppression of mTOR activity is associated with increased autophagy [48], it will be intriguing to discover whether ARD1 induces autophagy through Beclin 1, mTOR, or both. Since transcriptional and translational regulation by mTOR have been reported in the literature, it is possible that ARD1 regulates the transcription of Beclin and mTOR controls its translation, or that ARD1 induces RNA expression of Beclin 1 through mTOR signaling. Moreover, the identification of other autophagy-associated genes and proteins controlled by ARD1 will further our knowledge of ARD1-mediated autophagy regulation.
In addition to being a survival mechanism, autophagy is also the process responsible for the degradation of long-life proteins. Dysregulation of autophagy has been reported to contribute to neurodegenerative diseases, for example, Alzheimer's disease [49]. It is believed that accumulation of amyloid β-protein in the brain initiates a critical series of events, ultimately leading to Alzheimer's disease. The observation that co-expression of ARD1 and NATH proteins suppresses amyloid β-protein secretion [25] raises an intriguing question of whether autophagy is involved in this regulation process.
Recent studies have shown that β-catenin is a downstream substrate of ARD1. By inducing ε-acetylation of β-catenin, ARD1 upregulates cyclin D1 and promotes lung cancer cell proliferation [29]. On the other hand, we recently demonstrated that ARD1 induced α-acetylation of TSC2. In our study, ARD1 suppressed breast cancer cell growth by regulating TSC2/mTOR signaling [11]. Additional investigation may be needed to determine whether different types of acetylation mediated by ARD1 explain the differential role of ARD1 in tumorigenesis.
Analysis of gene expression profiles have revealed that ARD1 controls a vast array of genes involved in multiple cellular functions, including apoptosis, cell proliferation, metabolism, and cell-cell interaction [16], which is reasonable considering the extensive acetyltransferase activity and possible substrates of ARD1. Future challenges will be to elucidate the difference in ARD1 associated partners and regulated proteins/genes under different conditions and in different types of cells.
ARD1: tumor suppressor or oncoprotein?
Recently, we reported that loss of heterozygosity (LOH) occurs at the ARD1 locus on Xq28 in breast cancer specimens [11]. Since loss of genomic stability is believed to be a crucial molecular step in the early stage of cancer development, our findings may suggest that there is a tumor suppression role of ARD1 in breast cancer. Supporting this notion, we also provided evidence showing that ARD1 expression is correlated with a better clinical outcome in breast cancer patients, including smaller tumor size, fewer lymph node metastases, and longer relapse-free survival. In addition, based on the gene expression patterns of non-small cell lung carcinoma, ARD1 gene expression was found to be 50% lower in tumor tissues than in the adjacent normal tissues in five out six paired samples. Taken together, these findings support ARD1's role in tumor suppression.
By using immunohistochemical staining, Arne-sen et al. found that the level of ARD1 protein is downregulated in most thyroid neoplasm specimens compared to that in non-neoplastic tissues [50]. However, two research groups showed conflicting results with a higher expression of ARD1 in tumor tissues in colorectal and other types of cancer [26, 51]. This discrepancy may have come from the histological specificity of ARD1 in different tissues, making it complicated to interpret the functional consequences of ARD1 expression in mammalian cells. Another possible explanation for the discrepancy is that due to the higher translation rates in actively growing cells, ARD1 is expected to be co-operatively expressed with other genes involved in the protein translation process and further mediates co-translational acetylation. Thus, the elevated level of ARD1 in cancer cells is probably a secondary effect that is caused by its acetyltransferase role in the modification of newly synthesized proteins. Alternatively, the specificity and quality of antibodies used for the immunohistochemical staining might also contribute to the different outcome.
By analyzing ARD1's amino acid sequence, it has been suggested that ARD1 contains several potential phosphorylation sites in the C-terminal. We demonstrated that IKKp phos-phorylates ARD1 at Ser209 and that phosphorylation by IKKp decreases the growth suppression effect of ARD1 [39]. It will be of great interest to determine whether other phosphorylation/dephosphorylation events are involved in ARD1 regulation, thereby changing ARD1's functions in cancer development.
Conclusions and perspectives
In summary, ARD1 is an important molecule that may play a critical role in multiple biological systems including tumor progression. Determining the precise role of ARD1 in cancer development and identifying ARD1's substrates and upstream regulators will remain major challenges for the future studies. Further investigations will need to continue to clarify whether ARD1 could act as a tumor suppressor or an oncoprotein, or have a role in both capacity under different cancer types or different conditions.
Acknowledgments
We would like to thank the editors in the Department of Scientific Publications at The University of Texas M. D. Anderson Cancer Center for editing this article. This work was partially supported by National Institutes of Health (NIH) grant R01 CA109311; the M. D. Anderson Cancer Center/China Medical University Hospital Sister Institution Fund; and grants from the Kadoorie Charitable Foundations, the National Breast Cancer Foundation, Inc., and the Taiwan National Science Council (NSC-96-3111-B) to M.-C.H.; and a predoctoral fellowship from the U.S. Army Breast Cancer Research Program (grant W81XWH-08-1-0397) and the Andrew Sowell-Wade Huggins Scholarship from The University of Texas Graduate School of Biomedical Sciences at Houston to H.-P.K.
References
- 1.Polevoda B, Sherman F. N-terminal acetyl-transferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J Mol Biol. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- 2.Meinnel T, Peynot P, Giglione C. Processed N-termini of mature proteins in higher eukaryotes and their major contribution to dynamic proteomics. Biochimie. 2005;87:701–712. doi: 10.1016/j.biochi.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Hershko A, Heller H, Eytan E, Kaklij G, Rose IA. Role of the alpha-amino group of protein in ubiquitin-mediated protein breakdown. Proc Natl Acad Sci USA. 1984;81:7021–7025. doi: 10.1073/pnas.81.22.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 5.Persson B, Flinta C, von Heijne G, Jornvall H. Structures of N-terminally acetylated proteins. Eur J Biochem. 1985;152:523–527. doi: 10.1111/j.1432-1033.1985.tb09227.x. [DOI] [PubMed] [Google Scholar]
- 6.Polevoda B, Sherman F. Nalpha-terminal acetylation of eukaryotic proteins. J Biol Chem. 2000;275:36479–36482. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]
- 7.Gautschi M, Just S, Mun A, Ross S, Rucknagel P, Dubaquie Y, Ehrenhofer-Murray A, Rospert S. The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol Cell Biol. 2003;23:7403–7414. doi: 10.1128/MCB.23.20.7403-7414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park EC, Szostak JW. ARD1 and NAT1 proteins form a complex that has N-terminal acetyl-transferase activity. EMBO J. 1992;11:2087–2093. doi: 10.1002/j.1460-2075.1992.tb05267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeong JW, Bae MK, Ann MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 10.Arnesen T, Anderson D, Baldersheim C, Lanotte M, Varhaug JE, Lillehaug JR. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem J. 2005;386:433–443. doi: 10.1042/BJ20041071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo HP, Lee DF, Chen CT, Liu M, Chou CK, Lee HJ, Du Y, Xie X, Wei Y, Xia W, Zhang W, Yang JY, Yen CJ, Huang TH, Tan M, Xing G, Zhao Y, Lin CH, Tsai SF, Fidler IJ, Hung MC. ARD1 stabilization of TSC2 suppresses tumorigenesis via the mTOR signaling pathway. Sci Signal. doi: 10.1126/scisignal.2000590. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bilton R, Trottier E, Pouyssegur J, Brahimi-Horn MC. ARDent about acetylation and deacetylation in hypoxia signalling. Trends Cell Biol. 2006;16:616–621. doi: 10.1016/j.tcb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Puig N, Fersht AR. Characterization of the native and fibrillar conformation of the human Nalpha-acetyltransferase ARD1. Protein Sci. 2006;15:1968–1976. doi: 10.1110/ps.062264006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SH, Park JA, Kim JH, Lee JW, Seo JH, Jung BK, Chun KH, Jeong JW, Bae MK, Kim KW. Characterization of ARD1 variants in mammalian cells. Biochem Biophys Res Commun. 2006;340:422–427. doi: 10.1016/j.bbrc.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 15.Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1alpha and is not induced by hypoxia or HIF. J Biol Chem. 2005;280:31132–31140. doi: 10.1074/jbc.M504482200. [DOI] [PubMed] [Google Scholar]
- 16.Fisher TS, Etages SD, Hayes L, Crimin K, Li B. Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J Biol Chem. 2005;280:17749–17757. doi: 10.1074/jbc.M412055200. [DOI] [PubMed] [Google Scholar]
- 17.Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 2005;579:6428–6432. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun KH, Cho SJ, Choi JS, Kim SH, Kim KW, Lee SK. Differential regulation of splicing, localization and stability of mammalian ARD1235 and ARD1225 isoforms. Biochem Biophys Res Commun. 2007;353:18–25. doi: 10.1016/j.bbrc.2006.11.131. [DOI] [PubMed] [Google Scholar]
- 19.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 20.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 21.Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 22.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO's road. Sci STKE. 2003;172:RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- 23.Huang WC, Hung MC. Induction of Akt activity by chemotherapy confers acquired resistance. J Formos Med Assoc. 2009;108:180–194. doi: 10.1016/S0929-6646(09)60051-6. [DOI] [PubMed] [Google Scholar]
- 24.Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 25.Asaumi M, lijima K, Sumioka A, lijima-Ando K, Kirino Y, Nakaya T, Suzuki T. Interaction of N-terminal acetyltransferase with the cytoplasmic domain of beta-amyloid precursor protein and its effect on A beta secretion. J Biochem. 2005;137:147–155. doi: 10.1093/jb/mvi014. [DOI] [PubMed] [Google Scholar]
- 26.Ren T, Jiang B, Jin G, Li J, Dong B, Zhang J, Meng L, Wu J, Shou C. Generation of novel monoclonal antibodies and their application for detecting ARD1 expression in colorectal cancer. Cancer Lett. 2008;264:83–92. doi: 10.1016/j.canlet.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 27.Whiteway M, Szostak JW. The ARD1 gene of yeast functions in the switch between the mitotic cell cycle and alternative developmental pathways. Cell. 1985;43:483–492. doi: 10.1016/0092-8674(85)90178-3. [DOI] [PubMed] [Google Scholar]
- 28.Whiteway M, Freedman R, Van Arsdell S, Szostak JW, Thorner J. The yeast ARD1 gene product is required for repression of cryptic mating-type information at the HML locus. Mol Cell Biol. 1987;7:3713–3722. doi: 10.1128/mcb.7.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim JH, Park JW, Chun YS. Human arrest defective 1 acetylates and activates beta-catenin, promoting lung cancer cell proliferation. Cancer Res. 2006;66:10677–10682. doi: 10.1158/0008-5472.CAN-06-3171. [DOI] [PubMed] [Google Scholar]
- 30.Arnesen T, Gromyko D, Pendino F, Ryningen A, Varhaug JE, Lillehaug JR. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. On-cogene. 2006;25:4350–4360. doi: 10.1038/sj.onc.1209469. [DOI] [PubMed] [Google Scholar]
- 31.Yi CH, Sogah DK, Boyce M, Degterev A, Christofferson DE, Yuan J. A genome-wide RNAi screen reveals multiple regulators of caspase activation. J Cell Biol. 2007;179:619–626. doi: 10.1083/jcb.200708090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugiura N, Adams SM, Corriveau RA. An evolutionarily conserved N-terminal acetyltransferase complex associated with neuronal development. J Biol Chem. 2003;278:40113–40120. doi: 10.1074/jbc.M301218200. [DOI] [PubMed] [Google Scholar]
- 33.Ohkawa N, Sugisaki S, Tokunaga E, Fujitani K, Hayasaka T, Setou M, Inokuchi K. N-acetyltransferase ARD1-NAT1 regulates neuronal dendritic development. Genes Cells. 2008;13:1171–1183. doi: 10.1111/j.1365-2443.2008.01235.x. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki R, Atherton AJ, O'Hare MJ, Entwistle A, Lakhani SR, Clarke C. Proliferation and differentiation in the human breast during pregnancy. Differentiation. 2000;66:106–115. doi: 10.1046/j.1432-0436.2000.660205.x. [DOI] [PubMed] [Google Scholar]
- 35.Sellappan S, Grijalva R, Zhou X, Yang W, Bar Eli M, Mills GB, Yu D. Lineage infidelity of MDA-MB-435 cells: expression of melanocyte proteins in a breast cancer cell line. Cancer Res. 2004;64:3479–3485. doi: 10.1158/0008-5472.CAN-3299-2. [DOI] [PubMed] [Google Scholar]
- 36.Chambers AF. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res. 2009;69:5292–5293. doi: 10.1158/0008-5472.CAN-09-1528. [DOI] [PubMed] [Google Scholar]
- 37.Hollestelle A, Schutte M. Comment re: MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res. 2009;69:7893. doi: 10.1158/0008-5472.CAN-09-2396. [DOI] [PubMed] [Google Scholar]
- 38.Tribioli C, Mancini M, Plassart E, Bione S, Rivella S, Sala C, Torri G, Toniolo D. Isolation of new genes in distal Xq28: transcriptional map and identification of a human homologue of the ARD1 N-acetyl transferase of Saccharomyces cerevisiae. Hum Mol Genet. 1994;3:1061–1067. doi: 10.1093/hmg/3.7.1061. [DOI] [PubMed] [Google Scholar]
- 39.Kuo HP, Lee DF, Xia W, Lai CC, Li LY, Hung MC. Phosphorylation of ARD1 by IKKbeta contributes to its destabilization and degradation. Biochem Biophys Res Commun. 2009;389:156–161. doi: 10.1016/j.bbrc.2009.08.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hor-tobagyi GN, Tsai FJ, Tsai CH, Hung MC. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 41.Hu Y, Baud V, Oga T, Kim Kl, Yoshida K, Karin M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 42.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 43.Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, Huang H, Kuo HP, Lee DF, Li LY, Lien HC, Cheng X, Chang KJ, Hsiao CD, Tsai FJ, Tsai CH, Sahin AA, Muller WJ, Mills GB, Yu D, Hortobagyi GN, Hung MC. ERK promotes tumorigenesis by inhibiting F0X03a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 45.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 46.Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res. 2009;15:752–757. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullen JR, Kayne PS, Moerschell RP, Tsunasawa S, Gribskov M, Colavito-Shepanski M, Grunstein M, Sherman F, Sternglanz R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989;8:2067–2075. doi: 10.1002/j.1460-2075.1989.tb03615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarbassov DD, AN SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- 50.Arnesen T, Gromyko D, Horvli O, Fluge O, Lillehaug J, Varhaug JE. Expression of N-acetyl transferase human and human Arrest defective 1 proteins in thyroid neoplasms. Thyroid. 2005;15:1131–1136. doi: 10.1089/thy.2005.15.1131. [DOI] [PubMed] [Google Scholar]
- 51.Yu M, Gong J, Ma M, Yang H, Lai J, Wu H, Li L, Li L, Tan D. Immunohistochemical analysis of human arrest-defective-1 expressed in cancers in vivo. Oncol Rep. 2009;21:909–915. doi: 10.3892/or_00000303. [DOI] [PubMed] [Google Scholar]