

Abstract
Developing a quantifiable in vitro model of steatosis is critical in understanding the pathogenesis of nonalcoholic fatty liver disease (NAFLD) and searchingfor effective therapies. Using an ORO-based colorimetric measurement, we developed a convenient assay to qualify the degree of OA-induced steatosis in HepG2 cells. We demonstrated that in the absence of exogenous inflammatory mediators, OA-induced steatosis was associated with increased production and secretion of tumor necrosis factor alpha and decreased expression of peroxisome proliferators-activated receptor α in HepG2 cells. OA-induced steatosis was also associated with increased lipid peroxidation, apoptosis, but decreased proliferation in these cells. The increased lipid peroxidation was related to decreased SOD-1, a free radical scavenger enzyme; while increased apoptosis was related to increased active caspase-9. The decreased proliferation mediated by OA-induced steatosis was associated with increased production of p27 with unchanged alanine transaminase (ALT) level in the culture medium, indicating OA-induced steatosis alters cell cycle progression without direct toxicity to these cells. In conclusion, the present study developed a colorimetric assay that accurately quantifies OA-induced steatosis in HepG2 cells. In the absence of exogenous inflammatory mediators, OA-induced steatosis results in a series of pathophysilogical changes in HepG2 cells, indicating direct pathogenic roles of hepatocytes in NAFLD.
Keywords: Steatosis, tumor necrosis factor α, peroxisome proliferators-activated receptor α, apoptosis, lipid peroxidation, cell proliferation
Introduction
Non-alcoholic fatty liver disease (NAFLD) is one of the most common liver diseases in Western countries. NAFLD encompasses a spectrum of liver diseases, ranging from simple steatosis to steatosis combined with varying degrees of necroinflammation and fibrosis [1]. It is estimated that approximately 20% to 30% of adults in the United States and other Western countries have excess fat accumulation in the liver [2]. Hepatic steatosis can either be a benign, noninflammatory condition, or can be associated with non-alcoholic steatohepatitis (NASH), a condition that can result in end-stage liver disease [3]. A convenient and quantifiable in vitro model will be valuable in understanding the pathogenesis and evaluating the effects of future therapies of this common liver disease.
Hepatic steatosis in human beings is associated with accumulation of excess oleic acid (OA), a monosaturated omega-9 fatty acid and the end product of de novo fatty acid synthesis [4]. Treatment of HepG2 cells, a human hepa-toblastoma cell line, with OA induces morphological similarities to steatotic hepatocytes [5, 6], but quantification of OA-induced steatosis has not been well established. Oil red O (ORO) is a lysochrome (fat-soluble dye) diazo dye used for staining of neutral triglycerides and lipids on frozen sections. ORO stains protein bound lipids in paraffin sections. This biochemical technique has been predominantly used for triglyceride staining on tissue sections [7]. Quantification of ORO stained cell with light microscopy is inconvenient, and its accuracy is operator-dependent that makes reliable quantification difficult
Although studies demonstrated profound pathogenic changes in cytokines and signal transduction, lipid metabolism, and hepatocytic apoptosis and injury in both alcoholic liver disease (ALD) and NAFLD [8-12], the underlying mechanisms remain to be determined. Developing a human cell model in which steatosis is qualifiable will hold a very special value in understanding mechanisms of NAFLD from liver cell level, because such human cell model exclude the interference from the matrix and other non-hepatocytic cells.
In the present study, we used OA and HepG2 cells and developed a cell model of steatosis. Using an ORO-based colorimetric measurement developed in the present study, we were able to rapidly and accurately quantify the degree of OA-induced steatosis. This model was also used to determine the underlying mechanisms of OA-induced steatosis in HepG2 cells.
Materials and methods
Reagents
Dulbecco's modified eagle's medium (DMEM), fetal bovine serum (FBS), trypsin-EDTA, and penicillin-streptomycin-fungizone were purchased from Cambrex Bio Science Walkers-ville, Inc. (Walkersville, MD). The cell proliferation assay was performed using a Cell Titer 96 AQueous One Solution Reagent purchased from Promega Corporation (Madison, Wl). OA-conjugated BSA solution and ORO were purchased from Sigma Chemical Company (St. Louis, MO). Antibodies to p21waf1/cip1, p27kip1, Bcl-2, Bax, PPARα, cleaved caspase-9, TNF-α, SOD1, and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-alanine transaminase (ALT) peptide anti-body was produced in our laboratory and its specificity (54 kDa) was confirmed by Western blot. Protein A/G PLUS-agarose was from Santa Cruz Biotechnology (Santa Cruz, CA). Su-perSignal West Dura Extended Duration Substrate for Western blots was purchased from Pierce Biotechnology (Rockford, IL). The protein assay kit, gel electrophoresis buffers, 4%-15% gradient Tris-HCI gels and 10% Tris-HCI glycine gels, and the nitrocellulose transfer membrane were purchased from Bio-Rad Laboratories (Hercules, CA).
Cell culture
HepG2 cells were cultured in DMEM-based medium as described before. [7, 8]. The experiments were performed when cells reached about 80% confluence, and after cultured in FBS-free media for 24 hrs.
OA-induced steatosis
Once approximately 80% confluence and cultured in FBS-free medium for 24 hrs in 96-well culture plate, HepG2 cells were treated with 200 μl of different concentration (0.1-2.0 mM) of OA solution for 24 hours. After the medium was removed, 100 μl of fixative solution were added, and incubated at room temperature for 10 minutes. Control cells were treated with OA-free medium containing albumin.
ORO cell staining
ORO solution was made by mixing 2.4 ml of ORO stock solution with 1.6 ml of distilled water then undergoing filtering. After removing the fixative solution from each well of the 96-well culture plate, the cells were washed three times with ddH20. A 50 μl of the ORO solution was then added to each well and incubated at room temperature for 15 minutes. After removing the ORO solution from each well, the cells were washed multiple times with ddH20 until the solution became clear. After dried and mounted with glycerin, the cells were examined under a light microscope, and the red oil droplet in staining in the cells indicate OA-induced steatosis.
ORO-based steatosis quantification
To develop a rapid and convenient quantification assay for OA-induced steatosis in HepG2 cells, ORO-based cell staining was combined with a colorimetric assay using 96-well plate and optical densitometer. HepG2 cells were cultured in a 96-well microplate at 5000 cells/well and treated with OA at the concentration ranged from 0.1-2.0 mM, then undergoing ORO staining as described above. After washing and drying completely, 100 μl of extraction solution was then added to each well and incubated for 10 minutes, followed by gently vibration to release ORO from steatosis staining. The extract solution containing ORO released from steatosis staining was then transferred well by well to another 96-well plate that will undergoing OD measurement at a wavelength of 405 nm using ELX800 Universal Microplate Reader (Bio-Tek instruments, Inc, Winooski, VT). All the tests were performed in triplicate and the means were calculated as the final results.
Cell proliferation assay
Cell proliferation was determined using an MTT assay as previously reported [13, 14]. Briefly, 5×103cells were plated into a 96-well plate containing 100 μl of cell culture medium in triplicate and were treated with different concentrations of OA. The effects of OA on HepG2 cell growth were then determined by optical density absorbance as previously reported [13, 14].
Assay of apoptosis
Apoptosis was determined with two different methods, as previously reported [14]. Briefly, after treatment of HepG2 cells with respective doses of OA for 24 hrs, cell death detection was performed in duplicate and measured by calculating the ratio of absorbance of treated cells relative to the control cells. The activated caspase-9, Bax, and Bcl-2 were measured by Western blots [13, 14].
Western blots
Protein expression was determined by Western blot [7, 8]. Briefly, HepG2 cells were cultured with OA for 24 hours, and then the cell pellets were lysed with 1 ml of the lysis buffer. The lysates were centrifuged at 12,000 rpm for 10 minutes at 4°C. The supernatants were used to detect TNF-α, PPARα, ALT, S0D1, p27kip1, p21waf1/cip1, Bax, Bcl-2, and cleaved caspase-9. All the Western blots were repeated for three times. Western blot for β-actin was used as internal control. To quantify the results, the relative amount of each protein was determined by digital scanning of the hybridizing bands, as previously described [13, 14].
Assay for LPO in OA-induced HepG2 cells
Lipid peroxidation (LPO) levels were determined using a commercial lipid hydroperoxide assay kit from Cayman Chemical Co. (Ann Arbor, Ml). Medium and cellular homogenates (500 μl) were deproteinated and extracted under acidic conditions with 1 ml ice-cold deoxygenated chloroform, and the chloroform extract was removed following centrifugation (1500 × g for 5 minutes at 0°C) for LPO determination. After mixing with chromogen reagent, 200 μl of the extracted samples were transferred to a 96-well plate. The absorbance was determined at 500 nM. The 13-hydroxyperoxy-octadecadienoic acid was used as a lipid hydroperoxide standard to construct a standard curve (linearity from 0.5 to 5 nM hydroperoxide).
Statistical analysis
The student t test was used to compare the difference of the means. Linear regression was used to calculate correlation coefficient and determine the association between OA-induced steatosis and OD values measured by ORO colorimetric assay developed in the present study.
Results
OA-induced steatosis in HepG2 cells
The histological definition of steatosis is the visible accumulation of lipid droplets in more than 5% of hepatocytes [15]. To determine the optimal concentration of OA to induce steatosis, HepG2 cells were cultured at OA concentrations of 0.1 mM, 0.5 mM, 1 mM, and 2 mM for 24 hours. In untreated control of HepG2 cells, ORO staining revealed almost absence of intracellular lipid (Figure 1A). After treatment with OA, lipid droplets were accumulated in the cytoplasm of HepG2 cells (Figure 1B). OA at concentrations between 0.1 mM to 2 mM, reliably induced steatosis in HepG2 cells in a dose-dependent pattern as determined by ORO staining.
Figure 1.

OA-induced steatosis in HepG2 cells determined by ORO staining and ORO-based colorimetric assay. (A) Non-treated HepG2 cells did not show ORO staining. (B) Positive ORO staining as shown in red in cytoplasm of HepG2 cells treated with 1 mM OA for 24 hours. (C) OA (0.1-2.0 mM) treatment of HepG2 cells for 24 hours induced steatosis in a dose-dependent manner (r2 = 0.97, p=0.001), as determined by ORO-based colorimetric assay.
An ORO-based colorimetric quantitative assay for OA-induced steatosis in HepG2 cells
Steatosis staining with ORO and its quantification under microscopy can be inconvenient, and its accuracy is operator-dependent. To objectively measure the degree of OA-induced steatosis in HepG2 cells, we develop a colorimetric assay to quantify steatosis, based on the unique feature of high organic solubility of ORO that can be colorimetrically quantifiable after its release by an extract solution from stained steatosis in the cells.
After confirming the feasibility and optimizing the conditions of the ORO-based colorimetric assay, we then tested its sensitivity and repro-ducibility. As shown in Figure 1C, our ORO-based colorimetric quantitative assay revealed an excellent correlation of the measured optical density with OA-induced dose-dependent steatosis in HepG2 cells with a correlation coefficient (r2) as high as 0.97 (p=0.001). The tests were repeated three times with the same results. This method easily and reliably quantifies the difference of steatosis induced by OA at the dose ranged from 0.1 to 2 mM, as observed under microscopy. Thus, ORO-based colorimetric quantitative assay developed in the present study is not only convenient, quantifiable, but also highly sensitive and reproducible.
Effects of OA-induced steatosis on production of TNF-α and PPARα in HepG2 cells
It is well known that elevated TNF-α plays a key role in the pathogenesis and disease progression of NAFLD [16-18]. Overexpression of TNF-α mRNA in both liver and adipose tissue has been reported in severely obese patients with NASH [19]. It was also reported that treatment of HepG2 cells with free fatty acid (FFA) resulted in increased production of TNF-α mRNA [20]. To ascertain if this effect also occurs at the translational level, we tested if OA treatment of HepG2 cells alters production and secretion of TNF-α. We found that OA treatment significantly induced TNF-α expression in HepG2 cells and its secretion to the culture medium (Figure 2A and 2B).
Figure 2.
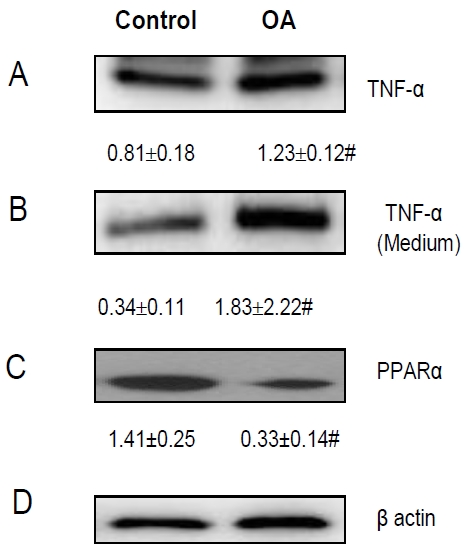
OA-induced Steatosis and Its Effects on TNF-α and PPARα Expression. OA-induced steatosis in HepG2 cells resulted in (A) increased production of TNF-α; (B) increased secretion of TNF-α from HepG2 cells to the culture medium; (C) inhibited PPARα expression. (D) β-actin was used as an internal control. “#” means p value < 0.05 compared to the untreated cells.
A growing body of literature implicates that peroxisome proliferators activated receptors (PPARs) play important roles in the pathogenesis of NAFLD [10]. For instance, PPARα may augment fatty acid oxidation and protects against steatosis [21-23]. To determine whether steatotic hepatocytes are a significant source of PPARα, we compared the difference of PPARα expression in HepG2 cells before and after OA treatment. We demonstrated that OA-induced steatosis was significantly associated with reduced expression of PPARα in HepG2 cells (Figure 2C).
Effects of OA-induced steatosis on lipid perox-idation in HepG2 cells
Many studies have indicated the important pathogenic role of oxidative stress in NAFLD. For instance, oxidative stress can initiate membrane lipid peroxidation and loss of cell viability in cultured hepatocytes [24-26]. The OA-induced steatosis in HepG2 cells provides a unique model to study the changes of lipid peroxidation in the hepatocytic level. To determine how OA-induced steotosis alters oxidative stress, we determined lipid peroxide formation by measuring lipid hydroperoxide in HepG2 cells after 24 hour exposure to OA. We found a significant rise in lipid peroxides in the OA-treated HepG2 cells (Figure 3A). Superoxide dismutase-1 (SOD-1) is a free radical scavenger enzyme that protects against cellular membrane injury mediated by lipid peroxidation. We observed that S0D1 expression was significantly reduced in HepG2 cells with OA-induced steatosis (Figure 4A).
Figure 3.

OA-induced steatosis and its effects on lipid peroxidation, cell proliferation and apoptosis. OA-induced steatosis in HepG2 cells resulted in (A) increased lipid peroxidation; (B) decreased cell proliferation, as determined by MTT assay; (C) no changes in ALT level in these cells and the culture medium, compared to untreated HepG2 cells and medium; and (D) increased apoptosis of HepG2 cells. “#” means p value < 0.05 compared to the untreated cells.
Figure 4.

OA-induced Steatosis and Its Effects on the Modulators Related to Lipid Peroxidation, Cell Proliferation and Apoptosis. OA-induced steatosis in HepG2 cells resulted in (A) increased SOD-1 expression; (B) increased p27; (C) no significant change in p21 expression; (D) increased production of activated cleaved caspase-9; and no significant change in of Bcl-2 (E) and Bax (F) expression. (G) β-actin was used as an internal control. “#” means p value < 0.05 compared to the untreated cells.
Effects of OA-induced steatosis on cell proliferation and cell Cycle progression in HepG2 cells
Since steatosis has been reported to alter cell proliferation [27], we determined whether OA-induced steatosis affects cell proliferation by measuring effects of OA on HepG2 cell viability using MTT assay [13, 14]. As shown in Figure 3B, we found OA-induced steatosis was associated with significant inhibition of HepG2 cell proliferation in an OA-dose dependent manner at the concentration from 0.2 mM to 1.4 mM. For instance, OA inhibited HepG2 cell proliferation by 21% at 1 mM, and 37.5% at 1.4 mM of the concentration.
ALT release from hepatocytes has been a traditional marker of liver cell injury. To determine if OA-induced steatosis could result in direct liver cell injury that might count for decreased cell proliferation, we measured ALT level in the cell lysates and the culture medium without treatment (as controls) and after treatment with various OA concentrations. As shown in Figure 3C, compared to the untreated HepG2 cells, treatment with OA at the concentration from 0.5 mM to 2 mM for 72 hours did not increase the concentration of ALT in these cells and the culture medium. Thus, OA-reduced proliferation of HepG2cells is not secondary to its direct toxic effects.
To further determine the effects of OA-induced steatosis on HepG2 cell proliferation, we assessed if OA-induced steatosis alters expression of several modulators involved in the progression of the cell cycle and cellular proliferation pathways. P21waf1/cip1 and p27kip1are two cyclin-dependent kinase (CDK) inhibitors involved in suppression of the cell cycle at the G1-S checkpoint. We found that OA-induced steatosis significantly increased the expression of p27kiP1, but not p21waf1/cip1 expression in HepG2 cells, as shown in Figures 4B and 4C.
Effects of OA-induced steatosis on apoptosis in HepG2 cells
Clinical studies have shown that NASH is associated with altered apoptosis activity [28, 29]. To evaluate the effects of OA-induced steatosis on apoptosis activity in HepG2 cell, we tested the cell death by measuring cytoplasmic histone-associated DNA fragments. We found that OA- induced steatosis significantly increased apoptosis activity in HepG2 cells in a dose-dependent manner at OA dose ranging from 0.1 mM to 1 mM (Figure 3D). At OA doses greater than 1 mM, the apoptosis activity measured in HepG2 cells appeared to reach a maximum level and began to plateau.
To determine the mechanisms involved in OA-induced apoptosis, we measured the expression of several apoptosis-related proteins. In OA-treated HepG2 cells, we found a significant increase in the expression of cleaved caspase-9, a marker of apoptosis signaling in the caspase activation pathway (Figure 4D). Although Bax, a pro-apoptotic marker, and Bcl-2 an anti-apoptosis marker, have been associated with apoptosis activity, we could not detect changes in their expression after OA induction (Figures 4Eand 4F).
Discussion
OA-induced steatosis in HepG2 cells may serve as an in vitro model for studying fatty liver disease. Consistent with previous reports [30], we revealed OA induces steatosis in HepG2 cells in a dose-dependent manner that can be assessed by ORO biochemical staining. However, this method is neither convenient, nor accurate in quantification, therefore, cannot be used as a routine steatosis assay, especially for the study of disease mechanisms and development of new therapy to NAFLD.
In the present study, we utilized the feature that after biochemical staining of steatosis, ORO can be released from the stained cells that could be further quantitatively measurable. Using this concept, we were able to develop an ORO-based colorimetric quantification assay to measure OA-induced steatosis in HepG2 cells. Our further analysis indicated that this novel method is not only convenient, and highly reproducible, but also accurate with a very high correlation between the dose of OA and the degree of steatosis as expressed as absorbance of optical density and histological ORO digital pixel measurement. Thus, the cell model of OA-induced steatosis, together with this novel ORO-based colorimetric quantitative assay, will provide valuable tools to study the pathogenesis and develop new treatment for NAFLD.
It is well known that TNF-α plays important pathogenic roles in both ALD and NAFLD [16-18]. For instance, TNF-α has been associated with insulin resistance and induce inflammatory cytokines formation. The mean plasma level of TNF-α was significantly higher in NAFLD patients with abnormal ALT than controls [31-33]. A key question is whether increased TNF-α in NAFLD is from hepatocytes and/or from other inflammatory cells. A recent study demonstrated that FFA treatment induces TNF-α mRNA in HepG2 cells [19], but it remains unknown if FFA-induced steatosis also promotes translation of TNF-α mRNA. In the present study, we demonstrated that OA-induced steatosis significantly increased TNF-α production and secretion from HepG2 cells. These in vitro results indicate that OA-induced steatosis promotes translation of TNF-α mRNA. Our data further support the pathogenic role of hepa-tocyte-derived TNF-α in NAFLD. Studies reported that TNF-α also stimulates ROS generation and induces lipid peroxidation [34]. We demonstrated that OA-induced steatosis is associated with increased lipid peroxides in HepaG2 cells. However, the pathogenic role of increased TNF-α in up regulation of lipid peroxides in OA-induced steatosis remains to be determined.
As HepG2 cells accumulate intracellular lipids, there is a significant rise in lipid peroxides. In previous studies, unsaturated fatty acids have been shown to induce the cytochrome P450 2E1 (CYP2E1) enzyme pathway and stimulate lipid peroxidation, which subsequently promotes apoptosis and cell toxicity [35]. Consistent with these findings, we found that OA-induced steatosis was associated with a significant rise in lipid peroxide formation in the OA-treated HepG2 cells. The pathogenic role of increased lipid peroxides in the cell injury of OA-induced steatosis in HepG2 cells is further supported by our findings that OA-induced steatosis in these cells was associated with a significantly decreased SOD-1, a free radical scavenger enzyme that protects against cellular membrane injury mediated by lipid peroxidation. These finding also indicated a potential therapeutic role of anti-peroxidation agents for NAFLD.
PPARα activates expression of a series of target genes involved in the uptake, transportation, and β-oxidation of fatty acids [36, 37]. Several studies have shown that PPARα increases fatty acid catabolism, and therefore, may prevent hepatic fat deposition [38-42]. Our findings that OA-induced steatosis resulted in reduced PPARα expression in HepG2 cells indicated its possible pathogenic role in NAFLD. Taken together, our results suggested that the association of OA-induced steatosis with increased lipid peroxidation may be mediated by decreased PPARα expression in these cells. This speculation is supported by the clinical and experimental evidence that PPARα antagonists may improve steatosis in patients and animal model [43, 44]. Further determining this association will be help in developing novel therapeutic approach to this disease.
Hepatocyte apoptosis is a feature of fatty liver disease [45], which may reduce cell regeneration or proliferation. We found that OA, at 0.1-lmM dose range, leads to increased apoptosis of HepG2 cells with OA-induced steatosis. This was associated with increased production of activated caspase-9 of the apoptosis cascade. Although apoptosis may be associated with decreased proliferation of the liver cells with steotosis, it may also contribute to the pathogenesis of the liver injury in non-alcoholic fatty liver disease. We could not demonstrate any significant effects of OA-treatment on Bcl-2 and Bax expression, the two other apoptotic modulators, indicating that they are unlikely regulating apoptosis in OA-induced steatosis in HepG2 cells.
The effects of steatosis on hepatocyte proliferation are unknown. Using MTT assay, we demonstrated that OA-induced steatosis in HepG2 cells was associated with inhibition of cell proliferation. These results are supported by a recent report of microarray analysis [27]. Since OA-increased apoptosis was saturated at the dose of 1 mM, the decreased HepG2 proliferation could not be solely explained by apoptosis. On the other hand, we demonstrated its association with increased p27 expression. Since p27 functions as a cyclin-dependent kinase (CDK) inhibitor involved in suppression of the cell cycle at the G1-S checkpoint, our results indicated that OA-induced steatosis decreases cell proliferation by inhibiting p27 expression, therefore, G1-S progression.
The clinical presentation of NAFLD could be very variable from persistently normal to significantly increased ALT. It is not known if steatosis results in direct liver injury and elevated ALT. Using HepG2 in vitro model, we demonstrated that baseline level of ALT was comparable in the culture medium of both untreated and OA-treated HepG2 cells. At a wide range of concentration, OA treatment does not alter the ALT level in the culture medium. These findings suggest that OA-induced steatosis itself does not result in liver cell injury. Instead, it is likely that hepatocytes’ direct response to steatosis results in generation of a series of inflammatory mediators that may cause liver cell injury. Further studies will be needed to detail theses underlying mechanisms.
In conclusion, the present study developed an ORO-based colorimetric assay to quantify lipid accumulation in OA-induced HepG2 cells, which provides a convenient tool for studying the pathogenesis and therapy for NAFLD. We also assessed the effects of OA-induced steatosis on HepG2 cell proliferation, apoptosis, and lipid peroxidation through a complicated signaling. These data are very valuable in understanding the pathogenesis of NAFLD.
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 3.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n -3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 5.Okamoto Y, Tanaka S, Haga Y. Enhanced GLUT2 gene expression in an oleic acid-induced in vitro fatty liver model. Hepatol Res. 2002;23:138–144. doi: 10.1016/s1386-6346(01)00172-3. [DOI] [PubMed] [Google Scholar]
- 6.Janorkar AV, King KR, Megeed Z, Yarmush ML. Development of an in vitro cell culture model of hepatic steatosis using hepatocyte-derived reporter cells. Biotechnology and Bioengineering. 2009;102:1466–1474. doi: 10.1002/bit.22191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carson FL. Amer Society of Clinical. 2nd. 1997. Histotechnology: A Self-instructional Text; p. l60. [Google Scholar]
- 8.Edmison J, McCullough AJ. Pathogenesis of non-alcoholic steatohepatitis: human Data. Clin Liver Dis. 2007;11:75–104. doi: 10.1016/j.cld.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 9.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in non-alcoholic fatty liver disease. Semin Liv Dis. 2008;28:370–377. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 10.Kallwitz ER, McLachlan A, Cotler SJ. Role of peroxisome proliferators-activated receptors in the pathogenesis and treatment of nonalcoholic fatty liver disease. World J Gastroenterol. 2008;14:22–28. doi: 10.3748/wjg.14.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.London RM, George J. Pathogenesis of NASH; animal Models. Clini Liver Dis. 2008;11:55–74. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Hoek JB, Pastorino JG. Cellular signaling mechanisms in alcoholic-induced liver damage. Semin Liv Dis. 2004;24:257–272. doi: 10.1055/s-2004-832939. [DOI] [PubMed] [Google Scholar]
- 13.Hu KQ, Yu CH, Mineyama Y, McCracken JD, Hillebrand DJ, Hasan M. Inhibited proliferation of cyclooxygenase-2 expressing human hepatoma cells by NS-398, a selective COX-2 inhibitor. Int J Oncol. 2003;22:757–763. [PubMed] [Google Scholar]
- 14.Cui W, Yu CH, Hu KQ. In vitro and in vivo effects and mechanisms of celecoxib-induced growth inhibition of human hepatocellular carcinoma cells. Clin Cancer Res. 2005;11:8213–8221. doi: 10.1158/1078-0432.CCR-05-1044. [DOI] [PubMed] [Google Scholar]
- 15.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCul-lough AJ, Sanyal AJ. Nonalcoholic Steatohepati-tis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 16.Tokushige K, Hashimoto E, Tsuchiya N, Kaneda H, Taniai M, Shiratori K. Clinical significance of soluble TNF receptor in Japanese patients with non-alcoholic steatohepatitis. Alcohol Clin Exp Res. 2005;29:298S–303S. doi: 10.1097/01.alc.0000191810.46000.37. [DOI] [PubMed] [Google Scholar]
- 17.Wong VW, Hui AY, Tsang SW, Chan JL, Tse AM, Chan KF, So WY, Cheng AY, Ng WF, Wong GL, Sung JJ, Chan HL. Metabolic and adipokine profile of Chinese patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:1154–1161. doi: 10.1016/j.cgh.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 18.Bahcecioglu IH, Yalniz M, Ataseven H, llhan N, Ozercan IH, Seckin D, Sahin K. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepatoga-stroenterology. 2005;52:1549–1553. [PubMed] [Google Scholar]
- 19.Crespo J, Cayon A, Fernandez-Gil P, Hernández-Guerra M, Mayorga M, Domínguez-Díez A, Fernśndez-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Srivastava S, Mittal S, Yang X, Sheng L, Chan C. A Three Stage Integrative Pathway Search (TIPS) framework to identify toxicity relevant genes and pathways. BMC Bioinformat-ics. 2007;8:202–218. doi: 10.1186/1471-2105-8-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo YS, Kim JH, Jo NY, Choi KM, Baik SH, Park JJ, Kim JS, Byun KS, Bak YT, Lee CH, Kim A, Yeon JE. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. Gastroenterol Hepatol. 2008;23:102–109. doi: 10.1111/j.1440-1746.2006.04819.x. [DOI] [PubMed] [Google Scholar]
- 22.Alwayn IP, Andersson C, Lee S, Arsenault DA, Bistrian BR, Gura KM, Nose V, Zauscher B, Moses M, Puder M. Inhibition of matrix metal-loproteinases increases PPAR-alpha and IL-6 and prevents dietary-induced hepatic steatosis and injury in a murine model. Am J Physiol Ga-strointest Liver Physiol. 2006;291:G1011–1019. doi: 10.1152/ajpgi.00047.2006. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka T, Masuzaki H, Nakao K. Role of PPARs in the pathophysiology of nonalcoholoic fatty liver disease. Nippon Rinsho. 2005;63:700–706. [PubMed] [Google Scholar]
- 24.Galli A, Svegliati-Baroni G, Ceni E, Milani S, Ridolfi F, Salzano R, Tarocchi M, Grappone C, Pellegrini G, Benedetti A, Surrenti C, Casini A. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP-2-mediated mechanism. Hepatology. 2005;41:1074–1084. doi: 10.1002/hep.20683. [DOI] [PubMed] [Google Scholar]
- 25.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1135–1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 26.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 27.De Gottardi A, Vinciguerra M, Sgroi A, Moukil M, Ravier-Dall'Antonia F, Pazienza V, Pugnale P, Foti M, Hadengue A. Microarray analyses and molecular profiling of steatosis induction in immortalized human hepatocytes. Lab Invest. 2007;87:792–806. doi: 10.1038/labinvest.3700590. [DOI] [PubMed] [Google Scholar]
- 28.Ramalho RM, Cortez-Pinto H, Castro RE, Solá S, Costa A, Moura MC, Camilo ME, Rodrigues CM. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. Eur J Gastroenterol Hepatol. 2006;18:21–29. doi: 10.1097/00042737-200601000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Ribeiro PS, Cortez-Pinto H, Sola S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 30.Gómez-Lechín MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O'Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Khoruts A, Stahnke L, McClain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, inter-leukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267–276. [PubMed] [Google Scholar]
- 32.McClain C, Hill D, Schmidt J, Diehl AM. Cyto-kines and alcoholic liver disease. Semin Liver Dis. 1993;13:170–182. doi: 10.1055/s-2007-1007347. [DOI] [PubMed] [Google Scholar]
- 33.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 34.Evereklioglu C, Turkoz Y, Calis M, Duygulu F, Karabulut AB. Tumour necrosis factor alpha, li-pid peroxidation and NO* are increased and associated with decreased free-radical scavenging enzymes in patients with Weill-Marchesani syndrome. Mediators Inflamm. 2004;13:165–170. doi: 10.1080/09511920410001713547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung M, Kim I, Park M, Whang Y, Lee M. Differential effects of dietary fatty acids on the regulation of CYP2E1 and protein kinase C in human hepatoma HepG2 cells. J Med Food. 2004;7:197–203. doi: 10.1089/1096620041224157. [DOI] [PubMed] [Google Scholar]
- 36.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 37.Schoonjans K, Staels B, Auwerx J. The peroxi-some proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 38.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 39.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 40.Ki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 41.Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A, Pacetti D, Sigmund S, Benedetti A, Casini A. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169:846–860. doi: 10.2353/ajpath.2006.050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shulman AI, Mangelsdorf DJ. Retinoid x receptor heterodimers in the metabolic syndrome. N Engl J Med. 2005;353:604–615. doi: 10.1056/NEJMra043590. [DOI] [PubMed] [Google Scholar]
- 43.Tonstad S, Retterstøl K, Ose L, Ohman KP, Lindberg MB, Svensson M. The dual peroxisome proliferator-activated receptor alpha/gamma agonist tesaglitazar further improves the lipid profile in dyslipidemic subjects treated with atorvastatin. Metabolism. 2007;569:1285–1292. doi: 10.1016/j.metabol.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Seo YS, Kim JH, Jo NY, Choi KM, Baik SH, Park JJ, Kim JS, Byun KS, Bak YT, Lee CH, Kim A, Yeon JE. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J Gastroenterol Hepatol. 2008;231:102–109. doi: 10.1111/j.1440-1746.2006.04819.x. [DOI] [PubMed] [Google Scholar]
- 45.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart U, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]