

Abstract
Proinsulin C-peptide has been found to exert beneficial effects in many tissues affected by diabetic microvascular complications, including the kidneys. Glomerular hyperfiltration and microalbuminuria are early markers of diabetic nephropathy. C-peptide at physiological concentrations effectively reduces diabetes-induced glomerular hyperfiltration via constriction of the afferent arteriole, dilation of the efferent arteriole, and inhibition of tubular reabsorption in experimental models of type 1 diabetes. The glomerular hypertrophy and mesangial matrix expansion seen in early diabetes can be reduced or prevented by C-peptide administration, possibly via interference with TGF-β1 and TNFα signaling. Several of C-peptide's reno-protective effects have been confirmed in human studies; reduced glomerular hyperfiltration and diminished urinary albumin excretion have been documented in type 1 diabetes patients receiving replacement doses of C-peptide for periods of up to 3 months. In this review, we critically summarize the current state of knowledge regarding C-peptide’s renal effects, and discuss possible mechanisms of its beneficial effects in diabetic nephropathy.
Keywords: diabetes, C-peptide, nephropathy, microalbuminuria, glomerular hypertrophy, capillary pressure
Abbreviations: ATP - adenosine triphosphate; Ca2+- bivalent ionic calcium; C-peptide - connecting peptide; ΔΠonc - oncotic pressure gradient; FF - filtration fraction; GFR - glomerular filtration rate; K+ - potassium ion; Kf - ultrafiltration coefficient; MD - macula densa; Na+ - sodium ion; Na+/K+-ATPase - sodium, potassium adenosintriphosphatase, also sodium-potassium pump; NO - nitric oxide; eNOS - endothelial nitric oxide synthase; NF-κB - nuclear factor-kappa light-chain enhancer of activated B cells; P - hydraulic pressure; Pcap - glomerular capillary pressure; Pnet - net driving force for filtration; Ptub - intratubular hydrostatic pressure; TGF - tubuloglomerular feedback; TGF-β1 - transforming growth factor beta, isoform 1; TNF-α - tumor necrosis factor alpha
Introduction
Diabetic nephropathy is typically defined as a progressive deterioration in kidney function, initially represented by augmented glomerular filtration rate (GFR), glomerular hypertrophy, and urinary leakage of albumin. A progressive increase in protein excretion, serum creatinine levels, and gradually diminished GFR are part of the subsequent clinical picture. Glomerulosclerosis and tubulo-interstitial fibrosis are commonly seen on microscopic examination of renal biopsies. Hypertension and cardiovascular disease are frequent co-morbidities, and correlate with angiopathy of the capillaries in the renal glomeruli.
There are several hypotheses for the pathogenic mechanisms that result in the progression of diabetes-induced renal dysfunction. In early diabetic nephropathy, glomerular pressure increases, causing an increased GFR [1]. Glomerular hypertension and hyperfiltration may play a crucial role in the subsequent development of overt diabetic nephropathy [1], and thus represent potential therapeutic targets for the prevention of diabetic nephropathy. During the past decade, proinsulin C-peptide has emerged as a bioactive peptide in its own right, and recent research indicates that it may play an important role in renal pathophysiology.
Exogenous C-peptide administration in type 1 diabetes has been shown to exert beneficial effects in many tissues affected by microvascular complications. This finding is supported by the fact that pancreas or islet transplantation is accompanied by significant amelioration of glomerular structural abnormalities after ten years of restored endogenous C-peptide levels and normoglycemia [2, 3]. Direct evidence confirms that C-peptide has the capacity to exert a series of beneficial effects on the diabetic kidney. A reduction in diabetes-induced glomerular hyperfiltration, diminished albumin excretion, and decreased glomerular hypertrophy have all been shown to accompany C-peptide administration in diabetic animals and type 1 diabetes patients [4-9]. In this report, the beneficial effects of C-peptide on the diabetic kidney will be reviewed, with regard to basic science and clinical aspects, with particular attention to the possible physiological mechanisms involved.
Effects on hyperfiltration
Early in the development of diabetic nephropathy, there is a marked increase in GFR. Diabetes-induced glomerular hyperfiltration has been proposed as an independent risk factor for the development of nephropathy in this disorder [10]. Therapeutic interventions aiming at reducing the augmented GFR delays the progression of kidney damage [11]. It is known that both the native C-peptide and its carboxy-terminal pentapeptide fragment normalize diabetes-induced glomerular hyperfiltration in experimental diabetes [7].
Regulators of glomerular filtration
The glomerular microcirculation and the glomerular capillary pressure (Pcap) are largely regulated via fine-tuning of the vascular resistances of the afferent and efferent glomerular arterioles. As defined by the Starling equation, alterations in these vascular resistances, and thus in Pcap, exert substantial effects on GFR (Figure 1). The connection between vascular resistance and GFR is Pnet, the net driving force for filtration. Pnet is determined by the hydraulic pressure (P) exerted by the fluid as well as the oncotic pressure gradient (ΔΠonc) generated by proteins in the blood. The hydrostatic pressure in the glomerular capillaries, Pcap, is the force driving fluid from the blood across the glomerular capillary and podocyte membranes into the tubular system; thus alterations in Pcap directly modify GFR. Changes in Pcap may be the result of alterations in afferent and efferent arteriolar diameter, which regulate the pressure transmitted into the glomerular capillaries [12, 13]. A constriction of the afferent arteriole or dilation of the efferent arteriole induces a reduction in Pcap, and lowers Pnet which in turn reduces GFR. Thus, the interplay between the vascular resistances of the afferent and efferent arterioles is a powerful determinant of GFR.
Figure 1. The Starling equation.
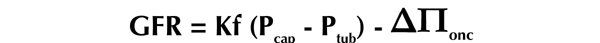
The equaltion includes the effect of hydrostatic and oncotic forces on the movement of fluid across capillary membranes. The equation above is modified for approximating GFR, but can be applied to all capillary flow.
The glomerular microcirculation and the glomerular capillary pressure (Pcap) are largely regulated via fine-tuning of the vascular resistances of the afferent and efferent glomerular arterioles. As defined by the Starling equation, alterations in these vascular resistances, and thus in Pcap, exert substantial effects on GFR (Figure 1). The connection between vascular resistance and GFR is Pnet, the net driving force for filtration. Pnet is determined by the hydraulic pressure (P) exerted by the fluid as well as the oncotic pressure gradient (ΔΠonc) generated by proteins in the blood. The hydrostatic pressure in the glomerular capillaries, Pcap, is the force driving fluid from the blood across the glomerular capillary and podocyte membranes into the tubular system; thus alterations in Pcap directly modify GFR. Changes in Pcap may be the result of alterations in afferent and efferent arteriolar diameter, which regulate the pressure transmitted into the glomerular capillaries [12, 13]. A constriction of the afferent arteriole or dilation of the efferent arteriole induces a reduction in Pcap, and lowers Pnet which in turn reduces GFR. Thus, the interplay between the vascular resistances of the afferent and efferent arterioles is a powerful determinant of GFR.
Pcap approximately equals the sum of the early intratubular hydrostatic pressure (Ptub) and ΔΠonc. Ptub is dependent on the balance between tubular reabsorption and the volume filtered across the glomerular capillary [14]. When augmented, Ptub will reduce GFR. ΔΠonc is regulated by the differences in protein concentration between the glomerular capillaries and the filtered fluid in Bowman's space and the proximal tubulus. Under normal circumstances, the protein concentration of the fluid in Bowman’s space is close to zero. Thus, both ΔΠonc and Ptub exert opposing forces on glomerular filtration, limiting the flux of filtrate into the tubular system. Intrarenal acute changes in ΔΠonc may be induced by alterations in the fraction of renal blood flow filtered to the tubular side of the nephron, termed the filtration fraction (FF) [15]. During normal conditions, FF largely reflects the balance between afferent and efferent arteriolar resistances. Thus, effects of FF on ΔΠonc will further increase the impact of afferent and efferent arteriole vascular resistances on Pnet and GFR. When FF is reduced, this decreases the protein concentration in the blood that leaves the glomerulus. This, in turn, reduces the driving force for reabsorption into the peritubular capillaries from the proximal part of the tubule, a chain of events referred to as the glomerulo-tubular balance.
Glomerular filtration is also affected by the biophysical characteristics of the capillary wall. The ultrafiltration coefficient (Kf) is dependent on the hydraulic conductivity and the size of the filtration surface area. GFR is dynamically regulated by tubuloglomerular feedback (TGF), a regulating function further described below. Finally, a regulatory mechanism has been proposed according to which alterations in early proximal tubular sodium transport may affect glomerular filtration through alterations in Ptub, which directly influences Pnet [16, 17]. This hypothesis is consistent with reports of a linear correlation between GFR and Na+/K+-ATPase activity in the kidney cortex in experimental diabetes [18, 19].
Glomerular afferent and efferent arteriolar tone
Due to their influence on ΔΠonc and Pcap, constriction of the afferent arteriole and dilation of the efferent arteriole, respectively, contributes to a lower glomerular filtration pressure, and may normalize GFR. The afferent glomerular arteriolar diameter is increased in animal models of type 1 diabetes [20], which contributes to a rise in GFR. Dilators of the efferent arteriole such as angiotensin-converting enzyme inhibitors, as well as afferent constrictors such as low protein diets, may reduce GFR, and tend to improve long-term renal function in a similar manner [11, 21-23].
C-peptide administration most likely influences the intraglomerular filtration pressure in the diabetic state in two ways. Firstly, the peptide simultaneously constricts the afferent arteriole, and induces a dilation of the efferent arteriole, resulting in a reduction of Pcap and GFR in diabetic rats. Secondly, C-peptide constricts isolated renal afferent arterioles from diabetic mice [24] (Figures 2 and 3). Also, in a recent in vivo study, it could be demonstrated that C-peptide simultaneously reduces GFR, tubular stop-flow pressure, and filtration fraction without altering renal blood flow [6]. These findings are consistent with a reduction in efferent arteriolar resistance. They are also in agreement with previous reports on C-peptide and renal blood flow [6, 25, 26]. Similar actions on renal afferent-efferent arteriole tone have previously been reported for Ca2+ channel blockers and nitric oxide synthase inhibitors [27, 28].
Figure 2. C-peptide-induced constriction of isolated renal afferent arteriole from a diabetic mouse.
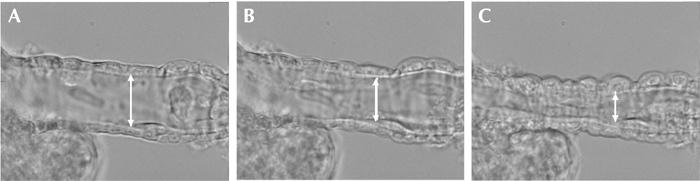
A: the arteriole at baseline. B: the arteriole after 15 minutes of C-peptide perfusion. C: the arteriole after 30 minutes of C-peptide perfusion.
Figure 3. Constriction of afferent arterioles.
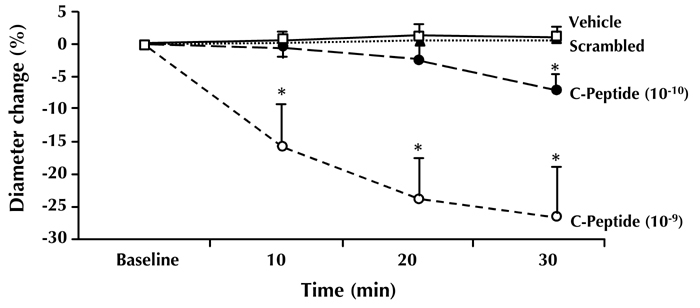
C-peptide induces dose-dependent constriction of afferent arterioles in diabetic mice. No effect was seen for scrambled C-peptide or vehicle. Via normalization of hyperfiltration, this may be one of the mechanisms in the renoprotective action of C-peptide. Modified from [24].
Tubuloglomerular feedback
Glomerular hyperfiltration in diabetes may in part be secondary to alterations in tubuloglomerular feedback (TGF) [29]. TGF is an intrarenal mechanism that stabilizes GFR and the tubular electrolyte load to match the tubular handling capacity. A prerequisite for TGF is the anatomical juxtaposition of each tubule to its corresponding glomerulus. The macula densa (MD) consists of specialized epithelial cells localized where the returning tubulus approaches its glomerulus. These cells represent a sensor mechanism for electrolytes. An increased tubular flow rate results in augmented tubular NaCl load, which may be sensed by the MD, and result in a feedback constriction of the afferent arteriole. However, it was recently shown that TGF is unlikely the mediator of diabetes-induced hyperfiltration, since hyperfiltration occurs in adenosine A1-receptor-deficient mice which lack a functional TGF mechanism [30, 31]. In addition, it has been demonstrated that C-peptide constricts the afferent arteriolar also in isolated arterioles that lack an intact tubulus. This suggests that a TGF-mediated mechanism is unlikely involved in the effects of C-peptide on the afferent arteriolar diameter in diabetes.
Tubular effects of C-peptide
Increased renal Na+/K+-ATPase activity and augmented oxygen consumption during hyperfiltration have been observed in several models of experimental diabetes [18, 19, 32, 33]. C-peptide is reported to inhibit Na+/K+-ATPase in isolated proximal tubular cells from diabetic animals [6], an effect accompanied by normalization of basal proximal tubular oxygen consumption [34]. The diabetes-specific C-peptide-induced inhibition of Na+/K+-ATPase is supported by the in vivo observation that C-peptide also alters lithium clearance and fractional sodium excretion secondary to a reduction in tubular sodium reabsorption in diabetic animals [6].
It is likely that C-peptide exerts a direct effect on renal sodium transport by activating Na+/K+-ATPase in tubular cells under normoglycemic conditions [35, 36] via phosphorylation of the α-subunit of Na+/K+-ATPase [36]. Interestingly, not only the native full-length C-peptide but also its C-terminal penta- and hexapeptides have the capacity to stimulate Na+/K+-ATPase activity [37]. In contrast, in hyperglycemic diabetic animals, C-peptide's effect on renal Na+/K+-ATPase is in the opposite direction, as also indicated by a lowering of tubular oxygen consumption by C-peptide in the diabetic state [6]. These observations suggest that C-peptide has state-specific effects. An analogous effect has been observed for endothelial nitric oxide synthase (eNOS) and C-peptide; although generally reported to be a stimulator of eNOS activity at normal glucose concentrations, C-peptide has been found to reduce diabetes-induced increases in renal eNOS levels [38, 39]. The background to these seemingly paradoxical effects of C-peptide is not apparent, but may be related to the profound alterations in intracellular signaling that accompanies diabetes and hyperglycemia.
Alterations in early proximal tubular sodium transport may in themselves influence glomerular filtration. In diabetes, proximal tubular Na+ reabsorption increases due to increased cortical Na+/K+-ATPase activity [19, 33, 40, 41]. Similarly, a decrease in Na+ reabsorption in the early proximal tubule increases Ptub, and thereby reduce Pnet and lower GFR. Accordingly, a C-peptide-induced reduction in Na+ reabsorption is accompanied by a decrease in GFR. Consequently, the C-peptide-induced inhibition of diabetic proximal Na+/K+-ATPase activity and sodium reabsorbtion seen in diabetic animals and cells from diabetic rats may contribute to a decrease in diabetic hyperfiltration [6].
C-peptide and kidney oxygenation
Despite accounting for only 0.5% of total body mass, the kidney consumes approximately 10% of the whole body oxygen uptake. Of the kidney oxygen consumption, approximately 80% is related to tubular electrolyte and glucose transport through activity of the sodium pump (Na+/K+-ATPase) [42]. The major renal consumer of energy is Na+/K+-ATPase, located basolaterally in proximal tubular cells within the renal cortex.
It has been shown that renal oxygen consumption is increased in diabetic cells, and that diabetic kidneys have reduced tissue oxygen availability [43, 44]. In several studies, increased renal Na+/K+-ATPase activity has been linked to hyperfiltration, and to the increase in oxygen consumption [18, 19, 32, 33]. Via inhibition of Na+/K+-ATPase, C-peptide may contribute to a normalization of the basal oxygen consumption of proximal tubules in diabetic animals [6].
C-peptide may also influence oxygenation through effects on nitric oxide release. In addition, C-peptide can improve diabetes-reduced erythrocyte deformability [45]. This has the potential to improve capillary blood flow, thus improving oxygen availability in the kidney, and other affected tissues.
C-peptide and renal structural abnormalities in diabetes
Profound changes of glomerular and tubular structures are hallmarks of diabetic renal disease. It is well established that glomerular mesangial matrix expansion and capillary basement membrane thickening occurs early in the development of nephropathy. This causes an increase in glomerular volume and augmented width of the podocyte interdigital processes [46]. At the same time, there is also an increase in tubular volume, due to expansion of the mesenchymal tissue. These abnormalities are followed by the gradual development of glomerulosclerosis and tubulointerstitial fibrosis as the disorder progresses. In this context, it is of interest that C-peptide has been found to exert a direct influence not only on renal function, as discussed above, but also on renal structural changes. Diabetes-induced glomerular hypertrophy is prevented or diminished in streptozotocin-diabetic rats treated with homologous C-peptide in physiological concentrations. At the same time, a reduction in GFR and urinary albumin excretion is observed in these rats [9]. The amelioration of glomerular changes following C-peptide administration is secondary to a diminished expansion of glomerular mesangial matrix [47]. No detailed information is available regarding C-peptide's possible effects on the tubular structural abnormalities in diabetes.
It is not fully clear how C-peptide exerts its beneficial effects in this regard. Transforming growth factor-β1 (TGF-β1), overexpressed in renal cells in diabetes, has been suggested as a major malefactor in the development of morphological changes in diabetes by stimulating the mesenchymal formation of collagen IV, and eliciting fibrosis in renal tissues [48]. It has been reported that C-peptide has a protective effect on early diabetic glomerular changes in response to TGF-β1 in streptozotocin-diabetic mice. In addition, an inhibitory effect of C-peptide on TGF-β1-induced gene expression has been demonstrated in a mouse podocyte cell line [49]. Likewise, C-peptide is reported to effectively reverse TGF-β1-induced structural changes in proximal tubular cells [50]. The mechanism by which this occurs is not fully understood, but there is evidence that C-peptide has an impact on TGF-β1 signaling [50]. It may also be that other mechanisms for C-peptide's beneficial effects are involved. The cytokine TNFα, capable of eliciting a wide spectrum of cellular responses including inflammation and cell death, is increased in diabetic nephropathy [51]. It has been reported that C-peptide, via activation of NF-κB-regulated survival genes, protects against TNFα-mediated renal tubular injury [52]. Irrespective of the mechanisms involved, the findings suggest a potential role for C-peptide in the therapy of diabetic nephropathy.
Studies in patients with type 1 diabetes
Short-term effects of C-peptide administration in type 1 diabetes have been studied in a group of young patients without signs of renal disease [53]. C-peptide was infused i.v. for 2 h to reach physiological concentrations. GFR decreased by 7%, renal plasma flow increased slightly, and renal FF decreased from 19% to 16%. This corresponds to reports from animal studies. In healthy subjects, C-peptide had no measurable effects on renal function.
These studies were extended to include more prolonged periods of C-peptide administration. In a double-blind randomized study, type 1 diabetic patients with incipient nephropathy received replacement doses of C-peptide for one month administered by subcutaneous infusion via a pump besides their regular insulin therapy [5]. All patients showed elevated GFR and mild microalbuminuria at study start. In C-peptide-treated patients, GFR fell by 6%, and urinary albumin excretion decreased by approximately half the initial value. At the same time, no significant change was observed in controls receiving insulin only. Indices of glycemic control improved slightly in the C-peptide group, but not in the controls. This possibly contributed to the positive outcome [5].
In another study with cross-over design involving type 1 patients with slightly more advanced renal dysfunction, C-peptide was administered subcutaneously for 3 months in replacement doses via 4 daily injections [4]. Pre-study albumin excretion was in the range 25-220 μg/min, and decreased gradually throughout the study period. At the end of the study, albumin excretion had fallen by approximately 40% in the C-peptide treated patients, but remained unchanged or was slightly increased in controls (Figure 4). Indices of glycemic control were similar in the C-peptide group and the controls, and all patients were normotensive throughout the study. The background to the observed reduction in albumin excretion is not apparent, but may in part be sought in a diminished intraglomerular pressure. The findings confirm that the results obtained in animal studies translate into beneficial effects in patients with type 1 diabetes. Further studies of longer duration and involving type 1 patients with more advanced renal disease are warranted.
Figure 4. Urinary albumin excretion.
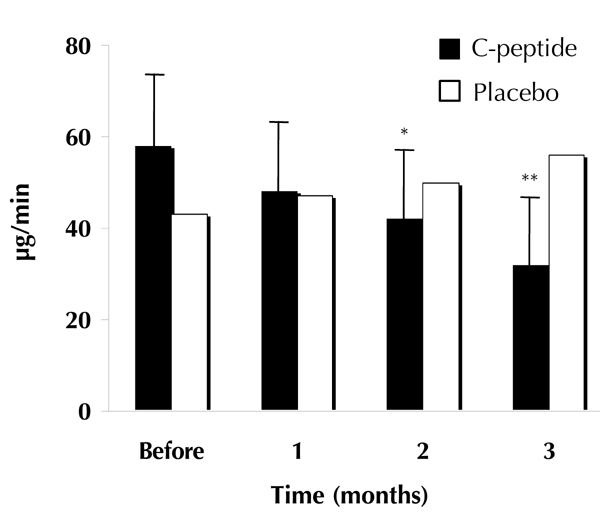
Geometric means of urinary albumin excretion in type 1 diabetic patients given C-peptide plus insulin (filled columns) or placebo plus insulin (open columns) for 3 months. Asterisks indicate significant differences from the baseline period. Modified from [4].
Concluding comments - what next?
Based on the currently available data it is apparent that C-peptide has the capacity to exert a number of beneficial effects on the functional and structural abnormalities of kidneys in diabetes. Further information on the mechanisms by which C-peptide reduces hyperfiltration and albuminuria would be helpful. Likewise, additional data characterizing C-peptide's modulatory influence on glomerular and tubular structural abnormalities are required. It should be noted that most of the present data have been generated in cell systems or animal experiments, while only few human studies have been carried out. Consequently, clinical trials involving C-peptide administration together with regular insulin therapy for prolonged periods of time (6-12 months) in type 1 patients at different stages of nephropathy are urgently needed to advance our knowledge of the peptide's therapeutic potential.
Finally, it should be considered that, at this time, little is known about possible C-peptide effects in type 2 diabetes. It may well be that in type 2 diabetic patients, who have reached a stage of relative insulin deficiency, C-peptide may exert a positive effect on the renal dysfunction that frequently accompanies this disorder. Thus, studies involving C-peptide administration in type 2 diabetes patients are also warranted.
Disclosures: Dr. Wahren reports that he is employed at Cebix Inc., CA, USA.
References
- 1.O'Bryan GT, Hostetter TH. The renal hemodynamic basis of diabetic nephropathy. Semin Nephrol. 1997;17(2):93–100. [PubMed] [Google Scholar]
- 2.Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–75. doi: 10.1056/NEJM199807093390202. [DOI] [PubMed] [Google Scholar]
- 3.Fiorina P, Folli F, Zerbini G, Maffi P, Gremizzi C, Di Carlo V, Socci C, Bertuzzi F, Kashgarian M, Secchi A. Islet transplantation is associated with improvement of renal function among uremic patients with type I diabetes mellitus and kidney transplants. J Am Soc Nephrol. 2003;14:2150–2158. doi: 10.1097/01.asn.0000077339.20759.a3. [DOI] [PubMed] [Google Scholar]
- 4.Johansson BL, Borg K, Fernqvist-Forbes E, Kernell A, Odergren T, Wahren J. Beneficial effects of C-peptide on incipient nephropathy and neuropathy in patients with Type 1 diabetes mellitus. Diabet Med. 2000;17:181–189. doi: 10.1046/j.1464-5491.2000.00274.x. [DOI] [PubMed] [Google Scholar]
- 5.Johansson BL, Kernell A, Sjoberg S, Wahren J. Influence of combined C-peptide and insulin administration on renal function and metabolic control in diabetes type 1. J Clin Endocrinol Metab. 1993;77:976–981. doi: 10.1210/jcem.77.4.8408474. [DOI] [PubMed] [Google Scholar]
- 6.Nordquist L, Brown RD, Fasching A, Persson P, Palm F. Proinsulin C-peptide reduces diabetes-induced glomerular hyperfiltration via efferent arteriole dilation and inhibition of tubular sodium reabsorption. Am J Physiol. 2009 doi: 10.1152/ajprenal.00228.2009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordquist L, Moe E, Sjoquist M. The C-peptide fragment EVARQ reduces glomerular hyperfiltration in streptozotocin-induced diabetic rats. Diabetes Metab Res Rev. 2007;23:400–405. doi: 10.1002/dmrr.704. [DOI] [PubMed] [Google Scholar]
- 8.Samnegard B. Danderyd Hospital and Department of Surgical Sciences. Karolinska Institute; Stockholm: 2005. Renal effects of C-peptide in experimental type-1 diabetes mellitus. [Google Scholar]
- 9.Samnegard B, Jacobson SH, Jaremko G, Johansson BL, Sjoquist M. Effects of C-peptide on glomerular and renal size and renal function in diabetic rats. Kidney Int. 2001;60:1258–1265. doi: 10.1046/j.1523-1755.2001.00964.x. [DOI] [PubMed] [Google Scholar]
- 10.Mogensen CE. Early glomerular hyperfiltration in insulin-dependent diabetics and late nephropathy. Scand J Clin Lab Invest. 1986;46(3):201–206. doi: 10.3109/00365518609083660. [DOI] [PubMed] [Google Scholar]
- 11.Dullaart RP, Beusekamp BJ, Meijer S, van Doormaal JJ, Sluiter WJ. Long-term effects of protein-restricted diet on albuminuria and renal function in IDDM patients without clinical nephropathy and hypertension. Diabetes care. 1993;16:483–492. doi: 10.2337/diacare.16.2.483. [DOI] [PubMed] [Google Scholar]
- 12.Persson AE, Gushwa LC, Blantz RC. Feedback pressure-flow responses in normal and angiotensin-prostaglandin-blocked rats. Am J Physiol. 1984;247:F925–F931. doi: 10.1152/ajprenal.1984.247.6.F925. [DOI] [PubMed] [Google Scholar]
- 13.Ren YL, Garvin JL, Ito S, Carretero OA. Role of neuronal nitric oxide synthase in the macula densa. Kidney Int. 2001;60:1676–1683. doi: 10.1046/j.1523-1755.2001.00987.x. [DOI] [PubMed] [Google Scholar]
- 14.Karlsen FM, Holstein-Rathlou NH, Leyssac PP. A re-evaluation of the determinants of glomerular filtration rate. Acta Physiol Scand. 1995;155(4):335–350. doi: 10.1111/j.1748-1716.1995.tb09984.x. [DOI] [PubMed] [Google Scholar]
- 15.Brenner BM, Troy JL. Postglomerular vascular protein concentration: evidence for a causal role in governing fluid reabsorption and glomerulotublar balance by the renal proximal tubule. J Clin Invest. 1971;50:336–349. doi: 10.1172/JCI106501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leyssac PP. Dependence of glomerular filtration rate on proximal tubular reabsorption of salt. Acta Physiol Scand. 1963;58:236–242. doi: 10.1111/j.1748-1716.1963.tb02644.x. [DOI] [PubMed] [Google Scholar]
- 17.Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest. 2001;107:217–224. doi: 10.1172/JCI10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korner A, Eklof AC, Celsi G, Aperia A. Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes. 1994;43:629–633. doi: 10.2337/diab.43.5.629. [DOI] [PubMed] [Google Scholar]
- 19.Scherzer P, Nachliel I, Bar-On H, Popovtzer MM, Ziv E. Renal Na-K-ATPase hyperactivity in diabetic Psammomys obesus is related to glomerular hyperfiltration but is insulin-independent. J Endocrinol. 2000;167:347–354. doi: 10.1677/joe.0.1670347. [DOI] [PubMed] [Google Scholar]
- 20.Ikenaga H, Bast JP, Fallet RW, Carmines PK. Exaggerated impact of ATP-sensitive K(+) channels on afferent arteriolar diameter in diabetes mellitus. J Am Soc Nephrol. 2000;11:1199–1207. doi: 10.1681/asn.v1171199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aurell M. Aspects of the role of intraglomerular pressure as a cause of progressive renal damage. Drugs. 1988;35(Suppl 5):42–47. doi: 10.2165/00003495-198800355-00007. [DOI] [PubMed] [Google Scholar]
- 22.Inman SR, Stowe NT, Nally JV Jr, Brouhard BH, Vidt DG. Dietary protein does not alter intrinsic reactivity of renal microcirculation to angiotensin II in rodents. Am J Physiol. 1995;268:F302–F308. doi: 10.1152/ajprenal.1995.268.2.F302. [DOI] [PubMed] [Google Scholar]
- 23.Myers BD, Deen WM, Brenner BM. Effects of norepinephrine and angiotensin II on the determinants of glomerular ultrafiltration and proximal tubule fluid reabsorption in the rat. Circ Res. 1975;37:101–110. doi: 10.1161/01.res.37.1.101. [DOI] [PubMed] [Google Scholar]
- 24.Nordquist L, Lai EY, Sjoquist M, Patzak A, Persson AE. Proinsulin C-peptide constricts glomerular afferent arterioles in diabetic mice. A potential renoprotective mechanism. Am J Physiol. 2008;294:R836–R841. doi: 10.1152/ajpregu.00811.2007. [DOI] [PubMed] [Google Scholar]
- 25.Huang DY, Richter K, Breidenbach A, Vallon V. Human C-peptide acutely lowers glomerular hyperfiltration and proteinuria in diabetic rats: a dose-response study. Naunyn Schmiedebergs Arch Pharmacol. 2002;365(1):67–73. doi: 10.1007/s00210-001-0502-1. [DOI] [PubMed] [Google Scholar]
- 26.Samnegard B, Jacobson SH, Johansson BL, Ekberg K, Isaksson B, Wahren J, Sjoquist M. C-peptide and captopril are equally effective in lowering glomerular hyperfiltration in diabetic rats. Nephrol Dial Transplant. 2004;19:1385–1391. doi: 10.1093/ndt/gfh163. [DOI] [PubMed] [Google Scholar]
- 27.Feng MG, Navar LG. Nitric oxide synthase inhibition activates L- and T-type Ca2+ channels in afferent and efferent arterioles. Am J Physiol Renal Physiol. 2006;290:F873–F879. doi: 10.1152/ajprenal.00042.2005. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi K, Wakino S, Sugano N, Ozawa Y, Homma K, Saruta T. Ca2+ channel subtypes and pharmacology in the kidney. Circ Res. 2007;100(3):342–353. doi: 10.1161/01.RES.0000256155.31133.49. [DOI] [PubMed] [Google Scholar]
- 29.Vallon V, Blantz RC, Thomson S. Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am J Physiol. 1995;269:F876–F883. doi: 10.1152/ajprenal.1995.269.6.F876. [DOI] [PubMed] [Google Scholar]
- 30.Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, Mizel D, Kajiyama H, Zerfas P, Briggs JP, Kopp JB, Schnermann J. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J Am Soc Nephrol. 2008;19:722–730. doi: 10.1681/ASN.2007060721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sallstrom J, Carlsson PO, Fredholm BB, Larsson E, Persson AE, Palm F. Diabetes-induced hyperfiltration in adenosine A(1)-receptor deficient mice lacking the tubuloglomerular feedback mechanism. Acta Physiol (Oxf) 2007;190(3):253–259. doi: 10.1111/j.1748-1716.2007.01705.x. [DOI] [PubMed] [Google Scholar]
- 32.Ku DD, Sellers BM, Meezan E. Development of renal hypertrophy and increased renal Na,K-ATPase in streptozotocin-diabetic rats. Endocrinology. 1986;119:672–679. doi: 10.1210/endo-119-2-672. [DOI] [PubMed] [Google Scholar]
- 33.Wald H, Scherzer P, Rasch R, Popovtzer MM. Renal tubular Na(+)-K(+)-ATPase in diabetes mellitus: relationship to metabolic abnormality. Am J Physiol. 1993;265:E96–E101. doi: 10.1152/ajpendo.1993.265.1.E96. [DOI] [PubMed] [Google Scholar]
- 34.Nordquist L, Stridh S. Effects of proinsulin C-peptide on oxygen transport, uptake and utilization in insulinopenic diabetic subjects - a review. Adv Exp Med Biol. 2009;645:193–198. doi: 10.1007/978-0-387-85998-9_30. [DOI] [PubMed] [Google Scholar]
- 35.Ohtomo Y, Aperia A, Sahlgren B, Johansson BL, Wahren J. C-peptide stimulates rat renal tubular Na+, K(+)-ATPase activity in synergism with neuropeptide Y. Diabetologia. 1996;39:199–205. doi: 10.1007/BF00403963. [DOI] [PubMed] [Google Scholar]
- 36.Zhong Z, Kotova O, Davidescu A, Ehren I, Ekberg K, Jornvall H, Wahren J, Chibalin AV. C-peptide stimulates Na+, K+-ATPase via activation of ERK1/2 MAP kinases in human renal tubular cells. Cell Mol Life Sci. 2004;61:2782–2790. doi: 10.1007/s00018-004-4258-x. [DOI] [PubMed] [Google Scholar]
- 37.Ohtomo Y, Bergman T, Johansson BL, Jornvall H, Wahren J. Differential effects of proinsulin C-peptide fragments on Na+, K+-ATPase activity of renal tubule segments. Diabetologia. 1998;41:287–291. doi: 10.1007/s001250050905. [DOI] [PubMed] [Google Scholar]
- 38.Kamikawa A, Ishii T, Shimada K, Makondo K, Inanami O, Sakane N, Yoshida T, Saito M, Kimura K. Proinsulin C-peptide abrogates type-1 diabetes-induced increase of renal endothelial nitric oxide synthase in rats. Diabetes Metab Res Rev. 2008;24:331–338. doi: 10.1002/dmrr.810. [DOI] [PubMed] [Google Scholar]
- 39.Kitamura T, Kimura K, Makondo K, Furuya DT, Suzuki M, Yoshida T, Saito M. Proinsulin C-peptide increases nitric oxide production by enhancing mitogen-activated protein-kinase-dependent transcription of endothelial nitric oxide synthase in aortic endothelial cells of Wistar rats. Diabetologia. 2003;46:1698–1705. doi: 10.1007/s00125-003-1232-3. [DOI] [PubMed] [Google Scholar]
- 40.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–2576. doi: 10.1681/ASN.V10122569. [DOI] [PubMed] [Google Scholar]
- 41.Vervoort G, Veldman B, Berden JH, Smits P, Wetzels JF. Glomerular hyperfiltration in type 1 diabetes mellitus results from primary changes in proximal tubular sodium handling without changes in volume expansion. Eur J Clin Invest. 2005;35:330–336. doi: 10.1111/j.1365-2362.2005.01497.x. [DOI] [PubMed] [Google Scholar]
- 42.Thaysen JH, Lassen NA, Munck O. Sodium transport and oxygen consumption in the mammalian kidney. Nature. 1961;190:919–921. doi: 10.1038/190919a0. [DOI] [PubMed] [Google Scholar]
- 43.Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46:1153–1160. doi: 10.1007/s00125-003-1155-z. [DOI] [PubMed] [Google Scholar]
- 44.Palm F, Hansell P, Ronquist G, Waldenstrom A, Liss P, Carlsson PO. Polyol-pathway-dependent disturbances in renal medullary metabolism in experimental insulin-deficient diabetes mellitus in rats. Diabetologia. 2004;47:1223–1231. doi: 10.1007/s00125-004-1434-3. [DOI] [PubMed] [Google Scholar]
- 45.Kunt T, Schneider S, Pfutzner A, Goitum K, Engelbach M, Schauf B, Beyer J, Forst T. The effect of human proinsulin C-peptide on erythrocyte deformability in patients with Type I diabetes mellitus. Diabetologia. 1999;42:465–471. doi: 10.1007/s001250051180. [DOI] [PubMed] [Google Scholar]
- 46.Osterby R. Lessons from kidney biopsies. Diabetes Metab Res Rev. 1996;12:151–174. doi: 10.1002/(SICI)1099-0895(199610)12:3<151::AID-DMR162>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 47.Samnegard B, Jacobson SH, Jaremko G, Johansson BL, Ekberg K, Isaksson B, Eriksson L, Wahren J, Sjoquist M. C-peptide prevents glomerular hypertrophy and mesangial matrix expansion in diabetic rats. Nephrol Dial Transplant. 2005;20:532–538. doi: 10.1093/ndt/gfh683. [DOI] [PubMed] [Google Scholar]
- 48.August P, Suthanthiran M. Transforming growth factor beta signaling, vascular remodeling, and hypertension. New Engl J Med. 2006;354:2721–2723. doi: 10.1056/NEJMcibr062143. [DOI] [PubMed] [Google Scholar]
- 49.Maezawa Y, Yokote K, Sonezaki K, Fujimoto M, Kobayashi K, Kawamura H, Tokuyama T, Takemoto M, Ueda S, Kuwaki T, Mori S, Wahren J, Saito Y. Influence of C-peptide on early glomerular changes in diabetic mice. Diabetes Metab Res Rev. 2006;22:313–322. doi: 10.1002/dmrr.612. [DOI] [PubMed] [Google Scholar]
- 50.Hills CE, Al-Rasheed N, Al-Rasheed N, Willars GB, Brunskill NJ. C-peptide reverses TGF-beta1-induced changes in renal proximal tubular cells: implications for treatment of diabetic nephropathy. Am J Physiol. 2009;296:F614–F621. doi: 10.1152/ajprenal.90500.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasegawa G, Nakano K, Sawada M, Uno K, Shibayama Y, Ienaga K, Kondo M. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991;40:1007–1012. doi: 10.1038/ki.1991.308. [DOI] [PubMed] [Google Scholar]
- 52.Al-Rasheed NM, Willars GB, Brunskill NJ. C-peptide signals via Galpha i to protect against TNF-alpha-mediated apoptosis of opossum kidney proximal tubular cells. J Am Soc Nephrol. 2006;17:986–995. doi: 10.1681/ASN.2005080797. [DOI] [PubMed] [Google Scholar]
- 53.Johansson BL, Sjoberg S, Wahren J. The influence of human C-peptide on renal function and glucose utilization in type 1 (insulin-dependent) diabetic patients. Diabetologia. 1992;35:121–128. doi: 10.1007/BF00402543. [DOI] [PubMed] [Google Scholar]