

Abstract
The increased burden of chronic kidney and end-stage kidney diseases (ESKD) in populations of African ancestry has been largely unexplained. To identify genetic variants predisposing to idiopathic and HIV-1–associated focal segmental glomerulosclerosis (FSGS), we carried out an admixture-mapping linkage-disequilibrium genome scan on 190 African American individuals with FSGS and 222 controls. We identified a chromosome 22 region with a genome-wide logarithm of the odds (lod) score of 9.2 and a peak lod of 12.4 centered on MYH9, a functional candidate gene expressed in kidney podocytes. Multiple MYH9 SNPs and haplotypes were recessively associated with FSGS, most strongly a haplotype spanning exons 14 through 23 (OR = 5.0, 95% CI = 3.5–7.1; P = 4 × 10−23, n = 852). This association extended to hypertensive ESKD (OR = 2.2, 95% CI = 1.5–3.4; n = 433), but not type 2 diabetic ESKD (n = 476). Genetic variation at the MYH9 locus substantially explains the increased burden of FSGS and hypertensive ESKD among African Americans.
The prevalence of chronic kidney disease (CKD) in the United States is currently estimated at 13% and is associated with significant morbidity and mortality1. Approximately 100,000 Americans develop end-stage kidney (renal) disease (ESKD) each year. The cumulative lifetime risk for ESKD varies by ancestry, and is approximately 7.5% for African Americans and 2.1% for European Americans2. African Americans have a disproportionate risk for several forms of CKD, among them diabetic nephropathy3, hypertensive nephrosclerosis4, lupus nephritis5, focal segmental glomerulosclerosis (FSGS)6 and HIV-associated nephropathy (a distinct form of FSGS, also termed collap–sing glomerulopathy)7,8. The disproportionate risk for CKD may be partially explained by differences in social-economic status, lifestyle factors and clinical factors such as blood pressure control, but most of the increased risk remains unexplained9.
FSGS is a clinical syndrome involving podocyte injury and glomerular scarring, and includes genetic forms with autosomal dominant or recessive mendelian inheritance, reactive forms associated with other illnesses (including HIV-1 disease) or medications, and a sporadic, idiopathic form, which accounts for the majority of cases10. Recent data suggest an increase in the incidence of FSGS, which currently accounts for up to 3% of ESKD cases6. African Americans have a fourfold increased risk for sporadic FSGS11 and an 18- to 50-fold increased risk for HIV-1–associated FSGS7,12. Individuals of African descent also have increased risk for FSGS in other geographic regions, further suggesting that genetic factors contribute to these disparities11.
A strategy for identifying genes underlying such ancestry-driven health disparities is mapping by admixture linkage disequilibrium (MALD). MALD has successfully identified a genomic region associated with prostate cancer13 subsequently replicated by a genome-wide association study14, as well as genes associated with hypertension15, multiple sclerosis16, white blood cell count17 and variable interleukin 6 levels18. The admixture method is most robust for diseases that have large ancestry-driven or racial disparities that are not fully explained by environmental factors and that occur in populations that have undergone recent admixture between ancestral populations, for example, Hispanic Americans and African Americans19,20.
The principle underlying admixture mapping is that, whereas most genetic variants are similar in allele frequencies across ancestral populations, a small subset of genetic variants shows considerable frequency differences, and these variants can be used to estimate local chromosomal ancestry. Recent admixture occurring between genetically distinct populations generates long-range linkage disequilibrium (LD) and chromosomal segments originating from the ancestral populations that can be exploited to map disease genes that are more frequent in one ancestral population relative to the other. For diseases or traits where the predisposing genetic variants occur at sufficiently different frequencies between the ancestral populations, admixture mapping has a distinct advantage over genome wide scans: both theoretically and empirically, considerably fewer informative SNP markers (<2,000) are required for complete genome coverage21.
We hypothesized that given the fourfold increased risk for idiopathic FSGS, the 18- to 50-fold increase in HIV-1–associated FSGS, and the higher rate of many kidney diseases in people of African ancestry, the underlying genetic variants would both be very frequent and have moderate to strong effects in African Americans but would be infrequent in European Americans. Here we present the results of a MALD study to discover genes associated with biopsy-proven, idiopathic FSGS and HIV-1–associated FSGS in 412 African Americans, followed by regional fine mapping to identify specific alleles and a haplotype associated with FSGS. Having identified MYH9 as a functional and positional candidate gene, we tested associated MYH9 SNPs in a larger group of 852 African American FSGS cases and controls. The associated MYH9 SNPs were also tested in a replication group of 346 European American FSGS cases and controls and in an extension group of 717 African American cases and controls with ESKD due to hypertension or type 2 diabetes mellitus.
RESULTS
Overview of MALD results
The MALD scan, consisting of 1,272 SNPs typed on African American FSGS cases (n = 190) and African American controls matched for age, sex and HIV-1 infection status (n = 222) (Table 1), yielded a genome-wide lod score of 9.2 (Fig. 1a,b); scores greater than 2 are considered significant. There was a single prominent peak located on chromosome 22, with a lod score of 12.4 (Fig. 1b), and a pronounced elevation of African ancestry in FSGS cases, with 92% carrying African-inherited chromosomes as compared to a genome average of 81% (Supplementary Fig. 1a online). To determine whether the peak was robust and not subject to fluctuation if a different marker set was used, we divided the markers into even and odd ordered SNPs and tested each set independently: the genome-wide and peak scores for the even set were 8.7 and 10.1, respectively, and 7.7 and 8.8 for the odd set, respectively (Fig. 1b). After additional MALD markers were added to the original scan, the genome-wide and peak scores were 10.5 and 13.7, respectively (Fig. 1b). The 95% credible interval for the MALD peak extended along chromosome 22q13.1 from coordinates 34.423 Mb to 35.656 Mb (Fig. 1b).
Table 1. Study subjects for discovery, replication and extension analysis.
| Analysis Ancestry |
Cases |
Controls |
||
|---|---|---|---|---|
| Type | n (age at diagnosis) | Type | n (age at enrollment) | |
| MALDa and regional mappingb discovery | ||||
| African American | Idiopathic FSGS | 188 (38 ± 14) | Normal donors | 370 (44 ± 8) |
| African American | HIV+ FSGS | 53 (38 ± 8) | Hypernormalc | 241 (44 ± 6) |
| MYH9 confirmation | ||||
| European American | Idiopathic FSGS | 125 (39 ± 18) | Normal donors | 221 (50 ± 9) |
| MYH9 extension | ||||
| African American | Hypertensive ESKD | 241 (54 ± 15) | Normal donorsd | 192 (50 ± 10) |
| African American | Diabetic ESKD | 284 (60 ± 10) | ||
| Totale | 891 | 1,024 | ||
MALD analysis was of 190 African American HIV-negative and HIV-positive FSGS cases randomly selected and matched by HIV-1 status, age and sex to 222 controls.
Regional mapping (Table 2) was done for the same 412 MALD participants with global and local ancestry estimates. All African American FSGS of 241 cases and 611 controls were used in the positional candidate gene interrogation of MYH9.
HIV-1–infected for 8 or more years with normal urine protein to creatinine ratio <0.5.
Shared donors for MYH9 extension geographically matched to hypertensive and diabetic ESRD cases.
Seventeen MYH9 SNPs were typed for 891 cases and 1,024 controls.
Figure 1.

Genome-wide admixture analysis and chromosome 22 gene localization. (a) Lod scores of the case-control statistic (blue) and the locus-genome score (green) across the genome from the ANCESTRYMAP analysis. (The case control statistic was converted to a lod score as described in Methods.) The x axis shows a concatenated set of chromosomes (gray) with locations of the MALD markers interrogated indicated below (black). (b) Lod scores for alternate runs of the ANCESTRYMAP analysis, and the 95% credible interval for the location of a genetic factor responsible for the chromosome 22 MALD peak spans from 34,422,950 to 35,655,902 bp (build 36). The locus-genome statistic, with the 95% credible interval shaded, is shown along with the percent African ancestry in cases and controls.
Fine-mapping association with MYH9
The apex of the MALD peak occurred at the MALD mapping SNP rs735853, located in an intron close to the 3′ end of MYH9 (Fig. 2), which encodes nonmuscle myosin heavy chain IIA. A total of 35 genes were present in the 95% credible interval of the MALD peak (Supplementary Fig. 1b). As MYH9 is expressed in podocytes22, cells essential for glomerular filtration, and as extremely rare codonchanging mutations in MYH9 have been previously associated with familial clusters of glomerulonephritis22, we considered MYH9 the most plausible positional and functional candidate gene. We hypothesized that one or more genetic variations in MYH9, with substantial allele frequency differences between Africans and Europeans, accounted for the chromosome 22 MALD association with FSGS, and we therefore tested additional SNPs in MYH9 for association with FSGS on the 412 subjects genotyped in the MALD scan, for whom chromosomal ancestry data were available.
Figure 2.

MYH9 linkage disequilibrium and associations. Shown (black, below r2 plot) is the 3′ two-thirds of MYH9, comprising exons 2 through 41. The top is a modified Haploview r2 plot for the known 120 SNPs that are polymorphic in YRI. Below in blue are the two known recombination hot spots (blue horizontal bars); SNPs defining and localizing the most highly associated haplotype region, E, along with their susceptible alleles are shown in red. The genomic order of the SNPs is rs7078, rs12107, rs735853, rs5756129, rs5756130, rs11549907, rs875725, rs2187776, rs4821480, rs2032487, rs4821481, rs3752462, rs5756152, rs1557539, rs1005570, rs16996674 and rs16996677. The table shows allele frequencies for African American idiopathic and HIV + FSGS, hypertensive and diabetic (type 2) ESKD, and idiopathic European American FSGS along with controls for each group. Frequencies in boldface were still significant after correction for multiple testing (see Methods) for the dominant or recessive analyses; underlined frequencies are for associations nominally significant (P < 0.05) and supporting (in the same direction as) the African American FSGS associations (Table 4).
We selected 20 MYH9 SNPs by one or more of the following criteria: (i) a pronounced frequency difference between reference West African Yorubans (YRI) and European (CEU) populations using data from the International HapMap Project23, (ii) possible functional significance, or (iii) strong LD with previously typed SNPs whose associations with FSGS remained highly significant after correction for local ancestry (Fig. 2a). As a negative control, we selected SNPs in four neighboring genes with substantially different allele frequencies in Yoruban (YRI) relative to European (CEU) populations23.
By carrying out a test for association of these SNPs with FSGS using a log additive model in a logistic regression, we found many significant associations in MYH9 (Table 2). After correction for local African ancestry, nine SNPs within MYH9 were strongly associated with FSGS (OR = 1.76–2.96; 0.0004 ≥ P ≥ 1 × 10−7) and were below the study significance threshold of P = 0.0005 (applying a Bonferroni correction for 102 tests). Four additional SNPs (rs710181, rs9619601, rs8137674 and rs7285745) had minor allele frequencies <3% in African Americans and were not analyzed further. For the four nearby genes, three SNPs were nominally significant (P < 0.05) after correction for ancestry but without considering the number of tests; the most significant, rs5750175 in RBM9 (P = 0.002), notably had no association (P = 0.4) without correction for ancestry.
Table 2. Regional mapping for FSGS associations with MYH9 and four nearby genes.
| SNP association (local ancestry correction) |
|||||
|---|---|---|---|---|---|
| Uncorrected |
Correcteda |
||||
| SNPb | Genomic position (Mb) | OR (95% CI)c | Pd | OR (95% CI) | P |
|
RBM9
| |||||
| rs5750175 | 34.479 | 0.89 (0.67,1.17) | 0.40 | 0.60 (0.44,0.83) | 0.002 |
| rs5755943 | 34.496 | 1.08 (0.81,1.44) | 0.59 | 0.84 (0.62,1.15) | 0.28 |
|
| |||||
|
MYH9
| |||||
| rs7078 | 35.008 | 2.03 (1.29,3.20) | 0.002 | 1.49 (0.91,2.44) | 0.11 |
| rs12107 | 35.008 | 2.51 (1.35,4.64) | 0.003 | 1.81 (0.93,3.52) | 0.08 |
| rs735853 | 35.009 | 10.75 (3.82,30.3) | 7 × 10−6 | 5.65 (1.89,16.92) | 0.002 |
| rs5756129 | 35.014 | 2.74 (1.74,4.30) | 1 × 10−5 | 2.38 (1.49,3.81) | 0.0003 |
| rs5756130 | 35.014 | 0.49 (0.29,0.82) | 0.007 | 0.43 (0.25,0.74) | 0.002 |
| rs11549907 | 35.015 | 0.55 (0.30,0.98) | 0.04 | 0.49 (0.27,0.89) | 0.02 |
| rs875725 | 35.022 | 0.46 (0.26,0.81) | 0.008 | 0.40 (0.22,0.73) | 0.002 |
| rs2187776 | 35.025 | 1.21 (0.89,1.65) | 0.23 | 1.09 (0.79,1.50) | 0.61 |
| rs4821480 | 35.025 | 4.18 (2.74,6.40) | 4 × 10−11 | 2.96 (1.78,4.94) | 3 × 10−5 |
| rs2032487 | 35.025 | 3.95 (2.66,5.86) | 9 × 10−12 | 2.91 (1.85,4.58) | 4 × 10−6 |
| rs4821481 | 35.026 | 4.13 (2.78,6.12) | 2 × 10−12 | 2.95 (1.81,4.81) | 1 × 10−5 |
| rs3752462 | 35.040 | 3.88 (2.50,6.02) | 1 × 10−9 | 2.75 (1.72,4.40) | 2 × 10−5 |
| rs5756152 | 35.042 | 2.77 (2.04,3.76) | 7 × 10−11 | 2.34 (1.71,3.21) | 1 × 10−7 |
| rs1557539 | 35.044 | 0.34 (0.13,0.92) | 0.03 | 0.41 (0.14,1.19) | 0.10 |
| rs1005570 | 35.045 | 2.85 (2.07,3.92) | 1 × 10−10 | 2.28 (1.63,3.19) | 1 × 10−6 |
| rs16996674 | 35.057 | 2.14 (1.58,2.90) | 9 × 10−7 | 1.76 (1.29,2.42) | 0.0004 |
| rs16996677 | 35.057 | 2.28 (1.69,3.06) | 6 × 10−8 | 1.86 (1.37,2.54) | 8 × 10−5 |
|
| |||||
|
PVALB
| |||||
| rs736721 | 35.533 | 1.58 (1.17,2.14) | 0.003 | 1.49 (1.10,2.04) | 0.01 |
| rs739031 | 35.538 | 1.57 (1.14,2.16) | 0.006 | 1.25 (0.89,1.75) | 0.19 |
| rs2269511 | 35.541 | 1.09 (0.83,1.44) | 0.53 | 0.89 (0.66,1.2) | 0.44 |
|
| |||||
|
TST
| |||||
| rs5756477 | 35.737 | 1.10 (0.82,1.47) | 0.53 | 1.03 (0.76,1.40) | 0.84 |
| rs130598 | 35.738 | 1.28 (0.92,1.79) | 0.14 | 0.97 (0.68,1.40) | 0.88 |
| rs470029 | 35.740 | 1.58 (1.11,2.26) | 0.01 | 1.42 (0.99,2.04) | 0.06 |
|
| |||||
|
TMPRSS6
| |||||
| rs855788 | 35.804 | 2.62 (1.69,4.05) | 2 × 10−5 | 1.65 (0.99,2.74) | 0.05 |
| rs855787 | 35.805 | 1.15 (0.83,1.60) | 0.39 | 1.01 (0.72,1.42) | 0.94 |
| rs2543519 | 35.810 | 1.39 (1.03,1.86) | 0.03 | 1.36 (1.00,1.85) | 0.05 |
| rs1421312 | 35.818 | 1.11 (0.84,1.47) | 0.44 | 0.99 (0.74,1.32) | 0.92 |
SNP association with the estimated local chromosomal ancestry, determined by including that SNP in ANCESTRYMAP, included as a covariate in the logistic regression.
Genomic positions refer to NCBI build 36.
Odds ratio and P from logistic regression under the log additive model, limited to the African American FSGS cases (n = 190) and controls (n = 222) included in the MALD scan.
P values in boldface are less than the Bonferroni correction cutoff of 0.0005.
To determine if the associations of MYH9 SNPs resulted from admixture disequilibrium (typically observed across 5–10 Mb) or from local parental population disequilibrium (typically seen across 50 kb) that can be used for fine mapping, we compared associations for SNP alleles alone with those for SNP alleles plus estimated local chromosomal ancestry. For three MYH9 intron 23 SNPs in strong LD (rs4821480, rs2032487 and rs4821481), 79–83% of the association was attributable to the SNPs alone, with the remaining fraction attributable to chromosomal ancestry (Supplementary Table 1 online). Although these results do not rule out chromosome 22 associations distant from MYH9, they do point to fine-mapping association implicating a functional polymorphism in or close to MYH9.
We also repeated the association analysis in a subgroup of the MALD panel, limited to 245 African American individuals carrying two African chromosomes with certainty >95% at each locus examined, as estimated by ANCESTRYMAP. By restricting the analysis to individuals with only African ancestry at the locus, we largely eliminated the effects of admixture in the association analysis. Supplementary Table 1 presents the analyses done for each of 17 MYH9 SNPs and for the 12 SNPs in the four flanking genes. We found that 6 of 17 MYH9 SNPs retained significant associations (P = 4 × 10−4 to 5 × 10−5) below the Bonferroni P < 0.0005 cut-off for significance, whereas none of the 12 SNPs in nearby genes were significant (P > 0.07).
In an effort to determine the genetic model that best explained the SNP associations, we compared individuals carrying one or two risk alleles to individuals homozygous for the nonrisk allele (12 versus 11 and 22 versus 11, where 2 is the risk allele) and homozygous individuals carrying two risk alleles to individuals heterozygous for the risk allele (22 versus 12), with African ancestry taken as a covariate (Table 3). Using the stringent correction for multiple tests, we found that eight comparisons were significant: five comparing 22 to 12 and three comparing 22 to 11. For the three intron 23 SNPs (rs4821480, rs2032487 and rs4821481), the association was clearly recessive, as the upper confidence interval for the OR for the 12 vs. 11 comparison was close to 1, whereas the ORs for the 22 vs. 12 comparisons ranged from 3.5 to 4.4, with narrow confidence limits (Table 3). In addition, three SNPs (rs5756152, rs1005570 and rs16996677) seemed additive, with ORs for the 22 vs. 11 comparisons ranging from 3.9 to 5.7, again with narrow confidence intervals. As the fine-mapping analysis suggested that the intron 23 SNPs best represent the local association, a recessive model is favored, but without knowledge of the causal sequence variation, choosing a definitive genetic model is premature.
Table 3. MYH9 genotype associations for FSGS for 412 African American cases and controls with ancestry data.
| Alleles |
Genotype frequencya |
12 vs. 11 |
22 vs. 11 |
22 vs.12 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Ref(1) | Risk(2) | 11 | 12 | 22 | OR (95% CI)b | Pc | OR (95% CI) | P | OR (95% CI) | P |
| rs7078 | G | A | 0.023 | 0.255 | 0.721 | 0.69 (0.14,1.52) | 0.64 | 0.89 (0.19,1.95) | 0.88 | 1.81 (1.01,2.43) | 0.05 |
| rs12107 | A | G | 0.010 | 0.190 | 0.800 | ∞ (0,∞)d | 0.99 | ∞ (0,∞) | 0.98 | 1.71 (0.86,2.44) | 0.13 |
| rs735853 | G | C | 0.018 | 0.194 | 0.787 | ∞ (0,∞) | 1.00 | ∞ (0,∞) | 0.98 | 5.48 (1.8,9.69) | 0.003 |
| rs5756129 | C | T | 0.053 | 0.302 | 0.644 | 3.08 (0.36,9.15) | 0.30 | 6.95 (0.81,20.88) | 0.08 | 2.33 (1.39,3.03) | 0.001 |
| rs5756130 | T | C | 0.016 | 0.214 | 0.769 | ∞ (0,∞) | 0.99 | ∞ (0,∞) | 0.98 | 2.17 (1.24,2.90) | 0.007 |
| rs11549907 | A | G | 0.005 | 0.135 | 0.860 | 0.61 (0.03,2.6) | 0.73 | 1.35 (0.08,5.57) | 0.83 | 2.27 (1.19,3.16) | 0.01 |
| rs875725 | C | T | 0.003 | 0.163 | 0.834 | 0 (0,∞) | 0.99 | 0 (0,∞) | 0.99 | 2.86 (1.55,3.92) | 0.0008 |
| rs2187776 | T | C | 0.500 | 0.407 | 0.093 | 1.05 (0.68,1.31) | 0.81 | 1.23 (0.57,1.81) | 0.60 | 1.17 (0.55,1.72) | 0.68 |
| rs4821480 | T | G | 0.130 | 0.394 | 0.476 | 0.61 (0.18,1.13) | 0.42 | 3.29 (0.8,6.76) | 0.10 | 4.41 (2.47,5.94) | 5.7 × 10−7 |
| rs2032487 | T | C | 0.134 | 0.454 | 0.412 | 0.78 (0.24,1.43) | 0.69 | 3.82 (0.95,7.76) | 0.06 | 3.62 (2.18,4.68) | 6.0 × 10−7 |
| rs4821481 | T | C | 0.135 | 0.476 | 0.389 | 0.77 (0.23,1.41) | 0.66 | 3.19 (0.59,7.53) | 0.18 | 3.5 (2.05,4.59) | 4.1 × 10−6 |
| rs3752462 | C | T | 0.064 | 0.421 | 0.514 | 1.07 (0.21,2.45) | 0.94 | 2 (0.36,4.82) | 0.43 | 3.09 (1.87,4) | 1.2 × 10−5 |
| rs5756152 | G | A | 0.537 | 0.397 | 0.066 | 2.32 (1.44,2.95) | 0.0006 | 5.71 (2.95,8) | 2.3 × 10−7 | 2.36 (1.26,3.26) | 0.008 |
| rs1557539 | C | G | 0.003 | 0.056 | 0.941 | ∞ (0,∞) | 0.99 | ∞ (0,∞) | 0.98 | 2.39 (0.83,4.1) | 0.11 |
| rs1005570 | G | A | 0.323 | 0.497 | 0.180 | 1.71 (0.91,2.36) | 0.09 | 5.12 (2.49,7.4) | 8.8 × 10−6 | 2.69 (1.65,3.46) | 7.1 × 10−5 |
| rs16996674 | C | T | 0.571 | 0.370 | 0.058 | 1.54 (0.98,1.94) | 0.06 | 3.65 (1.82,5.21) | 0.0003 | 2.17 (1.09,3.09) | 0.03 |
| rs16996677 | G | A | 0.543 | 0.381 | 0.076 | 1.59 (1,2.02) | 0.05 | 3.94 (2.04,5.52) | 4.6 × 10−5 | 2.35 (1.24,3.24) | 0.008 |
Association analysis for combined idiopathic and HIV-associated FSGS using MALD study subjects (190 cases and 222 controls).
Population frequencies for normal controls in European Americans (n = 221) and African Americans (n = 370).
Odds ratios, confidence intervals and probabilities calculated by logistic regression controlling for African chromosomal ancestry.
P values in boldface are less than the Bonferroni correction cutoff of 0.0005.
Odds ratios are shown as infinite (∞) for logistic regression results divergent due to limited numbers.
MYH9 associations with idiopathic and HIV-1–associated FSGS
A larger set of African American FSGS cases (n = 241) and controls (n = 611), which includes the group used in the MALD scan along with additional FSGS cases, supported the initial MYH9 associations presented in Tables 2 and 3. Because local ancestry was available for only the MALD subset, the results could not be corrected for local ancestry. Of the 17 SNPs typed, all but five showed association with FSGS, with seven having P < 10−12 (Supplementary Table 2 online). For the seven most highly associated SNPs, the susceptible allele was very frequent both in African Americans (29–91%) (Supplementary Table 2) and in Yorubans (40–97%, data from the International HapMap Project)23, but much less frequent (3–52%) in European Americans (Supplementary Table 2). The strongest associations were for the three SNPs located within intron 23 (OR = 4.5–4.8, P = 2 × 10−18–1 × 10−20) for the recessive model, presented in Figure 3 along with the genotype distribution for African- and European-American cases and controls.
Figure 3.
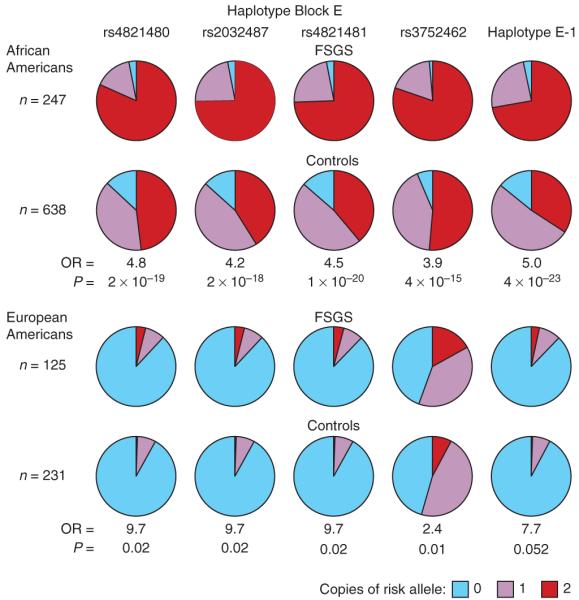
Frequencies of the three genotypes for the MYH9 SNPs rs4821480, rs2032487, rs4821481 and rs3752462, and haplotype E-1, the most frequent haplotype containing the four SNPs, in African American and European American FSGS cases and controls. Odds ratios and P values for the recessive model comparing FSGS cases and controls are shown.
The 17 MYH9 SNPs were also analyzed in the idiopathic FSGS and the HIV-1–associated FSGS subgroups, representing syndromes with distinct pathological features (Table 4). The pattern of MYH9 SNP associations is similar for both forms of FSGS in African Americans, showing consistent associations with FSGS (OR = 3.5–6.7) and significance below P = 0.0005 for 11 SNPs for idiopathic FSGS and 8 SNPs for HIV-1–associated FSGS for the recessive model. Results for the dominant genetic model are presented in Supplementary Table 3 online, showing that the significance and strength of the associations are weaker or absent in the dominant model.
Table 4. MYH9 associations for idiopathic FSGS, HIV-1-associated FSGS and hypertensive ESKD under a recessive model.
| African Americans |
European Americans |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Risk allele percenta |
Idiopathic FSGS (n = 587) |
HIV-associated FSGS (n = 298) |
Hypertensive ESKD (n = 433)b |
Idiopathic FSGS (n = 356) |
||||||
| SNP | EA | AA | OR | PFETc | OR | PFET | OR | PFET | OR | PFET |
| rs7078 | 72.9 | 85.2 | 2.12 (1.31,3.51) | 0.001 | 6.13 (1.88,31.8) | 0.0005 d | NTe | NT | 0.97 (0.62,1.54) | 0.91 |
| rs12107 | 85.3 | 89.1 | 2.81 (1.56,5.34) | 0.0002 | 10.7 (1.73,442) | 0.002 | 1.59 (0.9,2.83) | 0.10 | 1.24 (0.73,2.12) | 0.45 |
| rs735853 | 51.9 | 88.3 | 9.79 (3.91,31.6) | 2 × 10−10 | 6.55 (1.62,57.4) | 0.002 | 1.25 (0.74,2.11) | 0.38 | 1.23 (0.72,2.08) | 0.44 |
| rs5756129 | 77.9 | 79.3 | 2.32 (1.49,3.67) | 7 × 10−5 | 6.62 (2.3,26.2) | 2 × 10−5 | 2.18 (1.38,3.48) | 0.0006 | 0.68 (0.42,1.08) | 0.09 |
| rs5756130 | 97.6 | 87.5 | 0 (0,1.4) | 0.01 | 0 (0,11.3) | 0.01 | 2.45 (1.38,4.42) | 0.001 | 0.4 (0.16,0.98) | 0.03 |
| rs11549907 | 98.5 | 93.7 | 1.03 (0.02,19.94) | 0.78 | 0 (0,180) | 0.02 | NT | NT | 0.94 (0.23,4.45) | 1 |
| rs875725 | 98.5 | 92.8 | 1.01 (0.02,19.44) | 0.28 | 4.03 (1.22,21.1) | 0.01 | 1.49 (0.83,2.69) | 0.16 | 1.26 (0.28,7.67) | 1 |
| rs2187776 | 3.1 | 30.1 | 1.07 (0.58,1.94) | 0.88 | 1.71 (0.52,4.86) | 0.27 | 1.47 (0.77,2.9) | 0.23 | 1.7 (0.02,134) | 1 |
| rs4821480 | 4.3 | 67.4 | 4.54 (2.92,7.19) | 1 × 10−13 | 5.28 (2.4,12.9) | 2 × 10−6 | 1.89 (1.25,2.87) | 0.002 | 9.69 (1.07,461) | 0.02 |
| rs2032487 | 4.3 | 63.7 | 3.69 (2.47,5.55) | 7 × 10−12 | 6.67 (3.01,16.4) | 8 × 10−8 | NT | NT | 9.69 (1.07,461) | 0.02 |
| rs4821481 | 4.3 | 62.3 | 4.09 (2.74,6.18) | 1 × 10−13 | 6.55 (3.05,15.4) | 5 × 10−8 | 2.21 (1.47,3.33) | 7 × 10−5 | 9.73 (1.07,463) | 0.02 |
| rs3752462 | 31.0 | 72.0 | 3.49 (2.29,5.41) | 4 × 10−10 | 5.9 (2.5,16.2) | 3 × 10−6 | 1.75 (1.15,2.65) | 0.006 | 2.42 (1.17,5.04) | 0.01 |
| rs5756152 | 2.6 | 26.1 | 5.62 (3.16,10.3) | 2 × 10−10 | 4.22 (1.79,9.8) | 0.0004 | 3.31 (1.65,7.13) | 0.0002 | 0 (0,71.78) | 1 |
| rs1557539 | 99.6 | 97.5 | 0 (0,78.98) | 0.65 | 0 (0,179) | 0.22 | 0.88 (0.28,2.62) | 1.00 | 0.36 (0.03,3.16) | 0.35 |
| rs1005570 | 7.9 | 41.8 | 3.48 (2.3,5.3) | 7 × 10−10 | 5.2 (2.63,10.4) | 5 × 10−7 | 2.11 (1.33,3.38) | 0.0008 | 0 (0,72.7) | 1 |
| rs16996674 | <1 | 23.5 | 4.59 (2.48,8.76) | 1 × 10−7 | 3.3 (1.25,8.37) | 0.01 | 2.63 (1.32,5.57) | 0.004 | – f | – |
| rs16996677 | <1 | 26.2 | 4.08 (2.37,7.14) | 7 × 10−8 | 3.47 (1.42,8.23) | 0.005 | 2.95 (1.53,6.03) | 0.0006 | – | – |
| Haplotype percent | ||||||||||
| E-1 | 4.1 | 59.7 | 4.65 (3.11,7.02) | 9 × 10−16 | 5.92 (2.89,12.85) | 7 × 10−8 | 2.24 (1.48,3.43) | 7 × 10−5 | 7.66 (0.75,380.02) | 0.052 |
| E-2g | 69.0 | 20.5 | 0.24 (0.13,0.4) | 1 × 10−9 | 0.12 (0.02,0.4) | 3 × 10−5 | 0.63 (0.39,1.01) | 0.05 | 0.39 (0.19,0.8) | 0.008 |
| E-3 | 26.9 | 11.9 | 0.45 (0.24,0.78) | 0.003 | 0.42 (0.14,1.07) | 0.06 | 0.57 (0.35,0.94) | 0.02 | 0.99 (0.62,1.57) | 1 |
| E-4 | <1 | 4.0 | 0.88 (0.41,1.8) | 0.87 | 0.26 (0.01,1.71) | 0.21 | 0.35 (0.14,0.79) | 0.009 | – | – |
| E-5 | <1 | 3.2 | 0.42 (0.12,1.14) | 0.10 | 0.75 (0.14,2.72) | 1 | 0.97 (0.39,2.47) | 1 | – | – |
Results shown for cases and controls for African American idiopathic FSGS (188 cases, 370 controls), HIV-associated FSGS (53 cases, 241 controls), Hypertensive ESKD (288 cases, 192 controls) and European American FSGS (284 cases,192 controls).
Frequencies from controls.
Hypertensive ESKD and European Americans are replication cohorts.
Probabilities are calculated with Fisher’s exact test.
Comparisons significant after correction for multiple comparisons (see Methods) are shown in boldface, as are comparisons for ESKD or European American FSGS confirmatory of highly significant associations in African American FSGS.
Not typed.
Comparisons where a rare genotype or haplotype was absent in both cases and controls are shown with a dash (–).
For haplotypes E-2 to E-5, the dominant effect of the haplotype is shown (corresponding to taking absence of the protective haplotype as a recessive susceptible factor).
MYH9 haplotype associations
We analyzed inferred haplotypes for MYH9 SNPs, both within strictly defined local haplotype blocks (Supplementary Table 4a online) and within larger regions of elevated LD that are bounded by recombination hot spots23 (Supplementary Table 4b), for combined idiopathic and HIV-1–associated FSGS associations. The strongest risk association with FSGS (OR = 5.0, 95% CI = 3.5–7.1 and P = 4 × 10−23, recessive model) was for the most frequent haplotype (E-1) spanning a region indicated in Figure 2 comprising the three intron 23 SNPs and rs3752462 (Fig. 3 and Supplementary Table 2). The recessive E-1 haplotype association was also observed for idiopathic FSGS (OR = 4.7, 95% CI = 3.1–7.0, P = 9 × 10−16) and HIV-1–associated FSGS (OR = 5.9, 95% CI = 2.9–12.9, P = 7 × 10−8) as shown in Table 4. The E-1 haplotype has a frequency of 60% in African Americans but only 4% in European Americans.
MYH9 FSGS associations in European Americans
The study population also contained 125 European Americans with idiopathic, biopsy-confirmed FSGS and 221 European American normal donor controls, constituting an independent FSGS group for replication of the findings obtained in African Americans (Table 1). Testing a recessive model for SNP and haplotype FSGS associations for this group, uncorrected for population structure, we found that the four SNPs determining haplotype E-1 showed modestly significant associations, although the haplotype E-1 itself fell short of significance (Table 4). The haplotype most frequent (69%) in European Americans, E-2, showed a significant dominant protective effect (OR = 0.39, 95% CI = 0.19–0.80, P = 0.008), consistent with its effect in African Americans (Table 4). We considered whether FSGS risk factors rare in European Americans were associated with African ancestry in our self-identified European American subjects. Analysis of population substructure principal components using EIGENSOFT24 indicated that three of the four FSGS cases carrying two copies of haplotype E-1 had significant African ancestry. Eliminating these three individuals, and correcting for overall population substructure using the first EIGENSOFT eigenvector calculated from SNPs outside of chromosome 22, we found that the association with haplotype E-2 remained nominally significant (OR = 0.47, 95% CI = 0.23–0.97, P = 0.04), but none of the associations were significant (P > 0.07). Although African ancestry confounds the analysis of European American FSGS, the data support a replication of the FSGS signal in this independent group.
Extension to hypertensive ESKD
To determine whether MYH9 might harbor risk factors for ESKD attributed to hypertensive nephrosclerosis or type 2 diabetic nephrosclerosis in the African American population, we analyzed 14 MYH9 SNPs for association with hypertensive ESKD (n = 241) or type 2 diabetes–associated ESKD (n = 284) using a geographically matched control group (n = 192) (Table 1). Hypertensive ESKD associations (Table 4) were significant and consistent in direction with the FSGS associations for nine MYH9 SNPs and haplotype E-1. The strongest associations were for rs4821481 (OR = 2.2; 95% CI = 1.5–3.5, P = 7 × 10−5); the association for haplotype E-1 was virtually identical. SNP associations with hypertensive ESKD were independent of individual overall proportion of African ancestry (analysis not shown). No significant associations were observed for SNPs with type 2 diabetic ESKD (Supplementary Table 5 online).
We considered whether the MYH9 associations with FSGS might result from a specific association with ESKD due to FSGS. In a survival analysis of 161 African American FSGS cases, haplotype E-1 showed no association with progression to ESKD (relative hazard = 1.03, 95% CI = 0.55–1.94; P = 0.92, recessive model) (Supplementary Fig. 2 online).
DISCUSSION
The podocytopathies, including FSGS and collapsing glomerulopathy, involve genetic and acquired forms of podocyte injury10, and podocyte injury is proposed to play a central role in the pathogenesis of other progressive kidney diseases, including diabetic and hypertensive nephrosclerosis25. Over ten genes have been associated with FSGS; most are associated with disease forms with mendelian inheritance or with evidence for new mutations26. With regard to sporadic, non-familial FSGS, associations have been identified with WT1 and NPHS2 (refs. 27,28), but polymorphisms in these genes explain only a small portion of the disease burden.
We have used MALD to identify genetic variation in MYH9 as an important contributing factor to the development of idiopathic FSGS, HIV-associated FSGS (collapsing glomerulopathy) and hypertensive ESKD. The MALD scan revealed a single peak centered on MYH9 on chromosome 22q13.1 that showed strong association of African chromosomal ancestry with FSGS in African Americans. Fine mapping and functional considerations further implicated MYH9. MYH9 polymorphism does not differentially affect progression from FSGS to ESKD, indicating that the MYH9 association with FSGS is a general association for this disease, and is not limited to ESKD due to FSGS.
An independent study29 of an African American population composed of both diabetic and nondiabetic individuals with ESKD also found a MALD association with a similar, overlapping region of chromosome 22. As this study found a very similar peak on chromosome 22, and had overlapping collaborators with our study, we shared our finding of a strong association of MYH9 variants with FSGS. Kao et al. explain the admixture signal with the three MYH9 intron 23 SNPs that had the strongest FSGS associations in our study, and replicate our findings of an association of nondiabetic ESKD and the MYH9 locus29.
MYH9 risk alleles are more frequent among African Americans, and protective alleles are more frequent among European Americans (Fig. 3), providing a genetic explanation for the disparities in risk for FSGS and hypertensive ESKD in these populations.
Nonmuscle myosin, like muscle myosin, is assembled from separately coded heavy and light chains, and binds to actin to perform intracellular motor functions30. Mutations in MYH9 have been associated with four autosomal dominant clinical syndromes: May-Hegglin, Sebastian, Fechtner and Epstein31. All four syndromes involve autosomal dominant macrothrombocytopenia, with the variable accompaniment of sensorineural deafness, cataracts, neutrophil Döhle-like bodies and glomerular disease31. There are few histologic descriptions of MYH9-associated glomerular disease. One individual with a MYH9 mutation, undergoing kidney biopsy early in the disease course, had normal light microscopy and electron microscopic evidence of focal podocyte foot process effacement and loss of podocyte slit diaphragms, indicating podocyte injury32. The Myh9 knockout mouse shows embryonic lethality, whereas heterozgyous mice have apparently normal phenotype other than hearing loss with incomplete penetrance33. Myosin IIA has been localized to podocytes and possibly mesangial cells22,32. Other mutations affecting podocyte proteins that interact with the actin cytoskeleton, including alpha-actinin-4 (ref. 34), CD2-associated protein35 and synaptopodin36, have been associated with podocyte injury and FSGS in humans or experimental animals, suggesting the requirement for an intact actin cytoskeleton to maintain normal podocyte cytoarchitecture and filtration barrier function.
Our extension samples revealed that MYH9 was also associated with the common clinical syndrome of hypertensive nephrosclerosis in African Americans, but not with diabetic nephropathy. Individuals with hypertensive ESKD typically present with advanced nephropathy and secondarily elevated blood pressure with resultant left ventricular hypertrophy and retinal vascular changes. Some of these individuals may have occult glomerular diseases, often FSGS or global glomerulosclerosis presenting in non-nephrotic forms, as they do not typically undergo renal biopsy37,38. Renal biopsy studies typically reveal the presence of focal or global glomerulosclerosis in clinically diagnosed hypertensive nephrosclerosis39,40. Notably, the renal microvasculature changes that are typically attributed to hypertension fail to correlate with measured blood pressure, suggesting that factors other than hypertension caused nephropathy. In practice, cases are not often held to the strict diagnostic clinical criteria as in these reports, and hypertensive nephrosclerosis is a diagnosis of exclusion in many nondiabetic forms of nephropathy. It is possible that undiagnosed FSGS in those labeled with hypertensive nephrosclerosis contributed to the association, and studies involving biopsy-proven hypertensive nephrosclerosis are needed to clarify this issue. Alternatively, individuals with hypertension and with particular MYH9 alleles may be more susceptible to podocyte injury and progressive glomerular disease. The lack of demonstrated association of MYH9 with diabetic ESKD suggests that the underlying mechanisms leading to impaired kidney function in diabetics may be distinct from those of FSGS or hypertensive ESKD. It is possible that the podocyte is not the primary target of the metabolic abnormalities that are believed to cause diabetic nephropathy, or alternatively that those abnormalities are quantitatively much more important than MYH9 variation. Nevertheless, because of the disease burden, it should be determined whether MYH9 variation influences the rate of progressive kidney function loss in diabetic kidney disease.
Genetic studies of glomerular diseases such as FSGS are limited by the number of subjects available—typically hundreds rather than the thousands recommended for genome-wide association studies. Small sample sizes are generally unworkable for genome-wide association studies because of the need for statistical power to overcome correcting for the current 1,000,000 SNPs typed, and because the associations observed generally have modest odds ratios. However, there is a strong argument to be made for using admixture mapping for gene discovery in diseases such as FSGS with large ancestry-driven disparities that are not fully explained by non-genetic factors, because the gene or genes underpinning the disease may have strong effects and/or very frequent alleles, as observed in this study, and because the smaller number of markers needed simplifies the statistical issues.
A limitation of our study is that we have not yet identified the causal sequence variation in MYH9 that is associated with FSGS. Initial resequencing of the 40 exons and intron-exon junctions of MYH9 in 14 African Americans with FSGS and haplotype E-1 showed no obvious causal variation (data not shown). We hypothesize that the functional variation occurs in regulatory elements or splice-site determinants, and that these influence RNA expression or protein structure in podocytes. Further studies are needed to identify causal variants and to provide an understanding of what additional genetic or environmental factors must interact with MYH9 variants to cause podocyte injury leading to kidney failure.
Recently, results of genome-wide scans and MALD scans have been reported with generally modest odds ratios for the genotype-phenotype associations. These weak effects have been attributed to the complex nature of the diseases involving the interaction of multiple genetic and environmental factors. The present study using a MALD approach has securely identified a main effect gene that is involved in two histologically distinct forms of FSGS: idiopathic FSGS, which is characterized by depletion of podocytes, and HIV-1–associated FSGS (collapsing glomerulopathy), which is characterized by podocyte proliferation. Extrapolating to population data from our case-control study, the fractions of sporadic or HIV-associated FSGS among African Americans that can be explained41 by this haplotype are 4.7% and 12%, respectively. The large frequency difference for haplotype E-1 (60% in African Americans versus 4% in European Americans) combined with the extremely high FSGS susceptibility conferred by this factor (OR = 5.0 for carriers, 95% CI = 3.5–7.1, recessive model; Supplementary Table 2) provides a plausible cause for the higher incidence of FSGS, HIV-associated nephropathy and hypertensive nephrosclerosis among African Americans. For individuals not carrying haplotype E-1, the extrapolated population frequency of sporadic FSGS is very similar between African Americans (0.06%; 95% CI = 0.03–0.1%) and European Americans (0.05%; 95% CI = 0.04–0.06%), suggesting that the two populations have comparable background levels of susceptibility to FSGS in the absence of MYH9 genetic susceptibility factors, and that MYH9 factors may account for 66–100% of the disparity in susceptibility to FSGS. The identification of MYH9 genetic variation as a risk factor for FSGS and hypertensive ESKD underscores the importance of the actin cytoskeleton in podocyte biology and as a potential pharmacologic target.
METHODS
Human subjects
Kidney biopsy-confirmed idiopathic FSGS or HIV-1–associated FSGS cases were enrolled from 22 academic medical centers in the United States as part of the US National Institutes of Health (NIH) FSGS Genetic Study27,28, see Supplementary Note. The study enrolled 366 subjects with idiopathic or HIV-1–associated FSGS and 832 control subjects without known kidney disease. Cases with one or more family members with FSGS were excluded. Table 1 lists the number of participants included in the present study, self-reported ancestry, disease diagnosis and date of diagnosis for cases and date of enrollment for controls.
The FSGS study population (Table 1) consisted of three case groups: (i) idiopathic FSGS among African Americans (n = 188) without a family history of kidney disease, with biopsy-proven FSGS; (ii) HIV-1–associated FSGS among African Americans (n = 53) with HIV-1 infection and with kidney biopsies that were consistent with HIV-1–associated nephropathy, with collapse of glomerular capillaries and podocyte hyperplasia; (iii) idiopathic FSGS among European Americans with sporadic, biopsy-proven FSGS. Age at kidney biopsy, available for all cases, was taken as a proxy for age of FSGS onset. The mean age of onset of FSGS for European Americans was 39 ± 18 years, for African Americans the mean age of onset for HIV-1–associated FSGS was 38 ± 8 years and for idiopathic FSGS the mean age of onset was 34 ± 14 years.
Three groups of FSGS controls are listed in Table 1. African American normal controls (n = 370) are blood donors lacking a history of kidney disease from the NIH Clinical Center in Bethesda, Maryland and HIV-seronegative subjects enrolled in the AIDS Link to the Intravenous Drug Experience (ALIVE) cohort from Baltimore, Maryland. The mean age at enrollment was 44 ± 8 years. HIV-1–infected hypernormal controls (n = 241) are African Americans enrolled in the ALIVE cohort. The absence of kidney disease was defined as having normal serum creatinine (≥1.4 mg/dl) and lack of proteinuria (urine protein to creatinine ratio <0.5). This group represents a hypernormal sample, in that they are drawn from the African American population at risk for HIV-1–associated FSGS, have been HIV-1 infected for at least 8 years, and yet lack evidence of kidney disease. The mean age at enrollment was 44 ± 6 years. European American controls (n = 221) are normal blood donors lacking a history of kidney disease from the NIH Clinical Center in Bethesda, Maryland and the NCI-Frederick Blood Donor Program in Frederick, Maryland. The mean age at enrollment was 50 ± 9 years.
The extension cohort (Table 1) comprised unrelated African Americans with ESKD attributed to nondiabetic hypertension (n = 241) or type 2 diabetes (n = 284) and African American shared control subjects (n 192) without kidney disease from the southeastern United States, using previously reported diagnostic criteria42,43. For hypertensive ESKD, the mean age in years at enrollment was 56 ± 14 years and age of onset of ESKD was 54 ± 15 years. For diabetic ESKD, the mean age in years at enrollment was 64 ± 10 years, age at diagnosis for type 2 diabetes was 43 ± 11 years, and age of onset of ESKD was 60 ± 10 years. The shared controls were geographically matched with no signs or symptoms of kidney disease; the mean age in years at enrollment was 50 ± 10 years.
Institutional review boards at each collaborating medical center approved study protocols and each subject provided informed consent to participate. Additional information about the study subjects is provided in the Supplementary Note online.
Genotyping
The ParAllele platform (Affymetrix) was used to genotype 412 African American FSGS cases and controls for the MALD scan. The genotyping interrogated 1,331 highly informative MALD markers not known to be in LD in ancestral populations from the map that we previously published19. Of these, 59 were excluded from analysis because the controls either did not fit Hardy-Weinberg expectations or because the genotypes were inconsistent with estimates of ancestral population allele frequencies (both at P < 0.001). Ancestral population allele counts for admixture analysis were compiled from our MALD map construction data19, other studies13 and HapMap data44. Follow-up genotyping in the region of the chromosome 22 peak was done for the 412 MALD samples and MYH9 genotyping was conducted on all study participants (Table 1) using Taqman assays (Applied Biosystems). Duplicate samples (10%) were distributed across plates to assess for consistency between genotype calls. Plates with less than 95% complete call rate or with contaminated water controls were repeated. Genomic positions of SNPs are from NCBI build 36.
MALD genome-scan analysis
The ANCESTRYMAP program was used to scan the genome for regions of African ancestry that differ significantly from the genome average in cases and from the same locus in controls20. From current estimates of FSGS prevalence in African Americans and European Americans7,11, and the prevalence of HIV-1 infection in the study populations, we assumed an eightfold increase in the risk of FSGS in individuals of African ancestry (carrying two African chromosomes) compared to those of European ancestry. Under the log-additive model used by ANCESTRYMAP, this implies an approximately 2.8-fold increase in risk per African chromosome that was used in the analyses presented. Additional analyses considered alternate risk models ranging from 0.25- to 4-fold risk per African chromosome.
We assessed significance by lod scores reported by ANCESTRYMAP. The program was used with 100 burn-in and 200 follow-on iterations for all Markov chain–Monte Carlo runs as recommended20. Locus-genome lod scores were calculated on the basis of a comparison of the estimated percent ancestry at a locus and the genome-wide ancestry average of the cases. We converted the case-control T statistic to a lod score by calculating a P value assuming approximate normality due to the large number of degrees of freedom, and using lod = log10(P /(1 – P)). We also calculated a lod score for genome-wide significance; a score greater than 2.0 was considered significant45. The 95% credible interval for the location of the FSGS disease gene was derived from the locus-genome lod score–based relative probability distribution18. Calculations for the 95% credible interval were made using the maldmap package in R version 2.6.1.
Distinguishing short-range (fine mapping) from long-range (admixture-based) associations
We carried out three logistic regression analyses for each SNP in MYH9 or in nearby genes to separate the long-range effects of admixture linkage disequilibrium (5–10 cM) from the short-range (typically 10–50 kb) effects of local haplotype structure in the ancestral populations. The first two regressions used the number of African chromosomes carried by each individual at the given SNP, estimated by ANCESTRYMAP, as a regression covariate. The first analysis (Table 2) tested the strength of the genotype association for each SNP, comparing a model with the SNP alone (number of susceptible alleles carried) with a model including both the SNP and the ancestry (estimated number of copies of African chromosomes carried). In a second related analysis, we estimated the fraction of the associations due to chromosomal African ancestry by comparing the reduction of sample deviance in the model containing both the SNP genotype and the estimate of chromosomal ancestry, compared to the model containing the SNP genotypes alone18. A third regression analyzed the SNP effect alone, on a subset of individuals carrying two African chromosome segments at the tested locus with probability greater than 95%, based on the ANCESTRYMAP estimate. All three logistic regressions (function glm in R) used an additive model.
Adjusting for population substructure
We previously reported no evidence of population substructure among FSGS cases and controls for the African American subjects30. For 346 European American FSGS cases and controls, we used the EIGENSOFT26 program to test and correct for population stratification, using 11 markers on chromosome 22 outside of MYH9 and 88 markers on other chromosomes24. The first principal component captured most of the population variation, and, most relevantly, captured most of the variation differentiating African Americans and European Americans when both were included in the analysis; this variation was well resolved whether all SNPs, or only non–chromosome 22 SNPs, were used to generate the EIGENSOFT eigenvectors. To adjust for overall population substructure, as opposed to local chromosomal ancestry, we used each individual’s component of the first eigenvector calculated by EIGENSOFT from non–chromosome 22 SNPs as a covariate in logistic regression with explanatory SNP and haplotype variables. For 717 cases and controls in the ESKD replication and extension study, we estimated the overall admixture fractions of the individual subjects with frappe46 using 67 ancestry-informative markers, and tested whether admixture fraction contributed significantly to MYH9 associations with ESKD by including this estimate as a logistic regression covariate along with the SNP and haplotype variables.
Statistical analysis of SNP and haplotype associations
We tested for phenotype-genotype associations using dominant, recessive and additive models. P values were determined by Fisher’s exact test (function fisher.test in R); all tests are two-sided. To extrapolate from our case-control data to population data for estimation of the attributable fraction (AF) and explained fraction (EF)41, we assumed an FSGS prevalence of pAA = 0.2% for HIV-1–uninfected African Americans, and pEA = 0.05% for HIV-1–uninfected European Americans6; we assumed the 10% prevalence of FSGS among HIV-1–infected subjects that prevailed before effective antiretroviral therapy, as our HIV-1–infected cases and controls were collected in this era7. The estimate of the percent of difference in susceptibility, Δ, to FSGS explained by haplotype E-1 is based on the estimate prevalence p00 of FSGS in African Americans not carrying haplotype E-1, using Δ ≈ (pAA – p00)/(pAA – pEA).
For survival analysis for progression from FSGS to ESKD we used the R functions survfit and coxph. Time to ESKD was taken from the onset of kidney disease; where an onset date was not available the biopsy date was used.
Haplotype inference
We inferred haplotypes both within local haplotype blocks defined by the confidence interval method47, and within longer regions of LD bounded by defined recombination hot spots48. Haplotypes were inferred by an in-house implementation of the expectation-maximization (EM) algorithm49, which measures reliability of inferences as the uncertainty (calculated as entropy) of bootstrap-replicated haplotype inferences. Genetic associations with inferred haplotypes were confirmed with haplotypes inferred by PHASE 2.1 (ref. 50).
Corrections for multiple comparisons
For African American FSGS SNP associations, our analysis involved 102 MYH9 comparisons (17 SNPs examined for 6 genetic models); thus, using a Bonferroni correction that is conservative in that it ignores the non-independence of the tests, we considered P < 0.0005 to be significant after correction. We show nominal P estimates in the text and tables. We considered African American haplotypes for HapMap blocks, maximal robustly inferred blocks, and all robustly inferred blocks containing the highly associated intron 23 SNPs, for a total of 75 haplotypes considered for three genetic models (225 comparisons); thus, for African American haplotype associations, we considered P < 0.0002 to be significant (Table 3). For the ESKD and European American FSGS replication tests (Table 4), we considered P < 0.05 to be significant for associations confirming African American FSGS associations that were significant after correction.
Supplementary Material
ACKNOWLEDGMENTS
We thank the subjects and controls who participated in the NIH Genetic Study and the Wake Forest ESRD Study, and the physicians who assisted by identifying cases for these studies. M. Branton, D. Smith, L. Bartlett and L. Howard provided clinical support. E. Binns, Y. Zhou and M. Jamba provided laboratory assistance. The following members of the NIH FSGS Genetics Study Group also contributed samples: F. Hutchinson (University of South Carolina); R. Mannon (Duke University); P. Nachman (University of North Carolina, Chapel Hill); T.K. Rao (State University of New York, Downstate) and M. Smith (Cincinnati Children’s Hospital). Research blood donor samples were provided by S. Leitman (NIH). We thank S.J. O’Brien and B. Gold for discussions, N. Patterson for assistance with the ANCESTRYMAP program, and S. Leitman for assistance in identifying healthy volunteers.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US government. The publisher or recipient acknowledges right of the US government to retain a nonexclusive, royalty-free license in and to any copyright covering the article.
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400, the Intramural Research Programs of the National Institute for Diabetes, Digestive, and Kidney Diseases (ZO-1 DK043308), and by a grant from the NIH (RO1 DK 070941 (B.I.F.)).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
Note: Supplementary information is available on the Nature Genetics website.
References
- 1.Coresh J, et al. Prevalence of chronic kidney disease in the United States. J. Am. Med. Assoc. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 2.Kiberd BA, Clase CM. Cumulative risk for developing end-stage renal disease in the US population. J. Am. Soc. Nephrol. 2002;13:1635–1644. doi: 10.1097/01.asn.0000014251.87778.01. [DOI] [PubMed] [Google Scholar]
- 3.Cowie CC, et al. Disparities in incidence of diabetic end-stage renal disease according to race and type of diabetes. N. Engl. J. Med. 1989;321:1074–1079. doi: 10.1056/NEJM198910193211603. [DOI] [PubMed] [Google Scholar]
- 4.Toto RD. Proteinuria and hypertensive nephrosclerosis in African Americans. Kidney Int. Suppl. 2004;66:S102–S104. doi: 10.1111/j.1523-1755.2004.09224.x. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez M, et al. A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum. 2007;57:576–584. doi: 10.1002/art.22672. [DOI] [PubMed] [Google Scholar]
- 6.Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 2004;44:815–825. [PubMed] [Google Scholar]
- 7.Kopp JB, Winkler C. HIV-associated nephropathy in African Americans. Kidney Int. Suppl. 2003;63:S43–S49. doi: 10.1046/j.1523-1755.63.s83.10.x. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz EJ, Klotman PE. Pathogenesis of human immunodeficiency virus (HIV)-associated nephropathy. Semin. Nephrol. 1998;18:436–445. [PubMed] [Google Scholar]
- 9.Powe NR. To have and have not: health and health care disparities in chronic kidney disease. Kidney Int. 2003;64:763–772. doi: 10.1046/j.1523-1755.2003.00138.x. [DOI] [PubMed] [Google Scholar]
- 10.Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin. J. Am. Soc. Nephrol. 2007;2:529–542. doi: 10.2215/CJN.04121206. [DOI] [PubMed] [Google Scholar]
- 11.Kitiyakara C, Kopp JB, Eggers P. Trends in the epidemiology of focal segmental glomerulosclerosis. Semin. Nephrol. 2003;23:172–182. doi: 10.1053/snep.2003.50025. [DOI] [PubMed] [Google Scholar]
- 12.Eggers PW, Kimmel PL. Is there an epidemic of HIV infection in the US ESRD program? J. Am. Soc. Nephrol. 2004;15:2477–2485. doi: 10.1097/01.ASN.0000138546.53152.A7. [DOI] [PubMed] [Google Scholar]
- 13.Freedman ML, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. USA. 2006;103:14068–14073. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeager M, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, et al. Admixture mapping for hypertension loci with genome-scan markers. Nat. Genet. 2005;37:177–181. doi: 10.1038/ng1510. [DOI] [PubMed] [Google Scholar]
- 16.Reich D, et al. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 2005;37:1113–1118. doi: 10.1038/ng1646. [DOI] [PubMed] [Google Scholar]
- 17.Nalls MA, et al. Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am. J. Hum. Genet. 2008;82:81–87. doi: 10.1016/j.ajhg.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reich D, et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am. J. Hum. Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith MW, et al. A high density admixture map for disease gene discovery in African Americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson N, et al. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu X, Tang H, Risch N. Admixture mapping and the role of population structure for localizing disease genes. Adv. Genet. 2008;60:547–569. doi: 10.1016/S0065-2660(07)00419-1. [DOI] [PubMed] [Google Scholar]
- 22.Arrondel C, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J. Am. Soc. Nephrol. 2002;13:65–74. doi: 10.1681/ASN.V13165. [DOI] [PubMed] [Google Scholar]
- 23.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price AL, et al. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 26.Winn MP. Approach to the evaluation of heritable diseases and update on familial focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2003;18(Suppl. 6):vi14–vi20. doi: 10.1093/ndt/gfg1070. [DOI] [PubMed] [Google Scholar]
- 27.McKenzie LM, et al. NPHS2 variation in sporadic focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2007;18:2987–2995. doi: 10.1681/ASN.2007030319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orloff MS, et al. Variants in the Wilms’ tumor gene are associated with focal segmental glomerulosclerosis in the African American population. Physiol. Genomics. 2005;21:212–221. doi: 10.1152/physiolgenomics.00201.2004. [DOI] [PubMed] [Google Scholar]
- 29.Kao WHL, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat. Genet. 2008 September 14; doi: 10.1038/ng.232. advance online publication, doi:10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sellers JR. Myosins: a diverse superfamily. Biochim. Biophys. Acta. 2000;1496:3–22. doi: 10.1016/s0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- 31.Dong F, et al. Genotype-phenotype correlation in MYH9-related thrombocytopenia. Br. J. Haematol. 2005;130:620–627. doi: 10.1111/j.1365-2141.2005.05658.x. [DOI] [PubMed] [Google Scholar]
- 32.Ghiggeri GM, et al. Genetics, clinical and pathological features of glomerulonephritis associated with mutations of nonmuscle myosin IIA (Fechtner syndrome) Am. J. Kidney Dis. 2003;41:95–104. doi: 10.1053/ajkd.2003.50028. [DOI] [PubMed] [Google Scholar]
- 33.Matsushita T, et al. Targeted disruption of mouse ortholog of the human MYH9 responsible for macrothrombocytopenia with different organ involvement: hematological, nephrological, and otological studies of heterozygous KO mice. Biochem. Biophys. Res. Commun. 2004;325:1163–1171. doi: 10.1016/j.bbrc.2004.10.147. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan JM, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 35.Kim JM, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300:1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 36.Asanuma K, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 37.Freedman BI, Iskandar SS, Appel RG. The link between hypertension and nephrosclerosis. Am. J. Kidney Dis. 1995;25:207–221. doi: 10.1016/0272-6386(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 38.Zarif L, et al. Inaccuracy of clinical phenotyping parameters for hypertensive nephrosclerosis. Nephrol. Dial. Transplant. 2000;15:1801–1807. doi: 10.1093/ndt/15.11.1801. [DOI] [PubMed] [Google Scholar]
- 39.Fogo A, et al. AASK Pilot Study Investigators Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: a report from the African American Study of Kidney Disease (AASK) Trial. Kidney Int. 1997;51:244–252. doi: 10.1038/ki.1997.29. [DOI] [PubMed] [Google Scholar]
- 40.Marcantoni C, Ma LJ, Federspiel C, Fogo AB. Hypertensive nephrosclerosis in African Americans versus Caucasians. Kidney Int. 2002;62:172–180. doi: 10.1046/j.1523-1755.2002.00420.x. [DOI] [PubMed] [Google Scholar]
- 41.O’Brien SJ, Nelson GW. Human genes that limit AIDS. Nat. Genet. 2004;36:565–574. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- 42.Freedman BI, et al. A genome scan for ESRD in black families enriched for nondiabetic nephropathy. J. Am. Soc. Nephrol. 2004;15:2719–2727. doi: 10.1097/01.ASN.0000141312.39483.4F. [DOI] [PubMed] [Google Scholar]
- 43.Bowden DW, et al. A genome scan for diabetic nephropathy in African Americans. Kidney Int. 2004;66:1517–1526. doi: 10.1111/j.1523-1755.2004.00915.x. [DOI] [PubMed] [Google Scholar]
- 44.Thorisson GA, Smith AV, Krishnan L, Stein LD. The International HapMap Project web site. Genome Res. 2005;15:1592–1593. doi: 10.1101/gr.4413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reich D, Patterson N. Will admixture mapping work to find disease genes? Phil. Trans. R. Soc. Lond. B. 2005;360:1605–1607. doi: 10.1098/rstb.2005.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- 47.Gabriel SB, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 48.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 50.Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.