

Summary
The signalling pathways leading to the development of Helicobacter pylori-induced gastric cancer remain poorly understood. We tested the hypothesis that H. pylori infections involve the activation of Akt signalling in human gastric epithelial cancer cells. Immunoblot, immunofluorescence and kinase assays show that H. pylori infection of gastric epithelial cells induced phosphorylation of Akt at Ser 473 and Thr 308. Mutations in the H. pylori virulence factor OipA dramatically reduced phosphorylation of Ser 473, while the cag pathogenicity island mutants predominantly inhibited phosphorylation of Thr 308. As the downstream of Akt activation, H. pylori infection inactivated the inactivation of glycogen synthase kinase 3β at Ser 9 by its phosphorylation. As the upstream of Akt activation, H. pylori infection activated epidermal growth factor receptor (EGFR) at Tyr 992, phosphatidylinositol 3-OH kinase (PI3K) p85 subunit and PI3K-dependent kinase 1 at Ser 241. Pharmacologic inhibitors of PI3K or mitogen-activated protein kinase kinase (MEK), Akt knock-down and EGFR knock-down showed that H. pylori infection induced the activation of EGFR→PI3K→PI3K-dependent kinase 1→Akt→extracellular signal-regulated kinase signalling pathways, the inactivation of glycogen synthase kinase 3β and interleukin-8 production. The combined functions of cag pathogenicity island and OipA were necessary and sufficient for full activation of signalling at each level. We propose activation of these pathways as a novel mechanism for H. pylori-mediated carcinogenesis.
Introduction
Helicobacter pylori infection is now considered to be the major cause of gastric cancer. Although chronic H. pylori infection leads to the development of clinical gastroduodenal diseases in approximately 20% of infected patients, the risk of developing cancer varies widely among individual cases. This suggests that H. pylori–host interaction, host genetics, environmental factors and H. pylori factors may all be important determinants of the outcome of a particular H. pylori infection. H. pylori virulent factors, such as cag pathogenicity island (PAI) and outer inflammatory protein (OipA), are known to influence the risk of developing clinical H. pylori-related diseases, including gastric cancer (Yamaoka et al., 2000; 2002; 2006). The cag PAI encodes a type IV secretion system injecting virulence factors such as CagA and peptidoglycan into the host cells and interfere host signalling (Backert and Selbach, 2008). OipA is one member of a large outer membrane protein family. Both cag PAI and OipA are reported to be involved in the induction of pro-inflammatory cytokines, such as interleukin (IL)-8 and IL-6 from gastric epithelial cells through NF-κB and extracellular signal regulated kinase (Erk) signalling pathways (Yamaoka et al., 2004; Lu et al., 2005).
In a number of different cancers, including gastric cancer, the serine/threonine protein kinase Akt [also called protein kinase B (PKB)] is overexpressed, inappropriately activated, or expressed in phosphorylated form (Han et al., 2006; Manning and Cantley, 2007; Murakami et al., 2007). Akt is activated by various growth factors and survival factors and is a critical downstream effector of phosphatidylinositol 3-OH kinase (PI3K) signalling. Akt also plays a role as a regulator of diverse biological processes, including cell proliferation, apoptosis, glycogen metabolism, migration, cell survival and tumorigenesis (Vivanco and Sawyers, 2002; Chang et al., 2003; Dummler and Hemmings, 2007; Manning and Cantley, 2007). Previous reports also established a link between activation of NF-κB and PI3K/Akt, the upstream kinases of NF-κB, to modulate cell proliferation and antiapoptotic signalling (Zhong et al., 1998; Sizemore et al., 1999; Grandage et al., 2005). However, H. pylori-mediated regulation of cytokines through involvement of PI3K/Akt has not been well studied.
Complete activation of Akt requires activation of two key regulatory sites, Threonine (Thr) 308 in the catalytic domain and Serine (Ser) 473 in the C-terminal regulatory domain (Brazil and Hemmings, 2001; Sarbassov et al., 2005). PI3K-dependent kinase 1 (PDK1) has been identified as the upstream kinase responsible for Ser 308 phosphorylation; however, the identification of tentative kinase PDK2 responsible for Ser 473 phosphorylation remains controversial (Kawakami et al., 2004; Sarbassov et al., 2005; Manning and Cantley, 2007). Once activated, Akt changes its subcellular distribution to gain proximity to downstream target proteins, such as glycogen synthase kinase 3 (GSK3), a key enzyme involved in glycogen metabolism (Manning and Cantley, 2007). GSK3 is constitutively active under resting conditions, but becomes inactivated by phosphorylation of inhibitory serine residues, GSK3β (Ser 9) and GSK3α (Ser 21). Inactivation of GSK3 by Akt signalling has been linked to cell proliferation, inflammation, metabolism, apoptosis and the development of various cancers (Cohen and Frame, 2001; Frame and Cohen, 2001; Doble and Woodgett, 2003; Woodgett, 2005; Jope et al., 2007).
The present study was designed to test the hypothesis that H. pylori infection affects the pathogenesis of gastric cancer through the activation of Akt signalling pathways. We examined the effects of live H. pylori infection on the epidermal growth factor receptor (EGFR) and PI3K/Akt signalling pathways and IL-8 induction from human gastric epithelial cancer cells. We also examined the effects of H. pylori virulence factors, cag PAI and OipA, on these signalling pathways.
Results
Effects of H. pylori infection on phosphorylation of Akt in gastric cancer cell lines
Prior to H. pylori infection, total Akt protein was found to be highly expressed in multiple epithelial cancer cell lines (Fig. 1A). H. pylori infection of AGS cells caused a marked increase in Akt phosphorylation at both Thr 308 and Ser 473 (Fig. 1B and C) compared with uninfected cells incubated in infection media (mock-infected). Phosphorylation of Thr 308 was detected within 5 min and reached maximum levels within 15 min of H. pylori infection at a multiplicity of infection (MOI) of 100 (Fig. 1B). In contrast, phosphorylation of Ser 473 reached maximal levels after 30 min of H. pylori infection, suggesting the use of different upstream regulators for the activation of the two Akt phosphorylation sites. Full phosphorylation of Akt was achieved within 60 min of infection, suggesting that Akt activation is an early event in H. pylori infection. Further, the levels of phosphorylated Thr 308 and Ser 473 were observed to increase with increasing MOI (Fig. 1C). Transient infection of H. pylori did not affect total Akt protein levels at any time point or MOI (Fig. 1B and C, lower panel). Similar patterns of Akt phosphorylation were observed using MKN28 and MKN45 cells and/or different H. pylori strains (data not shown).
Fig. 1. Effect of H. pylori infection on phosphorylation of Akt in gastric epithelial cells.

A. Whole-cell lysates from AGS, MKN28 or MKN45 were analysed for total Akt expression by immunoblot using anti-Akt antibody. The blots were re-probed with β-actin to verify equal loading.
B. Cell lysates from mock-treated AGS cells or from cells infected with wild-type H. pylori for 5–60 min at an MOI of 100 were analysed by immunoblot using indicated phospho-specific Akt antibodies. Mock-infected control cells were incubated for 30 min.
C. Cell lysates from mock-infected AGS cells or from cells infected for 30 min with increasing MOI (12.5–200) of wild-type H. pylori were assayed by immunoblot with indicated phospho-specific Akt antibodies. The blots were re-probed with total Akt and β-actin antibodies to verify equal loading and used as internal control (B and C). At least three independent co-cultures were analysed (A, B and C).
In subsequent experiments, we used an MOI of 100 for H. pylori infection of AGS cells (unless otherwise indicated), as this level of infection eliminates the potential confounding effects of reduced adherence by the oipA mutants (Yamaoka et al., 2004) without affecting cell viability (Choi et al., 2007).
Effect of cag PAI and OipA on phosphorylation of Akt in gastric cancer cell lines
We found that infection of AGS cells with the oipA mutant H. pylori led to dramatically reduced levels of phosphorylation of Ser 473 compared with infection with wild-type H. pylori. In contrast, oipA mutants caused only a slight, though significant, reduction in the phosphorylation of Thr 308 (Fig. 2A). The cag PAI mutations of H. pylori had the opposite effects and completely abolished H. pylori-induced phosphorylation of Thr 308 compared with infection with wild-type H. pylori and only a slight, but significant, reduction in the phosphorylation of Ser 473 (Fig. 2A). The loss of OipA and CagA production in oipA mutants and cag PAI mutants was confirmed by re-probing the immunoblot with anti-OipA antiserum and anti-CagA antibody respectively (Fig. 2A). Infection with double cag PAI/oipA mutants resulted in complete inhibition of the phosphorylation of Ser 473, confirming that cag PAI also affects the phosphorylation of Ser 473. Importantly, these data suggested that the combined activities of cag PAI and OipA are sufficient for full activation of Akt.
Fig. 2. Effects of cag PAI and OipA on phosphorylation of Akt in AGS cells.

A. AGS cells were mock-infected or infected for 30 min with wild-type H. pylori, cag PAI mutants, oipA mutants or cag PAI/oipA double mutants at an MOI of 100. Whole-cell lysates were subjected to immunoblot analyses using indicated phospho-specific Akt antibodies. The blots were re-probed with β-actin to verify equal loading. Anti-CagA antibody or anti-OipA antiserum was used to assay CagA or OipA expression in H. pylori mutants. At least five independent co-cultures were analysed. The blots were re-probed with total Akt and β-actin antibodies to verify equal loading and used as internal control. For semiquantification, the density of phospho-specific sites was normalized to that of total Akt and the levels were expressed as fold increase compared with those of mock-infected control cells. Data are presented as average values ± SE (fold induction compared with mock-infected cells). **P <0.01 versus wild-type H. pylori-infected cells.
B. Akt kinase activity was evaluated using the same cell lysates used in the immunoblot analyses. Equal amount of whole-cell lysates were immunoprecipitated with immobilized anti-Akt antibody and incubated in kinase buffer containing GSK3 fusion protein and cold ATP. Akt kinase activity was analysed by detecting phosphorylation of GSK-3 using phospho-GSK3α/β (Ser 21/9) antibodies. Data are representative of three independent experiments.
C. AGS cells were mock-infected or infected for 15 min with wild-type H. pylori, cag PAI mutants, oipA mutants or cag PAI/oipA double mutants (an MOI of 100). Cells were subjected to identical conditions for fixation, staining and incubation with primary Akt Thr 308, Akt Ser 473 and secondary (FITC) antibody. DAPI was used for nuclear staining. Representative images of each sample were taken in triplicate.
We also used an Akt kinase assay to examine Akt kinase activity against its GSK3 substrate following H. pylori infection (Fig. 2B). Infection of cells with wild-type H. pylori induced Akt activity against GSK3α/β (Ser 21/Ser 9), and infection with cag PAI mutants led to marked reduction of H. pylori-induced Akt activity compared with wild-type H. pylori. The oipA mutations were also associated with decreased Akt activity; however, their effects were less dramatic than the cag PAI mutations. As expected, the combination of cag PAI and oipA mutants had a more profound effect on kinase activity of Akt than cag PAI mutants alone, confirming that both cag PAI and OipA affected kinase activity of Akt.
As OipA is an outer membrane protein, we examined whether other outer membrane proteins affected the phosphorylation of Akt. We evaluated the phenotypes of isogenic mutants of the H. pylori outer membrane proteins BabA, AlpAB, HorG and HorK. In contrast to the oipA mutants, none caused reduced levels of Akt phosphorylation (data not shown). Collectively, our results are consistent with cag PAI and OipA affecting site-specific activation of Akt.
These results were confirmed by immunofluorescence microscopy with phospho-specific antibodies to Akt. Mock-infected AGS cells had low fluorescence signals, indicating low basal levels of phosphorylated Akt (Fig. 2C). H. pylori infection markedly enhanced fluorescence signals for both phospho-Thr 308 and phospho-Ser 473 Akt in the cytoplasm of AGS cells. Infection of cells with oipA mutants and cag PAI mutants led to the same patterns of reduced Akt phosphorylation by immunofluorescence microscopy as were found by immunoblot analyses (Fig 2A and C).
Effect of H. pylori infection on inactivation of GSK3
The serine/threonine protein kinase GSK3 is constitutively active under resting conditions and becomes inactivated by phosphorylation of inhibitory serine residues, GSK3β (Ser 9) and GSK3α (Ser 21), by the kinases PKA, PKB and PKC (for review see Jope et al., 2007). H. pylori infection markedly enhanced phosphorylation of GSK3β (Ser 9) in an MOI-dependent manner without affecting total GSK3 protein levels (Fig. 3A). Infection with oipA mutants and cag PAI mutants resulted in significant reduction of GSK3β (Ser 9) phosphorylation compared with infection with wild-type H. pylori, although these effects were stronger with oipA mutants than with cag PAI mutants (Fig. 3B). Infection with double cag PAI/oipA mutants resulted in complete reduction of the phosphorylation of GSK3β (Fig. 3B), suggesting that the combined activities of cag PAI and OipA are involved in inactivation of GSK3 by inducing the full levels of phosphorylation of GSK3β and possibly GSK3α following H. pylori infection.
Fig. 3. Effect of H. pylori infection on phosphorylation of GSK3.

A. Cell lysates from mock-infected AGS cells or cells infected for 30 min with wild-type H. pylori at an MOI of 12.5–200 were analysed by immunoblot with phospho-specific GSK3β (Ser 9) antibody. The blots were re-probed with total Akt and β-actin antibodies to verify equal loading and used as an internal control. At least three independent co-cultures were analysed.
B. AGS cells were mock-infected, treated with PMA (100 nM) or infected for 30 min with wild-type H. pylori, cag PAI mutants, oipA mutants or double cag PAI/oipA mutants (an MOI of 100). PMA was used as a positive control. Whole-cell lysates were subjected to immunoblot analyses using phospho-specific GSK3β (Ser 9) antibody. At least five independent co-cultures were analysed. For semiquantification, the density of phospho-specific sites was normalized to that of total Akt and the levels were expressed as fold increase over mock-infected control cells. Data are presented as average values ± SE (fold induction compared with mock-infected cells). *P <0.05, **P <0.01 versus wild-type H. pylori-infected cells.
C. AGS cells were pre-treated for 1 h with 10 or 20 μM of pharmacologic PI3K inhibitor LY294002 or MEK1 inhibitor U0126, followed by infection for 30 min with wild-type H. pylori at an MOI of 100 (left panel). Similarly, AGS cells were pre-treated for 1 h with 5 μM PKA inhibitor H89 followed by infection for 30 min with H. pylori at an MOI of 100 (right panel). Whole-cell lysates were analysed by immunoblot with phospho-specific GSK3β antibody. The blots were re-probed with total Akt and β-actin antibodies to verify equal loading and used as internal control. At least three independent co-cultures were analysed.
In agreement with the consensus that GSK3β is located downstream of PI3K/Akt in the signalling pathway, pre-treatment of AGS cells with pharmacologic PI3K inhibitor completely blocked H. pylori-induced phosphorylation of GSK3β (Fig. 3C). Pre-treatment of cells with pharmacologic mitogen-activated protein kinase kinase (MEK) inhibitor also blocked the phosphorylation of GSK3β, suggesting that Erk is involved in the regulation of H. pylori-induced GSK3β phosphorylation and that Erk is located upstream of GSK3 or influencing H. pylori-induced GSK3 inactivation. Pre-treatment of cells with PKA inhibitor slightly reduced H. pylori-induced phosphorylation of GSK3β (Fig. 3C), suggesting that complete phosphorylation of GSK3β is regulated not only by PKA, but also by other factors, probably PKB and PKC.
Effects of H. pylori infection on phosphorylation of PI3K and PDK1
The possible activation of PI3K and PDK1, putative upstream mediators of Akt, in response to H. pylori infection has not been investigated. Phosphorylation of Akt Thr 308 has been reported to involve the activation of PDK1 (Kawakami et al., 2004; Sarbassov et al., 2005; Manning and Cantley, 2007). We found that H. pylori infection of AGS cells enhanced the phosphorylation of the p85 subunit of PI3K, with maximal levels detected 30 min after infection at an MOI of 100 (Fig. 4A). In addition, H. pylori infection dramatically induced the phosphorylation of PDK1 Ser 241; the levels increased at 15 min and then persisted for 60 min after infection. The total PDK1 protein levels remained unchanged (Fig. 4A).
Fig. 4. Effect of H. pylori infection on phosphorylation of PI3K and PDK1.

A. Cell lysates from mock-infected AGS cells or cells infected for 5–60 min with wild-type H. pylori at an MOI of 100 were analysed by immunoblot with phospho-specific PI3K p85 and PDK1 Ser 241 antibodies. The blots were re-probed with total PDK1 and β-actin antibodies to verify equal loading and used as internal control. At least three independent co-cultures were analysed.
B. AGS cells were mock-infected or infected with for 30 min wild-type H. pylori, cag PAI mutants, oipA mutants or double cag PAI/oipA mutants (an MOI of 100) and whole-cell lysates were subjected to immunoblot analyses using indicated phospho-specific antibodies. At least five independent co-cultures were analysed. For semiquantification, the density of phospho-specific sites was normalized to that of β-actin and the levels were expressed as fold increase over mock-infected control cells. Data are presented as average values ± SE (fold induction over mock-infected cells). *P <0.05, **P <0.01 versus wild-type H. pylori-infected cells.
C. Whole-cell lysates from mock-infected AGS cells or cells infected for 30 min with wild-type H. pylori at MOIs from 50 to 200 or AGS cells were pre-treated for 1 h with 5–20 μM of pharmacologic PI3K inhibitor LY294002, followed by infection for 30 min with H. pylori (an MOI of 100). Whole-cell lysates were analysed by immunoblot using indicated phospho-specific PDK1 Ser 241 antibodies. The blots were re-probed with total PDK1 and β-actin antibody to verify equal loading and used as internal control. At least three independent co-cultures were analysed.
Interestingly, infection with cag PAI mutants caused reduced H. pylori-induced phosphorylation of both the PI3K p85 subunit and PDK1 Ser 241 compared with infection with wild-type H. pylori (Fig. 4B). These results are in agreement with the hypothesis that Akt Thr 308 is regulated by the PI3K p85 subunit and PDK1. The oipA mutations reduced the H. pylori-induced phosphorylation of PI3K and PDK1 to a lesser extent than the cag PAI mutations; however, infection with the double cag PAI/oipA mutations resulted in complete inhibition of the phosphorylation of PI3K and PDK1 (Fig. 4B), suggesting that the combined activities of cag PAI and OipA are sufficient for full phosphorylation of PI3K p85 subunit and PDK1 Ser 241 in response to H. pylori infection.
As expected, pre-incubation with the pharmacological PI3K inhibitor, LY294002, abrogated H. pylori-induced phosphorylation of Akt at both sites (data not shown). PI3K inhibitor also inhibited H. pylori-induced phosphorylation of PDK1 (Fig. 4C), confirming that PDK1 is downstream of PI3K in the signalling pathway.
Effect of Akt knock-down by siRNA on H. pylori-induced phosphorylation of GSK3, PDK1 and mitogen-activated protein kinase
Various cell surface receptors can regulate mitogen-activated protein kinase (MAPK) signalling pathways. We have previously reported that H. pylori induced the phosphorylation of Erk and p38 MAPK (Yamaoka et al., 2004; Choi et al., 2007) and hypothesized that Akt signalling might be responsible for H. pylori-mediated regulation of Erk and p38. Transfection of the control siRNA plasmid did not affect H. pylori-induced phosphorylation of Akt, PDK1, GSK3β, Erk or p38 (Fig. 5). As expected, Akt knock-down by Akt-specific siRNA led to undetectable levels of GSK3β (Ser 9) phosphorylation after H. pylori infection, but had no effect on H. pylori-induced phosphorylation of PDK1 Ser 241. These results confirm that GSK3β is a downstream target of Akt, whereas PDK1 is upstream of Akt in the signalling pathway. Akt knock-down was also resulted in an absence of Erk Thr202/Tyr204 phosphorylation following H. pylori infection (Fig. 5). These data suggest that Akt plays essential roles in the H. pylori-mediated activation of Erk→GSK3β signalling pathways. In contrast, Akt knock-down had no effect on the H. pylori-induced phosphorylation of p38 Thr180/Tyr182, indicating that this protein is either upstream of Akt or is activated by Akt-independent mechanisms.
Fig. 5.

Effect of Akt Knock-down by Akt-specific siRNA on H. pylori-induced phosphorylation of GSK3, PDK1 and MAPK. AGS cells were transfected with negative control siRNA, or transfected with Akt-specific siRNA for 72 h followed by 30 min incubation with or without wild-type H. pylori. Equal amounts of protein from whole-cell lysates were subjected to immunoblot analyses, as described in Experimental procedures, using indicated phospho-specific antibodies. At least three independent co-cultures were analysed.
Involvement of EGFR in H. pylori-mediated phosphorylation of PDK1/AKT signalling
The EGFR is overexpressed in various types of tumours and is involved in cell migration, invasion and cell survival signalling (De et al., 2008). Recent reports indicate that H. pylori infection induces the transactivation of EGFR in gastric epithelial cells (Jorissen et al., 2003; Ashktorab et al., 2007; Keates et al., 2007). We hypothesized that site-specific phosphorylation of EGFR and possible subsequent activation of PI3K signalling pathways are necessary for the H. pylori-mediated activation of Akt. As expected, H. pylori infection induced the phosphorylation of EGFR Tyr 992 (Fig. 6A). Infection with cag PAI mutants markedly reduced the levels of H. pylori-induced phosphorylation of EGFR Tyr 992 as compared with infection with wild-type H. pylori. Infection with oipA mutants also decreased the levels of phosphorylation of EGFR Tyr 992 (Fig. 6B). Infection with a double cag PAI/oipA mutant completely blocked H. pylori-induced EGFR Tyr 992 phosphorylation, suggesting that the combined activities of cag PAI and OipA are sufficient for full phosphorylation of EGFR Tyr 992 triggered by H. pylori infection.
Fig. 6. Effect of EGFR on H. pylori -mediated Akt activation and downstream signalling.
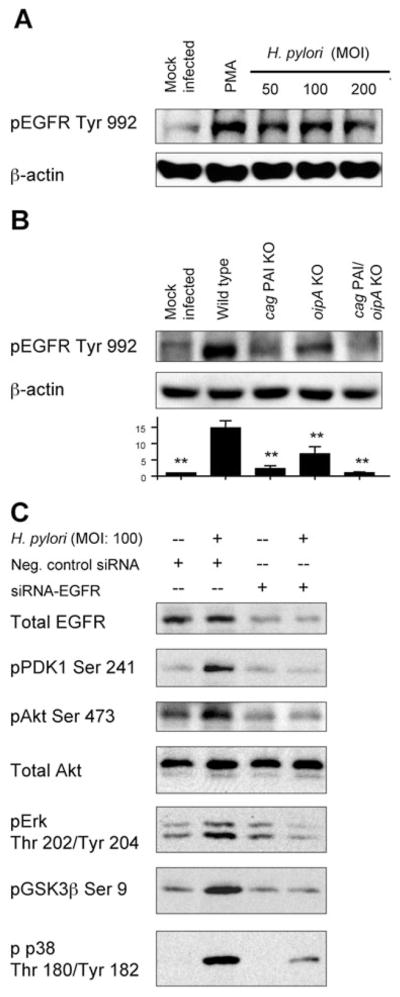
A. AGS cells were mock-infected, treated with PMA (100 nM) or infected for 30 min with wild-type H. pylori at an MOI of 50–200. PMA was used as a positive control. Whole-cell lysates were subjected to immunoblot analyses using phospho-specific EGFR Tyr 992 antibody. The same blots were re-probed with β-actin and used as an internal control. At least three independent co-cultures were analysed.
B. AGS cells were mock-infected or infected for 30 min with wild-type H. pylori, cag PAI mutants, oipA mutants or double cag PAI/oipA mutants (an MOI of 100). Whole-cell lysates were subjected to immunoblot analyses using phospho-specific EGFR Tyr 992 antibody. At least five independent co-cultures were analysed. For semiquantification, the density of phospho-specific sites was normalized to that of β-actin and the levels were expressed as fold increase over mock-infected control cells. Data are presented as average values ± SE (fold induction over mock-infected cells). **P <0.01 versus wild-type H. pylori-infected cells.
C. AGS cells were transfected with negative control siRNA, or transfected with EGFR-specific siRNA for 72 h followed by 30 min incubation with or without wild-type H. pylori. Equal amounts of proteins from whole-cell lysates were subjected to immunoblot analyses, as described in Experimental procedures, using indicated phospho-specific antibodies. At least three independent co-cultures were analysed.
We next examined the role of EGFR in H. pylori-induced activation of Akt signalling pathways using EGFR-specific siRNA. Transfection of the control siRNA plasmid did not affect H. pylori-induced phosphorylation of Akt, PDK1, GSK3β, Erk or p38 (Fig. 6C). After EGFR knock-down by EGFR-specific siRNA, detectable levels of phosphorylation of PDK1 Ser 241, Akt Ser 473, Erk Thr202/Tyr204 and GSK3β (Ser 9) were no longer induced by H. pylori infection, suggesting that EGFR signalling is essential for H. pylori-mediated regulation of the PDK1→Akt→Erk→GSK3β pathways. Immunofluorescence microscopy using a pharmacologic inhibitor of EGFR confirmed that inhibition of EGFR dramatically blocked H. pylori-induced phosphorylation of Akt Ser 473 and Thr 308 (data not shown). Overall, H. pylori-induced activation of PI3K/Akt signalling pathway and its downstream targets required EGFR-mediated signalling.
The EGFR knock-down also reduced the levels of H. pylori-mediated phosphorylation of p38; however, H. pylori infection still triggered some phosphorylation of p38 in the absence of EGFR expression (Fig. 6C), suggesting the existence of EGFR-independent mechanisms for activation of p38.
Role of EGFR and PI3K/Akt in H. pylori-induced IL-8 production in gastric epithelial cells
We previously reported that H. pylori induced IL-8 production through Erk and NF-κB involvement while isogenic mutants of both cag PAI and OipA significantly blocked IL-8 production in gastric epithelial cells (Yamaoka et al., 2004). We also showed that IL-8 levels reached maximal levels at 18–21 h after H. pylori infection at an MOI of 100 and plateaued until 30 h irrespective of H. pylori strain (Yamaoka et al., 2004). We therefore co-cultured H. pylori with AGS cells at an MOI of 100 for 18 h to evaluate the role of H. pylori-activated EGFR and PI3K/Akt on IL-8 production.
To test the hypothesis that EGFR signalling pathway plays a role in H. pylori-induced IL-8 production, we utilized pharmacologic inhibitor of EGFR (AG1478) to inhibit EGFR phosphorylation followed by infection with wild-type H. pylori, cag PAI mutants or oipA mutants. Pre-treatment of AGS cells with pharmacologic EGFR inhibitor blocked wild-type H. pylori-induced IL-8 by approximately 60%, while inhibition of EGFR activation suppressed cag PAI or oipA mutants’ mediated IL-8 production by only approximately 30% (Fig. 7A). Furthermore, pre-treatment of AGS cells with pharmacologic PI3K inhibitors blocked wild-type H. pylori-induced IL-8 by approximately 50%, while inhibition of PI3K activation suppressed cag PAI or oipA mutants’ mediated IL-8 production by only approximately 25% (Fig. 7B). Similar results were observed in MKN28 and MKN45 cells (data not shown). These results suggested that the activation of EGFR-PI3K signalling, at least in part, positively regulated H. pylori-mediated IL-8 production and both cag PAI and OipA are involved in EGFR-related IL-8 production.
Fig. 7.
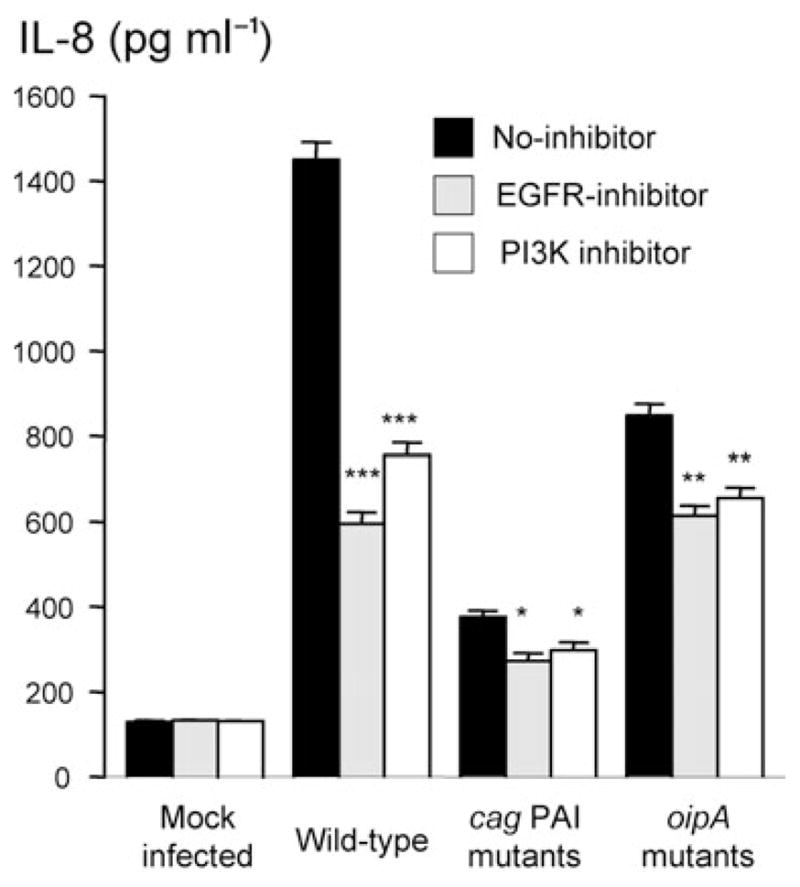
Role of the EGFR and PI3K in H. pylori-induced IL-8 production in gastric epithelial cells. AGS cells were mock-infected or infected with H. pylori for 18 h with or without pre-treating for 2 h with 5 μM EGFR inhibitor (AG1478) (A) or 10 μM of PI3K inhibitor (LY 294002) (B). IL-8 levels from the culture supernatants (pg ml−1) were measured by ELISA assay. Data are presented as mean ± SE from at least three independent experiments. *P <0.05, **P <0.01 and ***P <0.001 versus H. pylori-infected cells without inhibitors.
Discussion
Results from the current study have led to the conclusion that infection with live H. pylori induces EGFR- and PI3K-dependent phosphorylation of PDK1 and Akt, and the subsequent activation of Erk, inactivation of GSK3 and IL-8 production. Importantly, we found that the H. pylori virulence factors, cag PAI and OipA, differentially regulate phosphorylation of Akt on sites Thr 308 and Ser 473. The oipA mutation reduced the levels of Akt Ser 473, whereas the cag PAI mutation reduced activation of Akt Thr 308 in response to H. pylori infection. The specific functions of cag PAI or OipA in signalling the activation of Akt Ser 473 or Thr 308 might cause an imbalance in downstream proliferation and apoptotic signalling. Importantly, infection of cag PAI/oipA double mutants completely blocked H. pylori-mediated activation of both Akt sites, suggesting that both OipA and cag PAI are required for complete activation of Akt. Activated Akt signalling pathways are key mediators of diverse cellular responses, such as cell survival, proliferation and cell motility in other systems (Chang and Karin, 2001; Vivanco and Sawyers, 2002; Suthiphongchai et al., 2003; Liu et al., 2006). Upregulated Akt activation has also been observed in tissues adjacent to gastric tumours and Akt has been implicated in influencing the chemoresistance of gastric cancers (Vivanco and Sawyers, 2002; Ang et al., 2005; Oki et al., 2005). Taken together, phosphorylation of Akt mediated by the combination of cag PAI and OipA is suggested to be a regulator of intracellular signalling involved in the regulation of a number of cellular functions involved in gastric carcinogenesis.
We have observed for the first time that H. pylori infection causes the inactivation of GSK3 through the activation of Akt kinase activity against GSK3β. These findings are in agreement with previous studies of Akt-mediated signalling in other systems (Delcommenne et al., 1998; Stambolic and Woodgett, 2006). Inactivation of GSK3 has been linked to cell proliferation, inflammation, metabolism, apoptosis and the development of various cancers through the activities of Akt signalling pathways (Cohen and Frame, 2001; Frame and Cohen, 2001; Doble and Woodgett, 2003; Woodgett, 2005; Jope et al., 2007). We propose that inactivation of GSK3β during H. pylori infection may be important for IL-8 production and in the development of various pathologic conditions leading to gastric cancer. Regulation of IL-8 gene transcription by H. pylori appears to be mediated mainly through NF-κB and AP-1 (Yamaoka et al., 2004). Our preliminary data using uninfected gastric epithelial AGS cells shows that GSK3β inhibitor SB216763 induced upregulation of AP-1, but not of NF-κB, and also induced IL-8 production (F.H. Tabassam et al., unpubl. data). As H. pylori infection induced the inactivation of GSK3β, GSK3β→AP-1 pathway might regulate IL-8 production. GSK3β is one of the key elements of the Wnt pathway, which governs β-catenin homeostasis. Nuclear translocation of β-catenin and its association with LEF/TCF transcription factors are key steps in the transduction of Wnt signalling, which is aberrantly activated in a variety of human cancers (Frame and Cohen, 2001). There are several reports that β-catenin signalling induces IL-8 expression in other systems [e.g. umbilical endothelial cells (Masckauchan et al., 2005) and primary human hepatocytes (Levy et al., 2002)], and direct binding of β-catenin to the putative LEF/TCF site in the IL-8 promoter has been confirmed to play important roles in IL-8 induction in primary human hepatocytes (Levy et al., 2002). Our preliminary data show that following inactivation of GSK3β by H. pylori infection, β-catenin was released from a GSK3β/β-catenin complex, with subsequent nuclear translocation (F.H. Tabassam et al., unpubl. data). Overall, we suggest that GSK3β→AP-1 and GSK3β→β-catenin pathways will regulate IL-8 induction in H. pylori-infected gastric epithelial cells. Importantly, recent studies showed that both OipA and CagA are involved in the nuclear translocation of β-catenin (Franco et al., 2008), and OipA and cag PAI are involved in the activation of AP-1 (Choi et al., 2007). In the present study, although we did not focus on the each factor in the cag PAI and/or factors related to the cag PAI (e.g. peptidoglycan), interaction between CagA and β-catenin might play important roles in the Akt→GSK3β-related pathways. Further studies will be necessary to investigate the roles of CagA on the pathways. As was the case for Akt activation, maximal inactivation of GSK3β by H. pylori required functional cag PAI and OipA proteins, and the combination of cag PAI and OipA activities was sufficient for complete inactivation of GSK3β. It is well known that patients infected with cag PAI-positive/OipA-positive H. pylori strains are at a higher risk for developing gastric cancer than those infected with cag PAI-negative/OipA-negative strains (Yamaoka et al., 2002; 2006). However, how the functions of cag PAI and OipA combine to act as carcinogens has not been established, although several potentially carcinogenetic molecules interact with cag PAI and/or OipA, including SHP-2 (Higashi et al., 2002) and FAK (Tabassam et al., 2008). The results of our Akt knock-down experiments suggested that Akt is essential for H. pylori-mediated GSK3β signalling pathways. Overall, we propose a novel mechanism to explain H. pylori-induced cytokine production and a possible link to carcinogenesis, in which cag PAI and OipA combine to trigger the inactivation of GSK3 through activated PI3K/Akt signalling pathways.
To establish the upstream signalling events that direct H. pylori-mediated Akt phosphorylation, we confirmed that H. pylori infection increases the phosphorylation of the PI3K p85 subunit and PDK1. PI3K is a heterodimer composed of a p85-regulatory and a p110 catalytic subunit. Once activated, PI3K generates phosphatidylinositol-3,4, -bisphosphate and phosphatidylinositol-3,4,5-trisphos-phate to activate PI3K-dependent kinases, including PDKs and Akt, to regulate diverse cellular functions, such as cell growth, differentiation, cell motility and cell survival (Alessi and Cohen, 1998; Wang et al., 2000; Cantley, 2002; Vivanco and Sawyers, 2002; Woodgett, 2005; Manning and Cantley, 2007). In the present study, we found that cag PAI was most important in the phosphorylation of P13K p85 subunit and PDK1 Ser 241 after H. pylori infection. These results are in agreement with the hypothesis that PDK1 is the upstream kinase responsible for phosphorylation of Akt Thr 308 (Kawakami et al., 2004; Sarbassov et al., 2005; Manning and Cantley, 2007). However, OipA activity also contributed to the activation of PI3K and PDK1, and cag PAI/oipA mutants completely blocked H. pylori-mediated activation of P13K p85 subunit and PDK1 Ser 241. These results confirmed that the combination of cag PAI and OipA is sufficient for full activation of PI3K→PDK1 signalling pathways, as is the case for Akt→Erk→GSK3β downstream signalling.
Immunofluorescence images obtained using phospho-specific antibodies for Akt illustrated that activated Akt was localized to the cell periphery where activated cell surface receptors initiate H. pylori-mediated signalling. A role for EGFR as the surface receptor for H. pylori-induced signalling has been previously reported (Keates et al., 2007; Tabassam et al., 2007); however, the direct involvement of EGFR in activating Akt signalling in response to H. pylori infection has not been previously investigated. In the present study, EGFR was found to undergo rapid tyrosine phosphorylation at Tyr 992 following H. pylori infection; the kinetics of H. pylori-mediated EGFR activation coincide with phosphorylation of PI3K and Akt and are consistent with recent reports describing H. pylori-induced tyrosine phosphorylation of EGFR in gastric epithelial cells (Keates et al., 2007). EGFR knock-down completely inhibited H. pylori-induced phosphorylation of PDK1 and Akt, resulting in the inhibition of downstream effectors, such as Erk and GSK3β. Overall, our current study provides evidence to support the hypothesis that activation of EGFR is involved in the initiation of H. pylori-mediated PI3K/Akt signalling, followed by Erk→GSK3β signalling pathways. In addition, we have confirmed that the combination of cag PAI and OipA is sufficient for full activation of these signalling pathways. All lines of evidence in this study converge to show that activated EGFR is a critical mediator in H. pylori-mediated signalling and that activated Akt is a novel cellular mediator of H. pylori-induced signalling. In addition, we also found that the activation of EGFR-PI3K signalling, at least in part positively regulated H. pylori-mediated IL-8 production and both cag PAI and OipA are involved in EGFR-related IL-8 production. These results may have important implications for the development of drugs to counteract the effects of H. pylori infection. As EGFR and PI3K/Akt are overexpressed in gastric cancer, we propose that agents antagonizing these signalling pathways (e.g. blocking EGFR activation or silencing Akt expression) might provide more selective alternatives for the management of gastric cancer.
Finally, previous studies in other cell systems have suggested that the activation of Akt is required in order for phosphorylation of p38 MAPK to impair cell proliferation (Gratton et al., 2001; Parekh and Rao, 2007). Surprisingly, we could not confirm these results, as Akt knock-down did not block H. pylori-mediated p38 activation. These data suggest that p38 activation is divergent and that Akt-mediated interaction is either not required for p38 activation or upstream of Akt in the signalling pathway. We speculate that H. pylori activate cell surface receptors or non-receptor kinases, which activate p38 independent of Akt. Although the underlying mechanism is unknown, we propose that H. pylori-induced p38 activation is, at least in part, regulated by EGFR as EGFR knock-down blocked H. pylori-induced p38 activation.
Experimental procedures
Reagents
Site and phospho-specific affinity-purified polyclonal antibodies for Akt Thr 308 and Ser 473, GSK3α/βSer 9 and Ser 21, PDK1 Ser 241, Erk Thr 202/Tyr204, p38 Thr 180/Tyr 182 and EGFR Tyr 992; polyclonal purified anti-Akt, GSK3β, PDK1 and Erk; affinity-purified horseradish peroxidase-linked goat anti-rabbit/-mouse IgG (H and L) and LumiGLO reagent chemiluminescent substrate detection system were obtained from Cell Signalling Technology (Beverly, MA). Anti-OipA antiserum was described previously (Kudo et al., 2004). Anti-CagA antibody was purchased from Austral Biologicals (San Ramon, CA). FITC-conjugated anti-rabbit, mouse monoclonal antiβ-actin antibodies and protease inhibitor cocktail were purchased from Sigma-Aldrich (St Louis, MO). Selective nuclear probe DAPI, and SlowFade Antifade kit were obtained from Molecular Probes (Eugene, OR). Pharmacological inhibitors of EGFR (AG1478), PI3K (LY 294002), PKA (H-89, Dihydrochloride) and MEK1 (U0126) were purchased from Calbiochem (La Jolla, CA). Mammalian EGFR siRNA/siAB Assay Kits were purchased from Upstate Cell signalling Solution (Lake Placid, NY), and mammalian Akt siRNA kit expression plasmids were purchased from Cell Signalling Technology (Beverly, MA).
Cell culture
Human gastric epithelial cancer cell lines AGS (American Type Culture Collection, Manassas, VA), MKN28 (Riken Bank, Tsukuba, Japan) and MKN45 (Riken Bank) were grown in RPMI 1640 medium supplemented with penicillin, streptomycin and 10% FBS in a humidified 5% CO2 atmosphere at a density of 1 × 105 cells in six-well plates or 1 × 106 cells in 10 cm dishes. To avoid the influence of serum, gastric cells were serum-starved overnight before experiments. Cells with 80% confluence were left untreated in RPMI 1640 medium, co-cultured with H. pylori for specified times and/or with specified MOI as described in figure legends.
H. pylori
Three functional oipA-positive/cag PAI-positive H. pylori strains (TN2GF4, ATCC43504 and 26695), their isogenic oipA mutants, cag PAI totally-deleted mutants and oipA and cag PAI double mutants were used (Yamaoka et al., 2000; Kudo et al., 2005). We also used isogenic babA mutants, alpAB mutants, horG mutants and horK mutants (Yamaoka et al., 2000; Lu et al., 2007). H. pylori TN2GF4 was originally isolated from a Japanese gastric ulcer patient and has been shown to colonize Mongolian gerbils consistently for at least 1 year and to cause reproducible mucosal damage (Watanabe et al., 1998). H. pylori were cultured on brain heart infusion agar plates containing 7% horse blood and incubated at 37°C under microaerophilic conditions for 24–36 h. The cells were suspended in PBS and the density was estimated by spectrophotometry (A625) and by microscopic observation.
Protein extraction and immunoblot
Confluent gastric cells were co-cultured with H. pylori at the specified MOI or were co-cultured at an MOI of 100 for specified times as indicated in the figure legends. Cells were lysed with lysis buffer containing 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM Na3VO4, 1 mM PMSF sodium pyrophosphate, 1 μg ml−1 leupeptin and protease-inhibitor cocktail, and immunoblot was performed using standard techniques with indicated phospho-specific antibodies. In experiments using isogenic mutants, X-ray films were scanned and semiquantified as described previously (Tabassam et al., 2007). Statistical analysis was performed in immunoblot using isogenic mutants by non-paired t-test using SigmaStat 3.01 (SPSS, Chicago, IL). A P-value of less than 0.05 was accepted as statistically significant.
Kinase activity assay
Akt kinase assays were performed according to the manufacturer’s instructions. Briefly, confluent AGS cells were serum-starved over night and left untreated or co-cultured for 1 h with H. pylori at an MOI of 100. Cells were lysed with ice-cold lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM Na3VO4, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 μg ml−1 leupeptin, 1 mM PMSF and protease-inhibitor cocktail). Total proteins from cell lysates were immunoprecipitated with immobilized ant-Akt monoclonal antibody that recognizes all Akt isoforms. Immunoprecipitated pellets were washed and incubated in kinase buffer containing GSK3 fusion protein and cold ATP. The kinase activity of Akt was analysed by detecting GSK3 phosphorylation using phospho GSK3α/β (Ser 21/9) antibody, as recommended by the manufacturer.
Silencing Akt or EGFR protein expression
The AGS cells were plated in six-well plates and allowed to reach ~80% confluencecon on the day of transfection. To downregulate Akt expression, negative control siRNA or Akt-specific siRNA were introduced into the cells according to the manufacturer’s instructions. Similarly, cells were transfected with EGFR-specific siRNA or non-specific negative control siRNAs (100 pmol) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Briefly, 100 pmol of siRNAs diluted in 200 μl of Opti-MEM (Invitrogen, Carlsbad, CA) and 5 μl of Lipo-fectamine 2000 was diluted in 200 μl of Opti-MEM. Diluted siRNA and Lipofectamine 2000 were mixed and incubated for 15 min at room temperature for lipid complex formation. For subsequent transfection, complexes were added to each well and incubated for 4 h followed by incubation with 20% FBS. After 24 h, the media were changed to complete media and incubated for an additional 1–4 days after transfection, as indicated in the figure legends. Before infection with H. pylori, cells were suspended overnight in serum-free medium and infected for 1 h with H. pylori at an MOI of 100. Protein extraction and immunoblotting were performed according to established protocols.
Fluorescence microscopy
Helicobacter pylori-induced Akt Ser 473 phosphorylation in AGS cells was visualized by fluorescence microscopy (Olympus, America Melville, NY). H. pylori-infected or -uninfected cells were subjected to identical culture, fixation, staining and microscopy conditions for comparisons between samples. Images were captured at 1000× magnification obtained before substantial photo-bleaching occurred and representative images of each sample were taken in triplicate.
Determination of IL-8 productions
In vitro IL-8 production was measured as described previously (Yamaoka et al., 2004). Briefly, gastric epithelial cells (approximately 5 × 105 ml−1) were plated onto 24-well plates and cultured for 2 days. Serum-starved cells were pre-incubated with or without 5 μM EGFR inhibitor (AG1478) or 10 μM PI3K inhibitor (LY 294002) for 2 h followed by H. pylori infection at an MOI of 100 for 18 h. Culture supernatants were collected and assayed for IL-8 production by an ELISA (R and D Systems; Minneapolis, MN). Statistical analysis was performed by non-paired t-test using SigmaStat 3.01. A P-value of less than 0.05 was accepted as statistically significant.
Acknowledgments
This material is based upon work supported in part by the Office of Research and Development Medical Research Service Department of Veterans Affairs, by Public Health Service Grant DK56338 that funds the Texas Medical Center Digestive Diseases Center. The project described was also supported by Grant Number DK 62813 from NIH. Their contents are solely the responsibility of the authors and do not necessarily represent the official views of the VA or NIH.
References
- Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- Ang KL, Shi DL, Keong WW, Epstein RJ. Upregulated Akt signalling adjacent to gastric cancers: implications for screening and chemoprevention. Cancer Lett. 2005;225:53–59. doi: 10.1016/j.canlet.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Ashktorab H, Daremipouran M, Wilson M, Siddiqi S, Lee EL, Rakhshani N, et al. Transactivation of the EGFR by AP-1 is induced by Helicobacter pylori in gastric cancer. Am J Gastroenterol. 2007;102:2135–2146. doi: 10.1111/j.1572-0241.2007.01400.x. [DOI] [PubMed] [Google Scholar]
- Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10:1573–1581. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- Choi IJ, Fujimoto S, Yamauchi K, Graham DY, Yamaoka Y. Helicobacter pylori environmental interactions: effect of acidic conditions on H. pylori-induced gastric mucosal interleukin-8 production. Cell Microbiol. 2007;9:2457–2469. doi: 10.1111/j.1462-5822.2007.00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- De LA, Carotenuto A, Rachiglio A, Gallo M, Maiello MR, et al. The role of the EGFR signaling in tumor microenvironment. J Cell Physiol. 2008;214:559–567. doi: 10.1002/jcp.21260. [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007;35:231–235. doi: 10.1042/BST0350231. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandage VL, Gale RE, Linch DC, Khwaja A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia. 2005;19:586–594. doi: 10.1038/sj.leu.2403653. [DOI] [PubMed] [Google Scholar]
- Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem. 2001;276:30359–30365. doi: 10.1074/jbc.M009698200. [DOI] [PubMed] [Google Scholar]
- Han Z, Hong L, Wu K, Han S, Shen H, Liu C, et al. Reversal of multidrug resistance of gastric cancer cells by downregulation of Akt1 with Akt1 siRNA. J Exp Clin Cancer Res. 2006;25:601–606. [PubMed] [Google Scholar]
- Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Nishimoto H, Kitaura J, Maeda-Yamamoto M, Kato RM, Littman DR, et al. Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004;279:47720–47725. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- Keates S, Keates AC, Katchar K, Peek RM, Jr, Kelly CP. Helicobacter pylori induces up-regulation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Infect Dis. 2007;196:95–103. doi: 10.1086/518440. [DOI] [PubMed] [Google Scholar]
- Kudo T, Nurgalieva ZZ, Conner ME, Crawford S, Odenbreit S, Haas R, et al. Correlation between Helicobacter pylori OipA protein expression and oipA gene switch status. J Clin Microbiol. 2004;42:2279–2281. doi: 10.1128/JCM.42.5.2279-2281.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Lu H, Wu JY, Graham DY, Casola A, Yamaoka Y. Regulation of RANTES promoter activation in gastric epithelial cells infected with Helicobacter pylori. Infect Immun. 2005;73:7602–7612. doi: 10.1128/IAI.73.11.7602-7612.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L, Neuveut C, Renard CA, Charneau P, Branchereau S, Gauthier F, et al. Transcriptional activation of interleukin-8 by beta-catenin-Tcf4. J Biol Chem. 2002;277:42386–42393. doi: 10.1074/jbc.M207418200. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Yu JP, Yu HG, Lv P, Chen HL. Activation of Akt and ERK signalling pathways induced by etoposide confer chemoresistance in gastric cancer cells. Dig Liver Dis. 2006;38:310–318. doi: 10.1016/j.dld.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell. 2005;16:4954–4966. doi: 10.1091/mbc.E05-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wu JY, Beswick EJ, Ohno T, Odenbreit S, Haas R, et al. Functional and intracellular signaling differences associated with the Helicobacter pylori AlpAB adhesin from Western and East Asian strains. J Biol Chem. 2007;282:6242–6254. doi: 10.1074/jbc.M611178200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masckauchan TN, Shawber CJ, Funahashi Y, Li CM, Kitajewski J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. 2005;8:43–51. doi: 10.1007/s10456-005-5612-9. [DOI] [PubMed] [Google Scholar]
- Murakami D, Tsujitani S, Osaki T, Saito H, Katano K, Tatebe S, et al. Expression of phosphorylated Akt (pAkt) in gastric carcinoma predicts prognosis and efficacy of chemotherapy. Gastric Cancer. 2007;10:45–51. doi: 10.1007/s10120-006-0410-7. [DOI] [PubMed] [Google Scholar]
- Oki E, Baba H, Tokunaga E, Nakamura T, Ueda N, Futatsugi M, et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int J Cancer. 2005;117:376–380. doi: 10.1002/ijc.21170. [DOI] [PubMed] [Google Scholar]
- Parekh P, Rao KV. Overexpression of cyclin D1 is associated with elevated levels of MAP kinases, Akt and Pak1 during diethylnitrosamine-induced progressive liver carcinogenesis. Cell Biol Int. 2007;31:35–43. doi: 10.1016/j.cellbi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006;16:461–466. doi: 10.1016/j.tcb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Suthiphongchai T, Promyart P, Virochrut S, Tohtong R, Wilairat P. Involvement of ERK1/2 in invasiveness and metastatic development of rat prostatic adenocarcinoma. Oncol Res. 2003;13:253–259. doi: 10.3727/096504003108748302. [DOI] [PubMed] [Google Scholar]
- Tabassam FH, Graham DY, Yamaoka Y. OipA plays a role in Helicobacter pylori-induced focal adhesion kinase activation and cytoskeletal re-organization. Cell Microbiol. 2008;10:1008–1020. doi: 10.1111/j.1462-5822.2007.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005;17:150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA. 2000;97:7533–7538. doi: 10.1073/pnas.130079797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka Y, Kikuchi S, El-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123:414–424. doi: 10.1053/gast.2002.34781. [DOI] [PubMed] [Google Scholar]
- Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology. 2004;126:1030–1043. doi: 10.1053/j.gastro.2003.12.048. [DOI] [PubMed] [Google Scholar]
- Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, et al. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut. 2006;55:775–781. doi: 10.1136/gut.2005.083014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]