

Abstract
The gastric mucosal immune response is thought to be comprised predominantly of the Th1 type; however, there are limited data regarding the role of IL-18 in Helicobacter pylori-induced inflammation. We investigated IL-18 levels in gastric mucosal biopsy specimens as well as in isolated gastric epithelial cells and lamina propria mononuclear cells. We also investigated IL-18 levels in gastric epithelial cells and the monocyte cell line THP-1 cocultured with H. pylori. In both systems, IL-18 levels were markedly enhanced in H. pylori-infected epithelial cells and monocytes. IL-18 levels in H. pylori-infected gastric mucosa were well correlated with the severity of gastric inflammation, confirming that H. pylori-induced IL-18 plays an important role in gastric injury. Virulence factors of H. pylori; the cag pathogenicity island and OipA affected IL-18 induction in different manners. Up-regulation of IL-18 mRNA/protein in epithelial cells was dependent on both virulence factors. Interestingly, up-regulation of IL-18 mRNA in monocytes was independent of both factors, whereas IL-18 protein was OipA dependent – cag pathogenicity island independent, indicating that OipA regulates IL-18 induction in monocytes at the posttranscriptional level. IL-18 levels in the gastric biopsy specimens showed similar patterns to those in lamina propria mononuclear cells with respect to virulence factors, suggesting that submucosal monocytes/macrophages are the main source of IL-18 induced by H. pylori infection. H. pylori appeared to regulate the ERK/JNK→AP-1 pathway in both cell types. In addition, OipA and its related p38 pathway may be closely involved in IL-18 induction in H. pylori-infected gastric mucosa and may contribute to gastric injury.
The Helicobacter pylori infection is associated with infiltration of neutrophils, lymphocytes, monocytes, and plasma cells into the gastric mucosa. The gastric mucosal immune response is thought to consist predominantly of the Th1 type associated with a significant increase in IFN-γ secreting T cells (1– 4). This notion also is supported by the fact that H. pylori-infected IFN-γ-knockout mice develop minimal pathological changes (5–7).
IL-18, previously known as an IFN-γ-inducing factor, is a potent proinflammatory cytokine in the IL-1 superfamily (8, 9) that promotes the production of IFN-γ from Th1, B, and NK cells in synergy with IL-12 (10 –12). IL-18 also acts as a costimulant for Th1 cells to augment the production of IFN-γ, IL-2, and granulocyte macrophage CSF (11, 13). IL-18-deficient mice failed to develop protection after oral immunization with H. pylori lysates and cholera toxin adjuvant, indicating the importance of IL-18 in protection (14). Well-protected wild-type mice showed moderate gastric inflammation; whereas unprotected IL-18-deficient mice had less gastric inflammation. Therefore, we hypothesized that IL-18 plays an important role in the pathogenesis of H. pylori-induced gastroduodenal disease.
There are limited data regarding the role of IL-18 in H. pylori-induced inflammation in humans. Tomita et al. (15) reported that antral IL-18 mRNA levels were up-regulated in H. pylori infection, whereas mature IL-18 protein was present in mucosa of both infected and uninfected subjects as measured by immunoblot analyses. In contrast, Fera et al. reported that antral H. pylori infection was associated with IL-18 production as determined by immunohistochemistry; whereas IL-18 mRNA was expressed irrespective of H. pylori infection (16). The reason for the discrepancy is unknown; however, one reason might be due to the nonquantitative nature of analyses of mRNA and protein levels.
H. pylori strains containing an intact cag pathogenicity island (PAI)4, which encodes the type IV secretion system, and the outer inflammatory protein (OipA), which encodes the outer membrane protein, have been reported to increase the risk of more severe clinical outcomes (17, 18). Tomita et al. (15) reported that antral IL-18 mRNA levels were independent of the presence or absence of the cag PAI; however the relationship between OipA and IL-18 is not understood.
The current study was designed to test the hypothesis that IL-18 is biologically important in H. pylori-infected mucosa as determined by quantitative analyses using real-time RT-PCR and ELISA. We measured mucosal IL-18 levels in human gastric mucosa as well as in isolated gastric epithelial cells and lamina propria mononuclear cells (LPMC) in relation to H. pylori infection and its virulence factors (cag PAI and OipA), and correlated IL-18 levels with gastric mucosal inflammation. We also examined in vitro IL-8 expression in both gastric epithelial cells and the monocytes in response to H. pylori infection in addition to the signaling pathways regulating H. pylori-infection induction of IL-18.
Materials and Methods
Patients and samples
Antral gastric mucosal samples were collected from H. pylori-infected Colombian patients with nonulcer dyspepsia. The samples included patients infected with cag PAI-positive/OipA-positive H. pylori isolates, cag PAI-negative/OipA-positive isolates, cag PAI-positive/OipA-negative isolates, and cag PAI-negative/OipA-negative isolates. H. pylori-positive status was evaluated by culture using standard methods as previously described (19). Multiple colonies on the plates were harvested en masse and three to six single colonies also were harvested. Genomic DNA and total proteins were extracted using a commercially available DNA extraction kit (Qiagen) and bacterial protein extraction kit (Pierce), respectively. The cag PAI status was determined by PCR as previously described (17). OipA status was determined by immunoblot as previously described (18). Controls consisted of gastric mucosal biopsies without H. pylori infection. The patients were proven to be H. pylori-negative by a combination of serology, histology, rapid urease test, and culture. The biopsy specimens were obtained under protocols approved by the ethics committees in Universidad Nacional de Colombia (Bogota, Colombia) and informed consent was obtained from all patients.
Isolation of gastric epithelial cells and lamina LPMC
In addition to the whole antral biopsy specimens, isolated gastric epithelial cells and gastric LPMC from some Colombian patients were used for examining the IL-18 mRNA levels. Gastric epithelial cells were isolated from freshly obtained antral gastric biopsy specimens from patients with and without H. pylori infection as previously described (20). In brief, biopsy specimens were washed in HBSS at 4°C, cut into small pieces (~0.4 mm) and washed in 10 ml of HBSS with 0.5 mM DTT for 5 min at 4°C under continuous stirring. Tissue was transferred and incubated with chelating buffer (27 mM trisodium citrate, 5 mM Na2HPO4, 96 mM NaCl, 8 mM KH2PO4, 1.5 mM KCl, 55 mM D-sorbitol, and 44 mM sucrose) for 7 min at 4°C. The supernatant was collected and centrifuged at 1,000 × g for 5 min at 4°C. Cells were resuspended in 200 μl of PBS. Cell viability was >88% as determined by staining with 0.1% trypan blue. Approximately 96% of cells had periodic acid-Schiff-positive material in the cytoplasm confirming that the population consisted of mucus producing epithelial cells with minimal contamination by other cells.
Gastric LPMC were isolated by the DTT-EDTA-collagenase sequence as previously described in detail (21). Briefly, antral biopsy specimens were freed of mucus and epithelial cells in sequential steps with DTT and EDTA and then digested with collagenase (Invitrogen). After collagenase digestion, the supernatant containing the mononuclear cells was collected and centrifuged at 500 × g for 10 min. After washing in calcium- and magnesium-free HBSS (Invitrogen), the pellet was resuspended in a 40% Percoll solution for further purification. Cell viability was >90%.
Histology
Antral biopsy specimens were fixed in 10% buffered formalin, embedded in paraffin, cut in sequential 4-μm sections, and stained with H&E and modified Giemsa stains. The histological severity of gastritis was blindly graded from normal to severe based on the degree of mononuclear cell (MNC) and polymorphonuclear leukocyte (PMN) infiltration, and atrophy according to the Updated Sydney system (22).
Quantitative analyses for IL-18 protein in the gastric mucosa using ELISA and immunoblot
IL-18 protein levels were determined in antral mucosal biopsy specimens. Gastric antral mucosal biopsy specimens were placed immediately in 200 μl PBS (pH 7.4), frozen on dry ice and stored at −80°C until use. Samples were homogenized using a tissue homogenizer (Kontes), and aliquots of supernatants from the homogenized tissues were obtained by centrifugation (10,000 × g for 10 min); total protein in the supernatants was measured by BCA protein assay reagent (Pierce) and IL-18 protein levels in the supernatants were determined by ELISA (Medical & Biological Laboratories). In our laboratory, the ELISA sensitivity for IL-18 was ~10 pg/ml. The ELISA detects mature IL-18; pro-IL-18 showed <1% cross-reactivity. The mucosal IL-18 levels were expressed as pg/mg protein.
We also performed immunoblot for IL-18 protein in the purified gastric epithelial cells and LPMC using anti-rabbit IL-18 polyclonal Ab (Santa Cruz Biotechnology), which detects both the precursor 24 K pro-IL-18 and 18 K mature IL-18. Protein extraction and immunoblotting were performed using standard techniques. For semiquantitative analysis, x-ray films were scanned and quantified using Image J 1.36 software (http://rsbweb.nih.gov/ij/) from the National Institutes of Health. The density of mature IL-18 was normalized to β-actin for quantitative analyses.
Quantitative analyses for IL-18 mRNA in the gastric mucosa using real-time RT-PCR
Total RNA was isolated from whole gastric biopsy specimens using RNeasy Mini kits (Qiagen). An aliquot containing 0.2 μg of total RNA was used for the reverse transcription reaction, which was conducted using the Superscript first-strand synthesis system (Invitrogen) according to the manufacturer’s instructions. The quantification of IL-18 mRNA levels was performed using an ABI Prism 7300 Sequence detection system (Applied Biosystems). Specific primers and TaqMan Probes were designed with the aid of the Primer Express program (Applied Biosystems). Forward and reverse primers for IL-18 were 5′-GACCAAGGAAATCGGCCTCTA-3′ and 5′-CCATACCTCTAGGCTGGCTATCTT-3′, respectively. The TaqMan probe was 5′-6-carboxyfluorescein (FAM)-ATTCTGACTGTA GAGATAATGCACCCCGGAC-6-carboxy-N, N, N′, N′-tetramethylrhodamine (TAMURA)-3′, synthesized by Applied Biosystems. A standard curve was constructed with 10-fold serial dilutions of a partial IL-18 cDNA that was subcloned into pCR2.1 TOPO (Invitrogen). Reaction mixtures for PCR (50 μl) were prepared by mixing 5 μl of synthesized cDNA solution, 2× TaqMan Universal Master mix (Applied Biosystems), 500 nM of each primer, and 250 nM of the TaqMan probe. The samples were placed in the analyzer and PCR was conducted according to the manufacturer’s instructions. To normalize the IL-18 mRNA expression level, GAPDH mRNA also was quantified in the same reactions using TaqMan GAPDH control reagents (Applied Biosystems). The expression levels of IL-18 mRNA were expressed as the ratio of IL-18 mRNA to GAPDH mRNA.
Cell culture
The human gastric epithelial cancer cell line AGS and human monocyte cell line THP-1 were obtained from American Type Culture Collection. Human gastric epithelial cancer cell lines MKN1, MKN7, MKN28, MKN45, and KATOIII were obtained from Riken Cell Bank (Tsukuba, Japan). Cell lines, except for KATOIII, were routinely maintained in a RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen), penicillin G (100 U/ml), and streptomycin (100 μg/ml) in 5% CO2-air at 37°C to 80% confluence; 20% heat-inactivated FBS was used for KATO III cells.
We also used primary gastric epithelial cells that were isolated enzymatically from uninfected adult human stomachs (noncancer containing normal appearing mucosa) as previously described (23). The method for purifying epithelial cells for cell culture experiments using surgically obtained tissues was different from that for biopsy specimens as described above. In brief, a piece of gastric mucosa (~3 cm2) was obtained from the normal appearing mucosa of the stomach at surgery. The patients were proven to be H. pylori-negative by a combination of serology, histology, and culture. The surface mucosal layer was removed with a razor blade, immediately minced and then incubated in Ham’s F-12 culture medium containing collagenase (0.2 mg/ml) (Invitrogen) for 10 min. Cells from the final incubation were washed and cultured in Ham’s F-12 medium supplemented with 10% FBS and streptomycin (300 μg/ml) in 5% CO2-air at 37°C. Epithelial cells were cultured in a 24-well collagen-coated dish at a final concentration of 106 cells/ml for 24 h before use. Cultured cells formed subconfluent monolayers within 24 h of the inoculation. Approximately 99% of cultured cells in the monolayers had periodic acid-Schiff-positive material in the cytoplasm. The patients gave informed consent, and the study was approved by the Human Research Committee of Kyoto Prefectural University of Medicine (Kyoto, Japan).
H. pylori culture and human cells cocultured with H. pylori
H. pylori strain TN2GF4 was used. H. pylori strain TN2GF4 was isolated from a Japanese gastric ulcer patient and was reported to induce gastric cancer in Mongolian gerbils (24). We also used its isogenic oipA mutants (25) and the whole cag PAI deleted mutants (26). H. pylori were cultured on brain heart infusion agar plates containing 7% horse blood in a microaerobic atmosphere at 37°C. After a 24- to 36-h culture, H. pylori were suspended in autoclaved saline, and the concentration was estimated by measuring the OD at 625 nm. Exponentially growing suspension cultures of H. pylori were added to human cells at a multiplicity of infection (MOI) ranging from 10 to 500. To avoid the influence of serum, human cells were serum starved for 16 h before experiments.
In some experiments, heat-killed H. pylori were used at the same MOI. H. pylori were heat-killed by boiling for 15 min in saline, followed by centrifugation; the supernatant or pellet of killed H. pylori was used. Killing was confirmed by lack of growth on blood agar plates. In some experiments, the same concentration of live bacteria was added to the upper well of a transwell plate (Falcon); the lower well contained gastric epithelial cells or THP-1 cells. In some experiments, human cells were pretreated with U0126 (a specific inhibitor of MAPK/ERK1/2), SB203580 (a specific inhibitor of p38), or the proteasome inhibitor MG-132, which inhibits NF-κB activation. We used 2 μM of each inhibitor according to our previous determination of optimal concentrations (23) as well as from our preliminary experiments (data not shown). All inhibitors were purchased from Calbiochem.
Analyses for IL-18 protein levels from human cells cocultured with H. pylori using ELISA and immunoblot
Exponentially growing H. pylori were added to the human cells in 24-well plates for 0 to 36 h and IL-18 protein levels released from human cells were assessed in duplicate using an ELISA and immunoblot, as described above.
Quantitative analyses for IL-18 mRNA levels from human cells cocultured with H. pylori using real-time RT-PCR
Exponentially growing H. pylori were added to human cells in 6-well dish for 0 to 24 h and total RNA was isolated from cocultured cells using RNeasy Mini kits (Qiagen). An aliquot containing 1 μg of total RNA was used for the reverse transcription reaction and IL-18 mRNA levels was measured by real-time RT-PCR as above. The expression levels of IL-18 mRNA were expressed as the ratio of IL-18 mRNA to GAPDH mRNA.
Luciferase plasmid and luciferase reporter gene assays
The pGL3–1375, 1375 bp 5′ flanking region of the human IL-18 promoter, and its deletion constructs, linked to the luciferase reporter gene of the pGL3 basic plasmid (Promega) were gifts from Dr. Koyama (University Hospital Frankfurt, Frankfurt, Germany) (27–29). AGS cells (plated into 24-well dishes at ~1 × 106/well) were transiently transfected with 500 ng of the plasmid together with 25 ng of phRL-TK (Renilla) plasmid using Lipofectamine 2000 reagent (Invitrogen). After 4 h culture, the medium was changed to antibiotic free medium followed by stimulation with H. pylori (MOI of 100) or without H. pylori (negative control) for 18 h. We then lysed the cells and measured firefly and Renilla luciferase activities using the dual luciferase reporter assay system (Promega) according to manufacturer’s instructions. The value for luciferase activity was normalized against the luciferase activity of a cotransfected phRL-TK plasmid (Promega). A total of 1 mM of sodium butyrate (Sigma-Aldrich) was used for positive control of IL-18 induction as previously described (28, 29).
EMSA analysis
Nuclear extracts of infected and uninfected human cells were prepared using hypotonic/nonionic detergent lysis as previously described (30). The concentration of extracted nuclear proteins was determined using the BCA protein assay reagent (Pierce) and equal amounts were used to bind to duplex oligonucleotides corresponding to the IL-18 specific PU.1, NF-κB, and AP-1 binding sites. The oligonucleotides used in this study are summarized in Table I. Binding reactions contained 10 –15 μg total protein, 5% glycerol, 12 mM HEPES, 80 mM NaCl, 1 mM DTT, 5 mM Mg2Cl, 0.5 mM EDTA, 1 μg of poly (dI-dC), and 40,000 cpm of 32P-labeled double-stranded oligonucleotide in a total volume of 20 μl. The nuclear proteins were incubated with the probe for 30 min at room temperature and then electrophoresed on 6% nondenaturing polyacrylamide gels in Tris-borate-EDTA buffer (22 mM Tris-HCl, 22 mM boric acid, 0.25 mM EDTA, pH 8). After electrophoresis, gels were dried and exposed for autoradiography using Kodak XAR film at −80°C and intensifying screens.
Table I.
Probes used in electrophoretic mobility shift assays
| Transcriptional Regulatory Site of Human IL-18 Promoter | Sequence |
|---|---|
| AP-1 | sense, 5′-CTATTTCAACTTGAGTCACGTATGTATTC-3′ |
| AP-1 | anti-sense, 5′-GAATACATACGTGACTCAAGTTGAAATAG-3′ |
| NF-κB | sense, 5′-TTGGGAGGAAGGGGAAGTCCTGAATGAGG-3′ |
| NF-κB | anti-sense, 5′-CCTCATTCAGGACTTCCCCTTCCTCCCAA-3′ |
| PU.1 | sense, 5′-CTCCACCTTCTTCCTCATTCTCTCC-3′ |
| PU.1 | anti-sense, 5′-GGAGAGAATGAGGAAGAAGGTGGAG-3′ |
The competition assays were performed by the addition of 5.0 pmol/L unlabeled competitors into the reaction mixture at the time of probe addition. In the gel shift assays, commercial Abs (anti-p50, anti-p60, and antic-Jun) (Santa Cruz Biotechnology) were added to the reaction mixtures, followed by incubation on ice for 1 h.
Statistical methods
All experiments were performed at least three times. Parametric data are presented as mean and SE and nonparametric data are presented as medians (25–75% quartiles). Statistical analysis was performed by Mann-Whitney Rank Sum test or nonpaired t test depending on the data set. A p value of <0.05 was accepted as statistically significant.
Results
Mucosal IL-18 protein levels in gastric mucosa
We initially examined 124 H. pylori-infected Colombian patients with nonulcer dyspepsia including 111 patients whose cag PAI and OipA status as determined using multiple colonies was identical with the status determined using single colonies from the same patient. Among these patients, 82 were infected with cag PAI-positive/OipA-positive isolates, 6 with cag PAI-negative/OipA-positive isolates, 5 with cag PAI-positive/OipA-negative isolates, and 18 with cag PAI-negative/OipA-negative isolates. Of these, we arbitrarily selected 20 patients infected with cag PAI-positive/OipA-positive isolates and 10 with cag PAI-negative/OipA-negative isolates, as well as all 11 patients with cag PAI-negative/OipA-positive or cag PAI-positive/OipA-negative isolates. We also selected 20 H. pylori-negative patients with nonulcer dyspepsia.
Gastric mucosal IL-18 protein levels were significantly higher in H. pylori-positive specimens (243 (131–370) pg/mg protein) than in H. pylori-negative specimens (21.2 (6.0 –24.0)) (p < 0.001). Importantly, among H. pylori infected patients, mucosal IL-18 protein levels were similar between cag PAI-positive/OipA-positive specimens (344 (173– 452)) and cag PAI-negative/OipA-positive specimens (278 (232–765)) (Fig. 1A). In contrast, cag PAI-positive/OipA-negative specimens and cag PAI-negative/OipA-negative specimens induced less IL-18 (198 (45.1–225) and 232 (104 –326)) compared with cag PAI-positive/OipA-positive specimens (p = 0.09 for both). However, IL-18 levels were still significantly higher in cag PAI-negative/OipA-negative specimens than in H. pylori-negative specimens (p < 0.001), indicating that H. pylori factors other than OipA and cag PAI likely also are involved in IL-18 induction.
FIGURE 1.

IL-18 levels in the antral gastric mucosa. Mucosal IL-18 protein levels (pg/mg protein) (A) and mRNA levels (IL-18/GAPDH × 104) (C) are expressed as box plots. The ends of the bars indicate the 25 and 75 percentiles. The median is indicated with a line, and 10 and 90 percentiles are indicated with error bars. Data also are displayed using symbols. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 compared with specimens without H. pylori infection by Mann-Whitney Rank Sum text (A) or non-paired t test (C). Mucosal IL-18 protein levels in isolated gastric epithelial cells and LPMC were measured by immunoblot (B). The number below the images represents the fold induction of IL-18/β-actin levels as compared with the levels of samples infected with cag PAI/OipA-positive strains.
We next examined IL-18 protein levels in isolated gastric epithelial cells and LPMC. For this purpose, we used five patients infected with cag PAI-positive/OipA-positive isolates, two with cag PAI-negative/OipA-positive isolates, two with cag PAI-positive/OipA-negative isolates, five with cag PAI-negative/OipA-negative isolates as well as five without H. pylori infection. Our attempts to isolate gastric epithelial cells and LPMC from additional samples with cag PAI-negative/OipA-positive isolates and/or cag PAI-positive/OipA-negative isolates were unsuccessful due to low cell viability. Due to the small number of cells, it was difficult to measure secreted IL-18 levels by ELISA; therefore, we performed immunoblot. A faint band corresponding to mature IL-18 was observed in uninfected specimens in the gastric epithelial cells; whereas a strong band was observed in cag PAI-positive/OipA-positive specimens (Fig. 1B). Importantly, only a weak band corresponding to mature IL-18 was observed in specimens lacking cag PAI and/or OipA.
In LPMC, a faint band corresponding to mature IL-18 was observed in uninfected specimens, and the density of the band was markedly enhanced both in cag PAI-positive/OipA-positive and cag PAI-negative/OipA-positive specimens (Fig. 1B). In contrast, the density was decreased in both cag PAI-positive/OipA-negative and cag PAI-negative/OipA-negative specimens. Overall, IL-18 production in the gastric epithelial cells was both cag PAI and OipA dependent; whereas IL-18 production in LPMC was OipA dependent, but cag PAI independent. The patterns observed in LPMC were similar to those in the whole biopsy specimens measured by ELISA, indicating that IL-18 was produced mainly from LPMC in the gastric mucosa.
Mucosal IL-18 mRNA levels in gastric mucosa
Antral mucosal IL-18 mRNA levels from the whole biopsy specimens correlated well with protein levels irrespective of H. pylori status (R = 0.84; p < 0.001 for H. pylori-positive cases and R = 0.61; p = 0.005 for H. pylori-negative cases). Accordingly, mucosal IL-18 mRNA levels were significantly higher in H. pylori-positive specimens (102 (82.2–114)) than in H. pylori-negative specimens (23.2 (12.4 –39.8)) (p < 0.001) (Fig. 1C). However, among H. pylori infected patients, mucosal IL-18 mRNA levels were independent of the cag PAI and OipA status. Overall, IL-18 protein levels in the whole biopsy specimens were dependent of OipA status; whereas IL-18 mRNA levels were not.
Relationship between mucosal IL-18 levels and gastric injury
The above data clearly showed that H. pylori induced IL-18 in the gastric mucosa. We next examined whether the IL-18 induced by H. pylori infection was related to gastric injury. Because tissue samples from all uninfected patients studied had normal histology (i.e., no PMN infiltration, no presence of atrophy and no to very mild MNC infiltration), we only compared samples obtained from H. pylori infected patients. IL-18 protein levels strongly correlated with MNC and PMN infiltration (p < 0.001 and in trend) (Table II). Cellular infiltration was closely related to OipA and cag PAI status (data not shown); accordingly, IL-18 mRNA levels only weakly correlated with MNC and PMN infiltration (p = 0.004 and p = 0.028, respectively) (Table II). IL-18 levels did not correlate with gastric atrophy (data not shown).
Table II.
IL-18 levels and cellular infiltration in H. pylori-infected tissuesa
| Histology Score (no.) | IL-18 Protein (pg/mg protein) | IL-18 mRNA (IL-18/GAPDH × 104) |
|---|---|---|
| MNC infiltration | ||
| 1 (14) | 80 (23–232) | 84 (65–105) |
| 2 (16) | 233 (213–336) | 105 (88 –116) |
| 3 (11) | 468 (380 – 804) | 120 (102–134) |
| p value | <0.001 | 0.004 |
| PMN infiltration | ||
| 0 (15) | 104 (24 –232) | 90 (65–109) |
| 1 (10) | 222 (198 –326) | 108 (86 –118) |
| 2 (7) | 357 (314 – 683) | 115 (93–119) |
| 3 (9) | 468 (370 – 661) | 117 (98 –143) |
| p value | <0.001 | 0.028 |
The p value was calculated by Kruskal-Wallis test.
Induction of IL-18 from cells cocultured with H. pylori
The ability of six gastric epithelial cancer cell lines to produce IL-18 was examined following coculture with H. pylori. Surprisingly, IL-18 protein levels were below the level of detection with all six gastric epithelial cancer cells irrespective of the presence or absence of H. pylori infection (MOI of 0 –500) during observation periods of up to 36 h (data not shown). IL-18 is synthesized as a precursor 24 K pro-IL-18 and must be cleaved by capase-1 to produce the mature bioactive peptide (31, 32). Caspase-1 adaptors ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain) also are essential for maturation of IL-18 (33). For example, AGS cells lack ASC and they cannot produce the mature IL-18 protein (our unpublished data). Immunoblot analyses showed that all the gastric cell lines tested were able to produce pro-IL-18 but were not able to process the precursor to the bioactive peptide, except for KATOIII cells (data not shown). Therefore, we examined the effect of H. pylori on IL-18 production from primary (noncancer) gastric epithelial cells. The subconfluent primary epithelial cells were cocultured with H. pylori (MOI of 100) or kept uninfected in a 24-well collagen-coated dish for 20 h. Cell viability was ~94% during the coculturing experiments (data not shown). Wild-type H. pylori infection resulted in the induction of IL-18 from the primary gastric epithelial cells (Fig. 2A).
FIGURE 2.
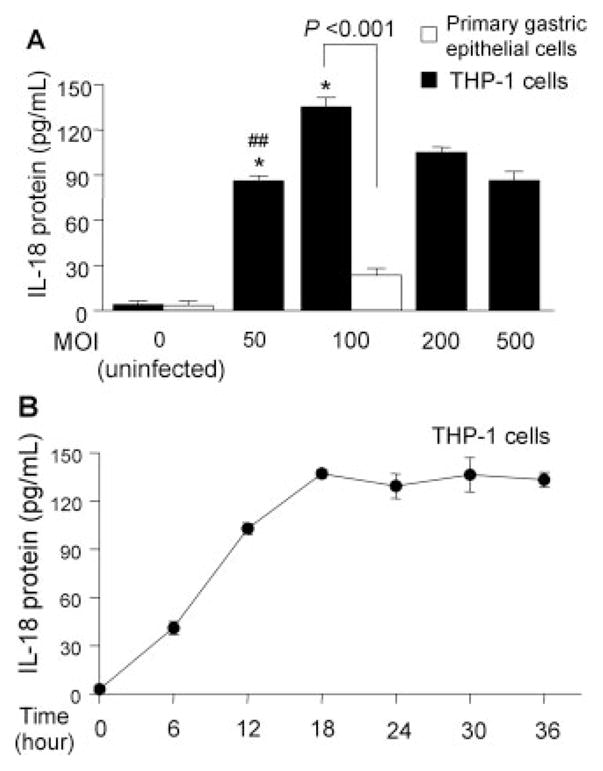
Induction of IL-18 protein expression by H. pylori infection from primary gastric epithelial cells and THP-1 cells. H. pylori were added to exponentially growing cells in 24-well plates at an MOI of 0–500 (THP-1 cells) or at an MOI of 100 (primary gastric epithelial cells) for 20 h (A); H. pylori were added to the exponentially growing THP-1 cells at an MOI of 100 for 0–36 h (B). IL-18 protein levels released from human cells were determined by ELISA. Three independent cocultures were performed and each was measured in duplicate. Data are expressed as mean ± SE. *, p < 0.001 compared with uninfected control; and #, p < 0.01 and ##, p < 0.001 compared with infected samples (MOI of 100) by nonpaired t test.
H. pylori infection also induced IL-18 protein from THP-1 cells and coculture resulted in much higher IL-18 levels compared with primary gastric epithelial cells (Fig. 2A). The increase in IL-18 production was directly proportional to the MOI peaking at an MOI of 100 and then decreasing at MOIs of 200 or greater, probably due to toxicity of H. pylori at high density. The induction levels were reproducible at an MOI of 100 and subsequent experiments used that MOI. IL-18 protein reached maximal levels at ~18 h post inoculation and then plateaued during an observation period of 36 h in THP-1 cells (Fig. 2B).
To investigate the effects of virulence factors of H. pylori on the IL-18 production, we cocultured cells with oipA mutants or with cag PAI mutants (Fig. 3). IL-18 was not induced from primary gastric epithelial cells cocultured with either the oipA mutants or the cag PAI mutants whereas IL-18 induction was similar to wild type in THP-1 cells using the cag PAI mutants (121.5 ± 12 pg/ml) and was slightly, but significantly decreased with the oipA mutants (94.3 ± 8 pg/ml) (p < 0.01 vs live wild-type strains). The data from in vitro coculturing experiments using gastric epithelial cells and THP-1 cells were identical with the data from in vivo measurement of IL-18 protein using isolated gastric epithelial cells and LPMC, respectively.
FIGURE 3.

Effect of viability and the H. pylori-cell interaction, OipA, and cag PAI on IL-18 protein production. H. pylori (wild-type H. pylori, heat-killed wild-type H. pylori strains, oipA mutants, or cag PAI mutants) (MOI of 100) were added to exponentially growing cells (primary gastric epithelial cells and THP-1 cells) in 24-well plates. We inhibited the interaction of wild-type H. pylori strains (MOI of 100) in both cell types using a polyester membrane to prevent direct bacteria-cell interaction. After 20 h coculture, IL-18 protein levels released from human cells were determined by ELISA. Three independent cocultures were performed and each was measured in duplicate. Data are expressed as mean ± SE. *, p < 0.001 compared with uninfected control; and #, p < 0.05, ##, p < 0.01 and ###, p < 0.001 compared with wild-type H. pylori infected samples by nonpaired t test.
To examine whether IL-18 induction required live H. pylori, we cocultured cells with heat-killed H. pylori for 18 h (Fig. 3). No IL-18 protein was produced from primary gastric epithelial cells but IL-18 protein was induced from THP-1 cells by heat-killed H. pylori (112.3 ± 6 pg/ml). The levels were slightly lower than those achieved with live wild-type H. pylori (135.4 ± 6 μg/ml) (Fig. 3), clearly showing that unknown heat-resistant factors in H. pylori unrelated to cag PAI and OipA also are involved in IL-18 induction by THP-1 cells.
We used a polyester membrane to separate the bacteria from the cells thus preventing direct H. pylori-cell interaction. Prevention of bacterial-cell attachment resulted in the prevention of IL-18 induction from both primary gastric epithelial cells and THP-1 cells (Fig. 3) suggesting that a direct interaction was necessary for IL-18 production irrespective of the cell type.
IL-18 mRNA expression from cells cocultured with H. pylori
We used RT-PCR to test whether IL-18 mRNA expression paralleled changes in IL-18 protein levels. Absolute quantitative RT-PCR on the six gastric epithelial cell lines and the primary gastric epithelial cells cocultured with H. pylori (MOI of 100) were performed 3 h post inoculation. IL-18 mRNA was significantly up-regulated by H. pylori infection in four of the six gastric cell lines and in the primary gastric epithelial cells (Fig. 4A). Cell viability of primary gastric epithelial cells was ~96% during the coculturing experiments (data not shown). Due to the limitations associated with primary gastric epithelial cells (e.g., difficulty obtaining sufficient cell numbers), we used AGS cells in subsequent experiments based on the fact that the mRNA in AGS cells was similar to those obtained from primary cells. An additional advantage to the use of AGS cells is that they are easily transfected with plasmids whereas this proved difficult with primary cells, MKN1, and MKN7 cells (data not shown). Transfection also was not successful in THP-1 cells (data not shown).
FIGURE 4.

IL-18 mRNA expression by H. pylori infection from gastric epithelial and THP-1 cells. H. pylori were added to the exponentially growing cells in 6-well plates at an MOI of 100 for 3 h (A) or for 0 –24 h (B). IL-18 mRNA levels were measured by absolute quantitative real-time RT-PCR. The expression levels of IL-18 mRNA were normalized by those of GAPDH mRNA. The expression levels of IL-18 mRNA were expressed as fold induction relative to uninfected control in AGS cells (A) or as the ratio of IL-18 mRNA to GAPDH mRNA (B). At least three independent cocultures were performed and each was measured in triplicate. Data are expressed as mean ± SE and p value was calculated by nonpaired t test. Primary, Primary gastric epithelial cells.
Similar to the protein levels, IL-18 mRNA levels in both AGS and THP-1 cells increased in proportion to the MOI and peaked at an MOI of 100, then decreased at MOIs of 200 or greater (data not shown). Although AGS cells spontaneously expressed IL-18 mRNA, IL-18 mRNA levels in both cell types significantly increased following H. pylori infection (MOI of 100) reaching maximal levels within 3– 6 h and then gradually decreasing to a baseline (Fig. 4B). Subsequent experiments examining IL-18 mRNA levels used an MOI of 100 and coculturing for 3 h.
As with IL-18 protein levels, IL-18 mRNA levels in AGS cells did not increase following infection with the oipA mutants (1.0 ± 0.2-fold induction of uninfected control) or the cag PAI mutants (1.0 ± 0.2-fold induction of uninfected control) (Fig. 5). IL-18 mRNA levels in THP-1 cells were similar with live H. pylori irrespective the presence or absence of functional OipA or the cag PAI. Overall, IL-18 protein levels, but not IL-18 mRNA levels, were lower using the oipA mutants in THP-1 cells, suggesting that OipA may play a role in regulating the posttranscriptional levels in THP-1 cells.
FIGURE 5.

Effect of H. pylori viability and H. pylori-cell interaction, OipA, and cag PAI on IL-18 mRNA expression. H. pylori (wild-type H. pylori, heat-killed wild-type H. pylori strains, oipA mutants, or cag PAI mutants) (MOI of 100) were added to exponentially growing cells in 6-well plates. We inhibited the interaction of wild-type H. pylori strains (MOI of 100) with cells using a polyester membrane to inhibit any direct bacteria-cell interaction. After 3-h coculture, IL-18 protein levels released from human cells were determined by absolute quantitative real-time RT-PCR. The expression levels of IL-18 mRNA were expressed as the ratio of IL-18 mRNA to GAPDH mRNA. At least three independent cocultures were performed and each was measured in triplicate. Data are expressed as mean ± SE. *, p < 0.001 compared with uninfected control and #, p < 0.001 compared with wild-type H. pylori infected samples by nonpaired t test.
Similar to the results with IL-18 protein production from primary gastric epithelial cells, H. pylori-induced IL-18 mRNA levels in AGS cells were significantly reduced when using killed H. pylori (0.9 ± 0.1-fold induction of uninfected control). In contrast, IL-18 mRNA was induced in THP-1 cells (3.9 ± 0.1-fold induction of uninfected control) at only slightly lower levels than those associated with live wild-type H. pylori (5.0 ± 0.2-fold induction of uninfected control; p = 0.2) (Fig. 5). Inhibition of direct H. pylori-cell interactions completely prevented induction of IL-18 mRNA synthesis in AGS cells and THP-1 cells (Fig. 5).
Effect of inhibition of MAPK and NF-κB pathways on IL-18 induction
To investigate the signal transduction pathways involved in H. pylori-associated IL-18 induction, we preincubated cells with chemical inhibitors or DMSO (for the infected control) at 37°C for 1 h followed by infection with wild-type H. pylori. U0126, an inhibitor of MEK1/2 and MG-132, an inhibitor of NF-κB significantly inhibited the induction of IL-18 protein and IL-18 mRNA in both gastric epithelial cells (primary cells for protein and AGS cells for mRNA) and THP-1 cells (Fig. 6). SB203580, an inhibitor of p38, also significantly inhibited the production of IL-18 protein both in primary gastric epithelial cells and THP-1 cells; whereas SB203580 had no effect on IL-18 mRNA levels in either AGS cells or THP-1 cells. These data suggest that p38 plays a role at the posttranscriptional level.
FIGURE 6.

Effect of inhibitors of signal transduction on IL-18 induction following H. pylori infection. Human cells were incubated with SB203580, U0126, or MG-132 for 1 h and subsequently cocultured with wild-type H, pylori strains at an MOI of 100 for 20 h (A) or 3 h (B). IL-18 protein levels released from human cells were determined by ELISA (A) and IL-18 mRNA expression levels by absolute quantitative real-time RT-PCR normalized to GAPDH mRNA. At least three independent cocultures were performed and each was measured in triplicate. Data are expressed as mean ± SE. *, p < 0.01 and **, p < 0.001 compared with wild-type H. pylori infected controls without inhibitors by nonpaired t test.
The fact that IL-18 protein levels, but not IL-18 mRNA levels in THP-1 cells were reduced following infection with the oipA mutants or the p38 inhibitor suggests a possible relationship between OipA and the p38 pathway. This result also is consistent with prior studies showing that in gastric epithelial cells, OipA, but not the cag PAI induced the p38 pathway (23, 26). To test the hypothesis that OipA plays a role in p38 induction in THP-1 cells, we preincubated cells with chemical inhibitors or DMSO at 37°C for 1 h followed by infection with the oipA mutants. IL-18 protein levels induced by the oipA mutants were unchanged by adding the SB203580 (102.6 ± 7 pg/ml for oipA mutants with DMSO and 96.0 ± 7 pg/ml for oipA mutants with SB203580) consistent with the proposed relationship between OipA and p38 pathway in THP-1 cells. We also used immunoblot analyses with phospho-p38 Ab and total p38 Ab (both from Cell Signaling Technology) to confirm that wild-type H. pylori but not the oipA mutants induced phosphorylation of p38 in THP-1 cells (our unpublished data).
Effect of H. pylori on the IL-18 promoter activity
To confirm our hypothesis that the induction in IL-18 mRNA associated with H. pylori infection was due to transcriptional regulation at the promoter level, we transfected the pGL3–1375 plasmid into AGS cells followed by the infection of wild-type H. pylori, oipA mutants or cag PAI mutants (Fig. 7A). Wild-type H. pylori significantly induced luciferase activity of the IL-18 promoter (1.6 ± 0.1-fold induction of uninfected control; p < 0.001). Similar to the results for IL-18 mRNA expression, the IL-18 promoter activity in AGS cells was not increased with heat-killed H. pylori or by the prevention of an H. pylori-AGS cells interaction. The promoter activity was significantly decreased using the cag PAI mutants and the oipA mutants (1.2 ± 0.3-fold and 1.3 ± 0.1-fold induction of uninfected control, respectively) compared with wild-type H. pylori. However, both mutants significantly induced luciferase activity compared with the uninfected control (p < 0.001 for oipA mutants and p < 0.05 for cag PAI mutants). This result differed from results of IL-18 mRNA expression suggesting that OipA and the cag PAI partially affect the expression of IL-18 mRNA after promoter activation. The basal promoter activity was relatively low. However, there were low interexperimental variations of IL-18 promoter activity and the addition of 1 mM sodium butyrate (positive control) resulted in a 2.1-fold induction of IL-18 promoter activity compared with nontreated AGS cells (data not shown).
FIGURE 7.

IL-18 promoter activation by H. pylori infection. A, AGS cells were transiently transfected with pGL3–1375 plasmid, together with Renilla plasmid by using Lipofectamine 2000 and subsequently incubated with saline (uninfected control) or H. pylori (wild-type H. pylori, heat-killed wild-type H. pylori strains, oipA mutants, or cag PAI mutants). We also inhibited the interaction of wild-type H. pylori strains with both cell types by a polyester membrane. After 18-h coculture, we analyzed luciferase activity by dual luciferase reporter assay. Luciferase activity induced by H. pylori infection is presented as normalized luciferase activity expressed as fold-increase in H. pylori-infected cells relative to uninfected controls. *, p < 0.01 and **, p < 0.001 compared with uninfected control, and #, p < 0.05, ##, p < 0.01 and ###, p < 0.001 compared with wild-type H. pylori infected samples. B, AGS cells were transiently transfected with the 5′ deletion mutants in the pGL3 plasmid backbone together with Renilla plasmid by Lipofectamine 2000; subsequently, the cultures were incubated with saline (uninfected control) or wild-type H. pylori. The relative lengths of the mutants are indicated by solid lines. After an 18-h coculture, we analyzed luciferase activity by the dual luciferase reporter assay system. The value for luciferase activity was normalized to the luciferase activity of a cotransfected Renilla plasmid. For experiments (A and B), at least three independent transfections, each done in triplicate, were performed. Data are expressed as mean ± SE and p value was calculated by nonpaired t test.
To confirm the signaling transduction pathways involved in H. pylori-induced IL-18 promoter activation, we preincubated the pGL3–1375-transfected AGS cells with chemical inhibitors or DMSO (for the infected control) at 37°C for 1 h followed by the wild-type H. pylori infection. Consistent with the results of real-time RT-PCR for IL-18 mRNA, H. pylori-inducible luciferase activity was not increased by preincubation of U0126 or MG-132, but was increased by that of SB203580 (data not shown).
Effect of the IL-18 promoter deletions on H. pylori-inducible luciferase activity
To define the necessary transcriptional regulatory region of the IL-18 promoter involved in the induction of IL-18 by H. pylori infection, a series of deletion constructs derived from pGL3–1375 were transfected into AGS cells followed by the infection of wild-type H. pylori. pGL3–972 (−972 to −1) contained binding sites for STAT, NF-κB, OCT-1, SP2, and PU.1 with deletion of the AP-1 binding site. pGL3–515 (−515 to −1) has a further deleted STAT binding site, pGL3–315 (−315 to −1) had a further deleted NF-κB binding site, pGL3–108 (−108 to −1) had a further deleted OCT-1 binding site, pGL3– 67 (−67 to −1) had a further deleted SP2 binding site, and pGL3–25 (−25 to −1) had an additionally deleted PU.1 binding site.
H. pylori inducible luciferase activity was reduced in all transfectants (Fig. 7B). Luciferase activity for pGL3–972 (−972 to −1) was independent of the H. pylori infection, suggesting that the AP-1 binding site may play an important role in the IL-18 gene transcription.
Analysis for the binding sites in IL-18 promoter of H. pylori infection induced DNA-binding proteins
We next performed EMSA using nuclear extracts from AGS cells or THP-1 cells cultured with or without wild-type H. pylori. Complexes of nuclear proteins (extracted from AGS cells) that bound to both the NF-κB and the AP-1 sites in the IL-18 promoter region were observed. The intensities of these complexes (indicated by arrow) were increased following H. pylori infection (Fig. 8, lane 2). These complexes were fully competed with the addition of unlabeled NF-κB or AP-1 oligonucleotide as shown by complete disappearance of the bands (lane 3). In addition, to confirm whether these sites in the IL-18 promoter were involved in the complex formation, we performed supershift analysis using specific Abs (anti-c-Jun for AP-1; anti-p50 and -p65 for NF-κB). The complexes were markedly supershifted by the addition of each specific Ab (lane 4 or later). THP-1 cells showed the similar results (data not shown). We also performed EMSA using a specific probe for PU.1 of the human IL-18 promoter gene; however PU.1 activation was independent of the H. pylori infection (data not shown).
FIGURE 8.
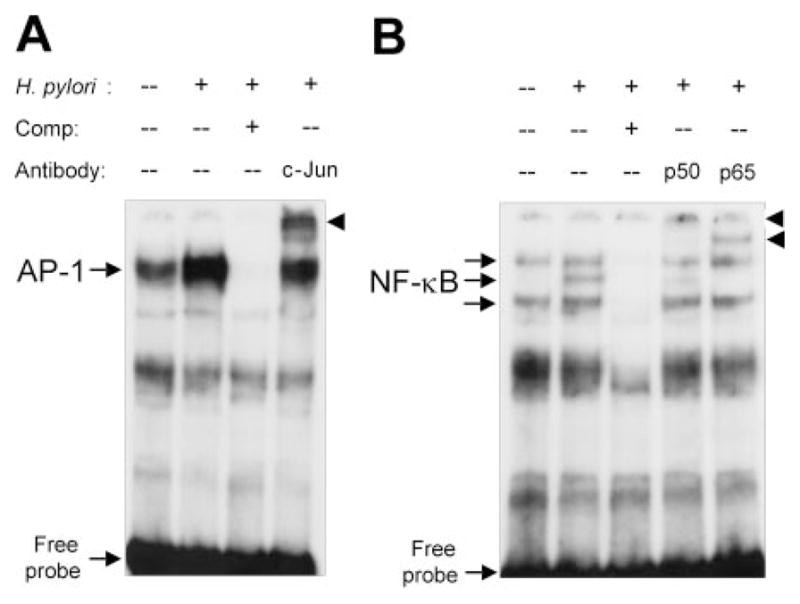
EMSA for binding of nuclear proteins to AP-1 (A) and NF-κB (B) sites in the human IL-18 promoter gene. EMSAs were performed using the nuclear proteins extracted from H. pylori-infected AGS cells as described in Materials and Methods. Lane 1, nuclear proteins from uninfected cells; lane 2, nuclear proteins from 2-h wild-type H. pylori infected cells; lane 3, competition analysis (presence of 100-fold excess of unlabeled competitors); lane 4, supershift analysis using anti-c-Jun (A) or anti-p50 (B); lane 5, supershift analysis using anti-p65.
Discussion
IL-18 normally is produced from both macrophage-like cells (8, 9, 34) and from some epithelial cells, such as intestinal epithelial cells (35). This study was designed to test the hypothesis that IL-18 expression as measured in H. pylori infected gastric mucosa reflected expression from both epithelial cells and from H. pylori-associated inflammatory infiltrates. We clearly confirmed that H. pylori induced both IL-18 mRNA and protein in the gastric mucosa using quantitative analyses. In addition, strong correlation between mucosal IL-18 levels and gastric inflammation suggest that H. pylori-induced IL-18 plays an important role in gastric inflammation via direct effects of elevated IL-18 or via indirect effects of IL-18 such that effects of IFN-γ, which is a well-known proinflammatory cytokine induced by IL-18.
We showed for the first time that IL-18 levels were up-regulated both in the gastric epithelial cells and LPMC isolated from H. pylori-infected gastric mucosa. Importantly, the behavior of gastric epithelial cells and monocytes differed markedly in response to the virulent factors of H. pylori; cag PAI and OipA. Previous studies (23, 25, 26) have suggested that both cag PAI and OipA were involved in the induction of various cytokines and chemokines in the gastric mucosa, such as IL-6, IL-8, and RANTES cells. Although the relationship between OipA and IL-8 remains unclear in epithelial cells (36), there is agreement that the cag PAI does not play a role in the induction of these cytokines in THP-1 cells (37). As shown here, both cag PAI and OipA were involved in IL-18 induction from gastric epithelial cells; whereas the cag PAI was not involved in IL-18 induction in monocytes, which was confirmed both by direct measurement of IL-18 in the isolated LPMC and measurement of IL-18 induced by coculturing of H. pylori with THP-1 cells. Of interest, OipA was involved in induction of IL-18 protein, but not in IL-18 mRNA in monocytes. In addition, we showed that OipA was involved in activation of the p38 pathway and that the p38 pathway also regulated IL-18 induction at a posttranscriptional level in both gastric epithelial cells and in monocytes. This effect possibly is related to p38 functioning as an mRNA stabilizer (38, 39). Further studies will be necessary to examine the details of the OipA→p38 pathways in mRNA stabilization as well as other posttranscriptional regulation events.
Of interest, the behavior of gastric epithelial cells and monocytes also differed markedly in response to heat-killed H. pylori. Haeberle et al. (40) previously reported both live and killed H. pylori were able to induce IL-12 and IL-10 from peripheral blood leukocytes whereas with H. pylori inducing significantly more IL-10 than with live organisms. We also previously reported the release of IL-6 not IL-8 following treatment of gastric cancer cell lines with heat-killed H. pylori (23). It remains unclear whether the different responses of epithelial and inflammatory cells to heat-killed H. pylori is restricted to IL-18 or whether the phenomenon is shared by other cytokines. One difference between gastric epithelial cells and THP-1 cells relates to the fact that THP-1 cells are professional phagocytes whereas gastric epithelial cells are not (41). Although a role for some soluble H. pylori proteins (e.g., urease) has been suggested in relation to IL-10 induction by killed H. pylori (40), separation of THP-1 cells and the bacteria using polyester membranes abrogated the response, providing compelling evidence that cell-bacteria interaction rather than soluble proteins mediate the response. Overall, our data suggest that direct attachment of H. pylori and cells along with the participation of the cag PAI and OipA are required to trigger H. pylori-associated IL-18 induction from gastric epithelial cells. In contrast, H. pylori appear to interact with monocytes differently although even both live H. pylori and OipA were required for maximal induction of IL-18 production.
We showed that IL-18 was produced mainly from LPMC in the gastric mucosa. Although H. pylori generally are attached to the gastric epithelial cells, but not to infiltrating cells, it is known that H. pylori and/or H. pylori products likely have many opportunities to come into direct contact with intraepithelial monocytes in the inflamed mucosa. For example, tight junctions are opened by the movement of polymorphonuclear cells, which is thought to result in increased permeability (42). H. pylori also are known to invade the gastric mucosa making whole cell Ags available to phagocytic cells (43). Finally, it has been postulated from in vitro studies that H. pylori itself may affect tight junction activity (44). The induction patterns of IL-18 in LPMC in H. pylori-positive patients were similar to those in THP-1 cells directly cocultured with H. pylori, especially in relation to the virulence factors, suggesting that H. pylori-associated IL-18 induction in the gastric mucosa in vivo occurs by direct contact with intraepithelial monocytes.
We showed that H. pylori up-regulated expression of IL-18 mRNA and the mature IL-18 protein in primary gastric epithelial cells whereas gastric epithelial cancer cell lines, with the exception of KATOIII cells, did not produce the mature IL-18 protein. However, we were unable to confirm recent studies suggesting that H. pylori could induce IL-18 from AGS cells (45). In fact, we were unable to detect the mature IL-18 protein from AGS cells either by ELISA or by immunoblot. Our preliminary studies showed that the AGS cells we used did not produce ASC (our unpublished data), which is essential for IL-18 maturation. IL-18 plays a critical role in the host defensive immune response against tumors (10, 46) and we speculate that mutations such as those blocking ASC function occur during carcinogenesis. Further studies will be necessary to determine whether the loss of ability to produce mature IL-18 in gastric cancer cells is biologically important.
One limitation of our study includes the use of primary gastric epithelial cells, which are unlikely to be perfectly pure cultures. Importantly, only ~1.5% of cultured cells in the monolayers did not express periodic acid-Schiff-positive material in their cytoplasm. It remains possible that the low levels of mature IL-18 produced from the primary gastric cell cultures originated from contaminating nonepithelial cells. Subsequent studies will be required that immunolocalize IL-18 within the gastric mucosa to define the relative contributions of the epithelial and inflammatory cells in terms of IL-18 expression.
We examined the transcriptional regulation of IL-18 promoter activity by luciferase reporter gene assay and EMSA. Luciferase reporter gene assay using serial deleted plasmids and EMSA showed that AP-1 played a major role in IL-18 transcription in AGS cells. It is well known that MAPK pathways, especially the ERK and JNK pathways, are involved in AP-1 activation. The fact that inhibition of the ERK pathway eliminated H. pylori-related IL-18 induction, and that supershift analysis showed that c-Jun, which is downstream of JNK, bound to the AP-1 site of the IL-18 promoter, support the hypothesis that H. pylori activated ERK/JNK→AP-1 pathways is followed by IL-18 induction in AGS cells. Interestingly, the same pathways also play important roles in THP-1 cells. EMSA and chemical inhibitor experiments showed that the NF-κB pathway also is involved in IL-18 induction in both AGS cells and THP-1 cells. Overall, although actual components of H. pylori (cag PAI, OipA, or unknown heat-resistant factors) may differ between gastric epithelial cells and THP-1 cells, the major signaling pathways appear to be similar.
Finally, we also examined the binding of the transcription factor PU.1 to the IL-18 promoter. Kalina et al. (28) previously reported that the regulatory region-related to the sodium butyrate-induced expression of IL-18 in THP-1 cells was the PU.1 consensus binding site. However, we failed to detect a relationship between PU.1 binding and H. pylori-induced IL-18 induction, suggesting that the signaling pathways of H. pylori-induced IL-18 differ from that of sodium butyrate induced IL-18 in THP-1 cells.
Acknowledgments
We thank Dr. Oscar Gutierrez and Diana Cittelly (Universidad Nacional de Colombia, Bogota, Colombia), Dr. Takeshi Okanoue, Dr. Shoji Mitsufuji, and Masakazu Kita (Kyoto Prefectural University of Medicine, Kyoto, Japan) for providing and handling clinical samples. We thank Dr. Hala M. El-Zimaity (Baylor College of Medicine, Houston, Texas) for histological analyses for Colombian samples. We also thank Dr. Noriko Koyama (University Hospital Frankfurt, Germany) for providing IL-18 reporter plasmids, and Dr. Tsutomu Katsuyama, Dr. Hiroyoshi Ota, and Junya Masumoto (Shinshu University School of Medicine, Nagano, Japan) for valuable suggestions.
Footnotes
Abbreviations used in this paper: PAI, pathogenicity island; LPMC, lamina propria mononuclear cell; DTT, dithiothreitol; MNC, mononuclear cell; PMN, polymorphonuclear leukocyte; MOI, multiplicity of infection.
This work was supported in part by the Office of Research and Development Medical Research Service Department of Veterans Affairs and by Public Health Service Grant DK56338, which funds the Texas Gulf Coast Digestive Diseases Center. Y.Y. is supported in part by National Institutes of Health Grant DK 62813.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, Luthra GK, Brooks EG, Graham DY, Reyes VE, Ernst PB. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology. 1998;114:482–492. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 3.D’Elios MM, Manghetti M, De CM, Costa F, Baldari CT, Burroni D, Telford JL, Romagnani S, Del PG. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158:962–967. [PubMed] [Google Scholar]
- 4.Karttunen R, Karttunen T, Ekre HP, MacDonald TT. Interferon γ and interleukin 4 secreting cells in the gastric antrum in Helicobacter pylori positive and negative gastritis. Gut. 1995;36:341–345. doi: 10.1136/gut.36.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawai N, Kita M, Kodama T, Tanahashi T, Yamaoka Y, Tagawa Y, Iwakura Y, Imanishi J. Role of γ interferon in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Infect Immun. 1999;67:279–285. doi: 10.1128/iai.67.1.279-285.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smythies LE, Waites KB, Lindsey JR, Harris PR, Ghiara P, Smith PD. Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-γ, gene-deficient mice. J Immunol. 2000;165:1022–1029. doi: 10.4049/jimmunol.165.2.1022. [DOI] [PubMed] [Google Scholar]
- 7.Akhiani AA, Pappo J, Kabok Z, Schon K, Gao W, Franzen LE, Lycke N. Protection against Helicobacter pylori infection following immunization is IL-12-dependent and mediated by Th1 cells. J Immunol. 2002;169:6977–6984. doi: 10.4049/jimmunol.169.12.6977. [DOI] [PubMed] [Google Scholar]
- 8.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, Hattori K, et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 9.Ushio S, Namba M, Okura T, Hattori K, Nukada Y, Akita K, Tanabe F, Konishi K, Micallef M, Fujii M, et al. Cloning of the cDNA for human IFN-γ-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J Immunol. 1996;156:4274–4279. [PubMed] [Google Scholar]
- 10.Coughlin CM, Salhany KE, Wysocka M, Aruga E, Kurzawa H, Chang AE, Hunter CA, Fox JC, Trinchieri G, Lee WM. Interleukin-12 and interleukin-18 synergistically induce murine tumor regression which involves inhibition of angiogenesis. J Clin Invest. 1998;101:1441–1452. doi: 10.1172/JCI1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Micallef MJ, Ohtsuki T, Kohno K, Tanabe F, Ushio S, Namba M, Tanimoto T, Torigoe K, Fujii M, Ikeda M, et al. Interferon-γ-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-γ production. Eur J Immunol. 1996;26:1647–1651. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- 12.Tone M, Thompson SA, Tone Y, Fairchild PJ, Waldmann H. Regulation of IL-18 (IFN-γ-inducing factor) gene expression. J Immunol. 1997;159:6156–6163. [PubMed] [Google Scholar]
- 13.Noguchi Y, Wada H, Marino MW, Old LJ. Regulation of IFN-γ production in granulocyte-macrophage colony-stimulating factor-deficient mice. Eur J Immunol. 1998;28:3980–3988. doi: 10.1002/(SICI)1521-4141(199812)28:12<3980::AID-IMMU3980>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Akhiani AA, Schon K, Lycke N. Vaccine-induced immunity against Helicobacter pylori infection is impaired in IL-18-deficient mice. J Immunol. 2004;173:3348–3356. doi: 10.4049/jimmunol.173.5.3348. [DOI] [PubMed] [Google Scholar]
- 15.Tomita T, Jackson AM, Hida N, Hayat M, Dixon MF, Shimoyama T, Axon AT, Robinson PA, Crabtree JE. Expression of interleukin-18, a Th1 cytokine, in human gastric mucosa is increased in Helicobacter pylori infection. J Infect Dis. 2001;183:620–627. doi: 10.1086/318541. [DOI] [PubMed] [Google Scholar]
- 16.Fera MT, Carbone M, Buda C, Aragona M, Panetta S, Giannone M, La TF, Giudice A, Losi E. Correlation between Helicobacter pylori infection and IL-18 mRNA expression in human gastric biopsy specimens. Ann NY Acad Sci. 2002;963:326–328. doi: 10.1111/j.1749-6632.2002.tb04125.x. [DOI] [PubMed] [Google Scholar]
- 17.Yamaoka Y, Kikuchi S, El-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123:414–424. doi: 10.1053/gast.2002.34781. [DOI] [PubMed] [Google Scholar]
- 18.Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity HM, Reddy R, Arnqvist A, Graham DY. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut. 2006;55:775–781. doi: 10.1136/gut.2005.083014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaoka Y, Kodama T, Kita M, Imanishi J, Kashima K, Graham DY. Relationship of vacA genotypes of Helicobacter pylori to cagA status, cytotoxin production, and clinical outcome. Helicobacter. 1998;3:241–253. doi: 10.1046/j.1523-5378.1998.08056.x. [DOI] [PubMed] [Google Scholar]
- 20.Sebkova L, Pellicano A, Monteleone G, Grazioli B, Guarnieri G, Imeneo M, Pallone F, Luzza F. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori-infected human gastric epithelial cells. Infect Immun. 2004;72:5019–5026. doi: 10.1128/IAI.72.9.5019-5026.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 22.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis: the updated Sydney System, international workshop on the histopathology of gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161–1181. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell. 2005;16:4954–4966. doi: 10.1091/mbc.E05-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 25.Yamaoka Y, Kwon DH, Graham DY. A M (r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA. 2000;97:7533–7538. doi: 10.1073/pnas.130079797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudo T, Lu H, Wu JY, Graham DY, Casola A, Yamaoka Y. Regulation of RANTES promoter activation in gastric epithelial cells infected with Helicobacter pylori. Infect Immun. 2005;73:7602–7612. doi: 10.1128/IAI.73.11.7602-7612.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koyama N, Hoelzer D, Ottmann OG. Regulation of human IL-18 gene expression: interaction of PU.1 with GC-box binding protein is involved in human IL-18 expression in myeloid cells. Eur J Immunol. 2004;34:817–826. doi: 10.1002/eji.200324420. [DOI] [PubMed] [Google Scholar]
- 28.Kalina U, Koyama N, Hosoda T, Nuernberger H, Sato K, Hoelzer D, Herweck F, Manigold T, Singer MV, Rossol S, Bocker U. Enhanced production of IL-18 in butyrate-treated intestinal epithelium by stimulation of the proximal promoter region. Eur J Immunol. 2002;32:2635–2643. doi: 10.1002/1521-4141(200209)32:9<2635::AID-IMMU2635>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 29.Koyama N, Koschmieder S, Tyagi S, Nurnberger H, Wagner S, Bocker U, Hoelzer D, Gerhard OO, Kalina U. Differential effects of histone deacetylase inhibitors on interleukin-18 gene expression in myeloid cells. Biochem Biophys Res Commun. 2002;292:937–943. doi: 10.1006/bbrc.2002.6753. [DOI] [PubMed] [Google Scholar]
- 30.Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology. 2004;126:1030–1043. doi: 10.1053/j.gastro.2003.12.048. [DOI] [PubMed] [Google Scholar]
- 31.Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, et al. Activation of interferon-γ inducing factor mediated by interleukin-1β converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 32.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, et al. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 33.Ozoren N, Masumoto J, Franchi L, Kanneganti TD, Body-Malapel M, Erturk I, Jagirdar R, Zhu L, Inohara N, Bertin J, et al. Distinct roles of TLR2 and the adaptor ASC in IL-1β/IL-18 secretion in response to Listeria monocytogenes. J Immunol. 2006;176:4337–4342. doi: 10.4049/jimmunol.176.7.4337. [DOI] [PubMed] [Google Scholar]
- 34.Akita K, Ohtsuki T, Nukada Y, Tanimoto T, Namba M, Okura T, Takakura-Yamamoto R, Torigoe K, Gu Y, Su MS, et al. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J Biol Chem. 1997;272:26595–26603. doi: 10.1074/jbc.272.42.26595. [DOI] [PubMed] [Google Scholar]
- 35.Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, Luzza F, Fusco A, Pallone F. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol. 1999;163:143–147. [PubMed] [Google Scholar]
- 36.Dossumbekova A, Prinz C, Mages J, Lang R, Kusters JG, van Vliet AH, Reindl W, Backert S, Saur D, Schmid RM, Rad R. Helicobacter pylori HopH (OipA) and bacterial pathogenicity: genetic and functional genomic analysis of hopH gene polymorphisms. J Infect Dis. 2006;194:1346–1355. doi: 10.1086/508426. [DOI] [PubMed] [Google Scholar]
- 37.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y, Omata M. Distinct mechanism of Helicobacter pylori-mediated NF-κB activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001;276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 38.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 39.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haeberle HA, Kubin M, Bamford KB, Garofalo R, Graham DY, El-Zaatari F, Karttunen R, Crowe SE, Reyes VE, Ernst PB. Differential stimulation of interleukin-12 (IL-12) and IL-10 by live and killed Helicobacter pylori in vitro and association of IL-12 production with γ interferon-producing T cells in the human gastric mucosa. Infect Immun. 1997;65:4229–4235. doi: 10.1128/iai.65.10.4229-4235.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26:171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 42.Goodgame RW, Malaty HM, El-Zimaity HM, Graham DY. Decrease in gastric permeability to sucrose following cure of Helicobacter pylori infection. Helicobacter. 1997;2:44–47. doi: 10.1111/j.1523-5378.1997.tb00057.x. [DOI] [PubMed] [Google Scholar]
- 43.Petersen AM, Krogfelt KA. Helicobacter pylori: an invading microorganism: a review. FEMS Immunol Med Microbiol. 2003;36:117–126. doi: 10.1016/S0928-8244(03)00020-8. [DOI] [PubMed] [Google Scholar]
- 44.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Day AS, Su B, Ceponis PJ, Jones NL, Yau E, Sieveking D, Sherman PM. Helicobacter pylori infection induces interleukin-18 production in gastric epithelial (AGS) cells. Dig Dis Sci. 2004;49:1830–1835. doi: 10.1007/s10620-004-9579-y. [DOI] [PubMed] [Google Scholar]
- 46.Osaki T, Peron JM, Cai Q, Okamura H, Robbins PD, Kurimoto M, Lotze MT, Tahara H. IFN-γ-inducing factor/IL-18 administration mediates IFN-γ- and IL-12-independent antitumor effects. J Immunol. 1998;160:1742–1749. [PubMed] [Google Scholar]