

Abstract
Hirschsprung disease is a serious disorder of enteric nervous system (ENS) development caused by the failure of ENS precursor migration into the distal bowel. We now demonstrate that retinoic acid (RA) is crucial for GDNF-induced ENS precursor migration, cell polarization and lamellipodia formation, and that vitamin A depletion causes distal bowel aganglionosis in serum retinol-binding-protein-deficient (Rbp4–/–) mice. Ret heterozygosity increases the incidence and severity of distal bowel aganglionosis induced by vitamin A deficiency in Rbp4–/– animals. Furthermore, RA reduces phosphatase and tensin homolog (Pten) accumulation in migrating cells, whereas Pten overexpression slows ENS precursor migration. Collectively, these data support the hypothesis that vitamin A deficiency is a non-genetic risk factor that increases Hirschsprung disease penetrance and expressivity, suggesting that some cases of Hirschsprung disease might be preventable by optimizing maternal nutrition.
Keywords: Enteric nervous system, Migration, Retinoic acid, Lamellipodia, Pten, Hirschsprung disease, Mouse
INTRODUCTION
The enteric nervous system (ENS) is a complex network of neurons and glia within the bowel that controls most aspects of intestinal function (Furness, 2000). Hirschsprung disease (HSCR), a partially penetrant life-threatening defect affecting 1:5000 children, is caused by the failure of ENS precursor colonization of the distal bowel (Skinner, 1996). ENS precursors originate in the vagal, sacral and upper thoracic neural tube before migrating extensively, proliferating and then differentiating to form the ENS (Heanue and Pachnis, 2007). Most ENS precursors arise from the vagal neural tube, enter the foregut and colonize the bowel in a rostral to caudal progression (Newgreen and Young, 2002a; Newgreen and Young, 2002b). Because molecular mechanisms required for ENS precursor migration, proliferation and differentiation are complex, many distinct mutations cause HSCR. Furthermore, HSCR penetrance and the extent of aganglionosis are governed by genetic interactions (Amiel et al., 2008; Cantrell et al., 2004; McCallion et al., 2003; Owens et al., 2005).
We now present data supporting the hypothesis that non-genetic factors, including vitamin A, crucially determine the penetrance of HSCR-like distal bowel aganglionosis. Vitamin A (retinol), an essential nutrient, is a precursor to retinoic acid (RA), which is generated by short-chain dehydrogenases/reductases and retinaldehyde dehydrogenases (Raldh) (Maden, 2007; Napoli, 2004). RA binds Rar and Rxr receptors that heterodimerize and then bind RA response elements (RARE) to regulate target genes and influence tissue morphogenesis. Recent data suggest that both excess RA (Pitera et al., 2001) and reduced RA synthesis cause abnormal ENS development, but the molecular and cellular mechanisms that RA influences are not known. Indeed, although Raldh2–/– mice develop HSCR-like aganglionosis (Niederreither et al., 2003), these mice have severe RA deficiency starting at conception, so it is unclear at what stage RA is required for ENS morphogenesis. Our new data demonstrate that RA is essential for efficient ENS precursor migration through the developing bowel and for lamellipodia formation in migrating cells.
Cell migration is complex, requiring polarization and coordinated activation of many proteins including RhoA-related GTPases that regulate the actin cytoskeleton. In particular, Rac1 activation at the leading edge of migrating cells is crucial for lamellipodia formation. Recent work (Jaffe and Hall, 2005; Kawauchi and Hoshino, 2008; Vohra et al., 2007a; Yoshimura et al., 2006) demonstrated that Rac1 activation occurs via a positive feedback loop initiated by conversion of phosphatidylinositol-2-phosphate (PIP2) to phosphatidylinositol-3-phosphate (PIP3) via phosphatidylinositol-3 kinase (Pik3) (Wang et al., 2002; Wedlich-Soldner and Li, 2003). This could be initiated by glial cell line-derived neurotrophic factor (GDNF) binding to its receptor Ret, a kinase commonly mutated in human HSCR and required for ENS precursor migration (Takahashi, 2001).
Because RA deficiency prevents efficient ENS precursor migration, we hypothesized that RA is required to maintain signals needed for cell migration. We show that, in response to GDNF, Pik3 and PIP3 accumulate at the leading edge of migrating enteric crest-derived cells and Pten (phosphatase and tensin homolog), an enzyme that reverses the Pik3 activity, is absent from most actively migrating ENS precursors. When Rar signaling is blocked, however, polarized distribution of Pik3, PIP3 and Pten is abolished and Pten levels dramatically increase in actively migrating cells. This is accompanied by loss of lamellipodia and reduced migration toward GDNF. This suggests that RA is required in the developing ENS to reduce Pten and enhance ENS precursor migration. Collectively, these data suggest that some cases of Hirschsprung disease might be preventable by ensuring adequate maternal vitamin A levels during early gestation.
MATERIALS AND METHODS
Mice
Rbp4–/– (Quadro et al., 2005) and Ret+/– mice (Enomoto et al., 2001) were bred >10 generations to C57BL/6. For timed breeding studies, the day of the vaginal plug was considered as embryonic day 0.5 (E0.5). Wild-type CF-1 mice were from Charles River. Mice were maintained on Purina PICO irradiated mouse diet 5058 until E7.5. Food was changed to synthetic chow (El Mel, St Charles, MO, USA) containing sufficient vitamin A (TestDiet 5755; 22.1 IU vitamin A/g) or to vitamin A deficient food (TestDiet 5822; <0.4 IU vitamin A/g) colored blue or yellow to avoid confusion. Mice were maintained on synthetic diets until analysis.
Retinoid measurements
RA was quantified using an API-4000 (Applied Biosystems) LC/MS/MS with atmospheric pressure chemical ionization in positive ion mode. Retinol and retinyl esters were quantified by HPLC/UV (Kane et al., 2008a). Tissues were harvested under yellow light, immediately frozen in liquid N2 and kept at –80°C until assay. Samples were homogenized on ice, extracted and analyzed as described (Kane et al., 2005; Kane et al., 2008b). Total protein was determined by Bradford (Bio-Rad).
Slice culture
E12.5 CF-1 midgut sections (300-400 μm length) from 400 μm proximal to the cecum were cultured on fibronectin-coated (250 μg/ml) dishes in Opti-MEM (Invitrogen), glutamine (2 mM), penicillin 100 IU/ml and streptomycin 100 μg/ml. Immediately after plating, cells were treated with RA (10–7 M, Sigma, St Louis, MO, USA) or BMS493 (10–5 M, generously provided by Dr Chris Zusi at Bristol-Myers Squibb). GDNF (100 ng/ml) was added to cultures three hours later. Cultures were maintained for 16 hours before fixation [4% paraformaldehyde (PFA), 10 minutes, 25°C].
Boyden chamber
Transwell supports (8.0 μm pore size; 0.33 cm2 area; Corning 3422; Fisher Scientific) were coated on both sides with 10% Matrigel (Fisher Scientific) in PBS (4°C, 18 hours) and rinsed with PBS. Neural crest medium [Dulbecco's modified Eagle medium (DMEM), 10% chick embryo extract, 1% N2 supplement, 2% B27 supplement, penicillin 100 IU/ml, streptomycin 100 μg/ml, β-mercaptoethanol 50 μM, all-trans-RA 35 ng/ml (∼10–7 M), bFGF 20 ng/ml, EGF 20 ng/ml] was added to the bottom and top chambers with 50,000 dissociated E12.5 CF-1 gut cells/well in the top chamber, prepared as previously described (Fu et al., 2006). Immediately after plating, BMS493 (10–5 M) or vehicle (2 μl ethanol) was added to the top well. Cells were incubated 2 hours before adding GDNF (100 ng/ml) to the top or bottom chambers and then 16 hours to allow migration. Cells on top of the membrane were removed with Kimwipes. Cells on membrane bottoms were fixed (4% PFA, 20 minutes, 25°C), blocked (1% BSA/PBS, 40 minutes, 25°C) and processed for immunohistochemistry.
Whole gut culture
The entire E11.5 CF-1 gastrointestinal tract was pinned to 2.5% agarose with 4-0 stainless steel filaments (Ethicon) and cultured in DMEM, 10% fetal calf serum, penicillin 100 IU/ml and streptomycin 100 μg/ml (Fu et al., 2006) with or without all-trans-RA (10–7 M, Sigma) or BMS493 (10–5 M). Cultures were maintained for 48 hours before fixing (4% PFA, 30 minutes, 25°C).
Dissociated cell culture
E12.5 CF-1 ENS precursor cells were maintained in culture as previously described (Sato and Heuckeroth, 2008a). For Ret pixel intensity measurements, ENS precursors from E13.5 Rbp4–/– mice deprived of vitamin A starting at E7.5, or from wild-type C57BL/6 on a vitamin A-containing diet, were cultured briefly (2 hours) before fixation.
Immunohistochemistry
After fixation, cells and organs were kept in TBST (100 mM Tris, 150 mM NaCl, 0.2% Triton X-100) for 20 minutes at 25°C, blocked with 4% donkey serum/TBST for 1 hour at 25°C and then incubated with primary antibody for 18 hours at 4°C. The primary antibodies used were: TuJ1 (rabbit, Covance, 1:100), Ret (goat; Neuromics, 1:100), P75NRT antibody (rabbit, G323A Promega, 1:700), Alexa Fluor 488 phalloidin (mouse, Invitrogen, 1:40), Pten (mouse, Covance, 1:40), PIP3 (mouse, Echelon Biosciences, 1:40), p85α (B-9) (mouse, Santa Cruz, 1:40) and Phox2b (rabbit, a generous gift from Jean-François Brunet, 1:1000). Antibodies were visualized using donkey anti-goat Alexa 594 (1:200) and donkey anti-rabbit Alexa 488 (1:100, Molecular Probes). Images were obtained with an Olympus BX60 microscope and Axiocam and AxioVision software (Zeiss). NIH Image J was used for pixel intensity measurements of Ret immunohistochemistry. Measurements were made in the cell body of p75NTR+ cells and we subtracted background signal intensity.
Pten overexpression vector
Full-length Pten cDNA (Open Biosystems, Clone ID 5038272) obtained using XbaI and XmaI was cloned into a modified version (FM) of the FUIV [ubiquitin promoter – gene of interest-IRES-enhanced YFP (Venus)] (Araki et al., 2004) using AgeI and NheI sites. Virus was produced by Washington University Hope Center Viral Vectors Core using 293T cells, pCMV-G, pMD-Lg, and RSV-REV (Li and Rossi, 2005). Viral titers were 1.4×108 (TU/ml) for FM-Pten and 1×108 (TU/ml) for FM.
Cell migration assay for virus-infected cells
E12.5 CF-1 whole gut was digested with collagenase (0.2 mg/ml) and dispase (0.2 mg/ml) for 10 minutes at 37°C, triturated, centrifuged and resuspended in the neural crest medium (see Boyden Chamber). Three microlitres of virus was added to 106 cells in 50 μl media. To prepare the collagen gel, 0.25 volume of 5× DMEM was added to 1 volume of type I rat tail collagen (4 mg/ml) in 0.02 N acetic acid (BD Biosciences). pH was adjusted using 0.8 M NaHCO3 (final concentration 8 mM) and 200 mM NaOH (final concentration 1.5 mM) to form gel and diluted with neural crest medium to 2 mg/ml collagen. Fifty microliters of gel was added to 50 μl cells/virus mixture and the gel was cultured in 96-well U-bottom plates (BD Biosciences) for 48 hours at 37°C. The cell-gel pellet was then transferred to 4-well dishes and embedded in 1% collagen gel containing GDNF (200 ng/ml). After 24 hours, Venus+ cells that migrated from the cell-gel pellet were counted after fixation in 4% PFA for 20 minutes at 25°C and immunohistochemistry.
Statistical analyses
All experiments were performed at least three times and analyzed by t-test or ANOVA analysis for multiple comparisons. P<0.05 was considered significant. Data show mean ± s.e.m.
RESULTS
Vitamin A deficiency causes HSCR-like distal bowel aganglionosis in vivo
To determine whether vitamin A (retinol) is required for efficient bowel colonization by ENS precursors, we used serum retinol-binding-protein-deficient (Rbp4–/–) mice (Quadro et al., 1999; Quadro et al., 2005). Rbp4 is required for transport of hepatic retinol to peripheral tissues, so tissue retinoids in Rbp4–/– mice are primarily derived from dietary retinyl ester incorporated into chylomicrons. Thus, unlike wild-type mice that have large hepatic retinol stores (Moore and Holmes, 1971) and are difficult to deplete (Clagett-Dame and DeLuca, 2002; Wolf, 1984), Rbp4–/– mice can be rapidly depleted of peripheral tissue retinoids by removing vitamin A from the diet. For these studies, Rbp4–/– mice were deprived of dietary retinol starting at E7.5, 48 hours before crest-derived precursors enter the bowel. Mice were analyzed at E14.5, shortly after ENS precursors normally completely colonize the colon. The entire small bowel and colon were stained using antibodies to Ret and neuron-specific beta-III tubulin (TuJ1, a neuronal lineage marker; Fig. 1). These studies demonstrated striking distal bowel aganglionosis in Rbp4–/– mice deprived of dietary retinol, but an apparently normal ENS in Rbp4–/– mice maintained on retinol containing food (Fig. 1A). In fact, under vitamin A-sufficient conditions, only 2 out of 34 Rbp4–/– failed to completely colonize the colon with ENS precursors at E14.5. By contrast, almost all Rbp4–/– mice fed vitamin A-deficient food starting at E7.5 had distal bowel aganglionosis (31/33). Some animals had total colonic aganglionosis (4/33). Thus, simple diet-induced retinol deficiency appears to cause an HSCR-like phenotype.
Fig. 1.

Vitamin A deficiency causes distal bowel aganglionosis in Rbp4–/– mice. (A) Rbp4–/– mice fed vitamin A-containing food (top), had a dense network of neurons and fibers in the distal bowel and almost all had ganglion cell bodies at the end of the colon. Diagrams indicate positions of the most distal TuJ1+ ganglion cell body or nerve fiber at E14.5 after feeding vitamin A-sufficient or -deficient diets starting at E7.5. Each oval represents one mouse. Representative TuJ1-stained colon images are shown. Vitamin A-deprived mice have distal colon aganglionosis and reduced ENS density in the colonized colon (bottom). (B) Ret/TuJ1 double-label immunohistochemistry at the wavefront of ENS precursors of an E13.5 Rbp4–/– mouse maintained on vitamin A-deficient food starting at E7.5. The Ret signal is less intense than TuJ1, but staining overlaps significantly and the wavefront visualized by Ret or TuJ1 antibodies is at the same position (n=4 vitamin A-sufficient mice, n=8 vitamin A-depleted mice). (C) Even when maintained on vitamin A, many Rbp4–/–;Ret+/– mice have distal bowel aganglionosis. When deprived of vitamin A starting at E7.5, there is more extensive aganglionosis. (D) Summary of bowel colonization in mutant mice. (E) Double-label immunohistochemistry for Ret and Phox2b at the migration wavefront of E13.5 Rbp4–/– mice maintained on a vitamin A-containing or -deficient diet starting at E7.5 shows colocalization of Ret and Phox2b-expressing cells. In all images, distal bowel is to the right. Scale bars: 100 μm in A; 50 μm in B,E.
Because retinol depletion causing loss of Ret and/or TuJ1 immunoreactivity in distal bowel ENS precursors is a potential concern, we performed double-label immunohistochemistry with antibodies to Ret and Phox2b in Rbp4–/– mice fed either retinol-deficient or -sufficient diets. Phox2b and Ret were expressed in the same cells at the migration wavefront under both conditions (Fig. 1E). Furthermore, Ret immunoreactivity in ENS precursors measured by pixel intensity demonstrated equal Ret abundance in cells from wild-type C57BL/6 versus retinol-deprived Rbp4–/– mice (WT: 26.6±1.3; retinol-deprived Rbp4–/–: 24.9±0.7, P=0.21). Therefore, immunohistochemistry for Ret, TuJ1 and Phox2b all indicate that the distal bowel is not colonized in E14.5 Rbp4–/– mice deprived of retinol starting at E7.5.
Mild vitamin A deficiency increases HSCR-like disease in genetically predisposed mice
To test the hypothesis that retinol depletion could increase HSCR penetrance in genetically predisposed mammals, we analyzed the ENS in Rbp4–/–;Ret+/– double-mutant mice. Ret mutations are the most commonly identified cause of human HSCR and Rbp4–/– animals have mild vitamin A deficiency even when maintained on retinol containing food (Wendler et al., 2003). Furthermore, Ret+/– mice are predisposed to aganglionosis (McCallion et al., 2003), but have almost normal ENS anatomy (Gianino et al., 2003). Rbp4–/–;Ret+/– mice were switched to vitamin A-containing or -deficient synthetic food starting at E7.5. Even when maintained on vitamin A containing food, many Rbp4–/–;Ret+/– animals have distal bowel aganglionosis (10/20) at E14.5 (Fig. 1C,D). Furthermore, dietary vitamin A deprivation caused more extensive bowel aganglionosis in Rbp4–/–;Ret+/– than in Rbp4–/– mice. Collectively, these data strongly support the hypothesis that vitamin A deficiency is a nutritional risk factor for HSCR. To evaluate the severity of retinoid depletion needed to cause ENS defects, we measured fetal retinoids in Rbp4–/– mice at E12.5, when migrating ENS precursors have reached the mid-colon and are entering distal bowel. These analyses demonstrated that switching to vitamin A-deficient diets at E7.5 caused 42-53% reductions in fetal retinoids at E12.5 (Table 1). Based on these findings, we investigated molecular and cellular mechanisms to explain our observations.
Table 1.
E12.5 fetal retinoid levels

Rar signaling is required for ENS precursor migration
To determine whether Rar signaling affects ENS precursor migration, we used a modified Boyden chamber (Boyden, 1962) to study ENS precursors at E12.5, the time that the distal colon is colonized. Dissociated gut cells placed in the top chamber were treated for 2 hours with control media containing RA or media containing a well-characterized pan-Rar antagonist, BMS493. GDNF was then added to the bottom chamber to induce ENS precursor migration through the porous membrane. Cells were cultured 16 hours before Ret immunohistochemistry (Fig. 2). BMS493 competitively blocks all Rar isotypes (α, β and γ) and has been used in embryo culture and in vivo to block retinoid action (Chazaud et al., 1999; Chazaud et al., 2003; Hochgreb et al., 2003; Mollard et al., 2000; Sato and Heuckeroth, 2008; Wendling et al., 2000; Wendling et al., 2001). Animals treated with BMS493 develop the same spectrum of defects seen in vitamin A deficiency. GDNF in the bottom chamber markedly increased Ret+ ENS precursor migration through the membrane. BMS493 completely blocked chemoattractive effects of GDNF (Fig. 2E). Control experiments with GDNF in the top chamber only did not increase Ret+ cell migration through the membrane, suggesting that GDNF acts as a chemoattractant rather than simply increasing motility or proliferation.
Fig. 2.
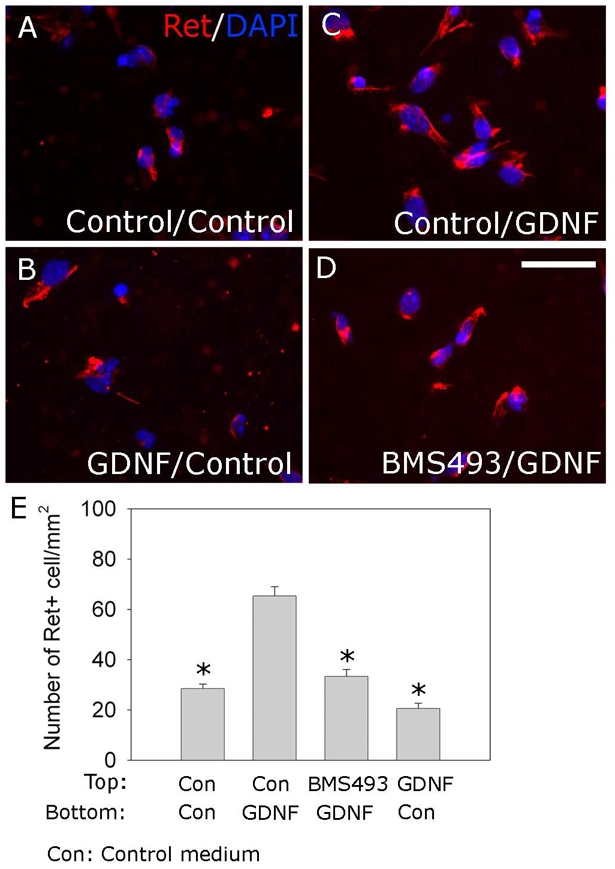
RA is required for efficient GDNF-induced ENS precursor migration. (A-D) E12.5 dissociated gut cells were placed in the top wells of Boyden chambers in retinoic acid (RA)-containing media with (D) or without (A-C) BMS493. After 2 hours, media with (C,D) or without (A,B) GDNF (100 ng/ml) was added to the bottom chamber. As a control (B), GDNF was also added to the top, but not the bottom chamber. After 16 hours, ENS precursors that migrated through membranes were visualized using DAPI (blue) and Ret immunohistochemistry (red). (E) Analysis of Ret+ cells that migrated through membranes (n=9 for each condition). Scale bar: 100 μm. *, P<0.0001 compared with GDNF in the bottom chamber.
Rar antagonism slows ENS precursor migration in organ culture
To further evaluate Rar effects on ENS precursor migration, E12.5 mid-small bowel slices were cultured for 16 hours in media with GDNF plus RA, or GDNF plus the Rar antagonist BMS493. Under these conditions, ENS precursors migrate from gut explants onto culture dishes and were visualized by video microscopy before Ret immunohistochemistry. Treatment with BMS493 considerably reduces migration of Ret+ ENS precursors from gut explants in response to GDNF (Fig. 3A-D). Adding RA had little effect on ENS precursor migration, suggesting that explants make enough RA to support migration in this assay. Time-lapse microscopy confirmed that BMS493 did not influence cell survival in these short-term studies. Double-label immunohistochemistry for Ret and TuJ1 demonstrated no effect of Rar signaling on neuronal differentiation or the level of Ret expression (see Fig. S2 in the supplementary material). Parallel studies of the effects of BMS493 on ENS precursor migration in whole gut culture confirmed that Rar antagonism reduces distal bowel colonization. In this case, the position of the most distal ganglion cell is the same in the presence or absence of BMS493, but the density of ENS precursors in the distal bowel is dramatically reduced by Rar antagonism (Fig. 4). BMS493 also appears to affect organization of the distal bowel ENS, but these changes are difficult to quantify. Collectively, these data support the hypothesis that RA is required for efficient GDNF-induced ENS precursor migration.
Fig. 3.

BMS493 reduces ENS precursor migration and lamellipodia formation in slice cultures. (A-C) E12.5 mouse mid-gut slices were cultured to allow crest-derived cells to migrate onto the dish in response to GDNF. Cultures were maintained for 16 hours without added retinoic acid (RA) (A), with added RA (B), or with BMS493 (C) before Ret immunohistochemistry (red) and DAPI staining. (D) Distance from the edge of the explant to the most distal Ret+ cells [4 measurements/explant (see Fig. S1 in the supplementary material); n=40 explants for each condition]. (E-L) Ret antibody (red) and Phalloidin (green) double-labeling demonstrate lamellipodia at the leading edge of crest-derived cells migrating from E12.5 mid-gut slices maintained in GDNF (E,I) or GDNF plus RA (F,J). Culture with GDNF plus BMS493 (G,K) or GDNF plus Pik3 inhibitor LY294002 (H,L) dramatically reduces lamellipodia formation and causes Ret+ cells to adopt very unusual shapes. The arrow in I shows a typical lamellipodium (n>320 cells analyzed; 12 slices per condition). (I-L) Higher magnification images of E-H. (M) Percentage of Ret+ cells at the leading edge of the migration wavefront with lamellipodia. Scale bars: 100 μm in A-C,E-H; 25 μm in E′-H′. *, P<0.0001 versus GDNF.
Fig. 4.

Rar signaling is required for colon colonization in organ culture. (A-C″) E11.5 mouse gut cultured for 48 hours in control media (A), with added RA (B) or added BMS439 (C) was stained with Ret (A-C) and PGP9.5 (A′-C′) antibodies. (A″-C″) Merged images. (D,F) Ret+ cell density in the terminal 200 μm of colonized colon was measured using a grid. (E) Position of the most distal Ret+ cell in colon relative to colon length. n=15 control, 15 RA- and 27 BMS493-treated gut explants. *, P<0.001 versus control. Scale bar: 100 μm.
Rar antagonism causes lamellipodia loss in migrating ENS precursors
To determine why Rar antagonism reduces ENS precursor migration, we examined Ret+ cells that migrated furthest from E12.5 mid-small bowel slices. Cells maintained with RA had well-defined lamellipodia at the leading edge (Fig. 3F,J), but lamellipodia were uncommon in BMS493-treated cultures (Fig. 3G,K). Analysis of Ret antibody and Phalloidin-stained cells confirmed that BMS493 markedly reduces lamellipodia in rapidly migrating ENS precursors (Fig. 3M). Furthermore, BMS493-treated cells often had strikingly abnormal morphology with filopodia-like projections, suggesting that RA critically regulates the signaling machinery required for lamellipodia formation (Fig. 3K).
Antagonizing Rar increases Pten accumulation in migrating ENS precursors and alters Pik3r1 and PIP3 distribution
Many signaling components required for ENS precursor migration and lamellipodia formation are known (Fig. 8) (Fukuda et al., 2002; Hall, 2005; van Weering and Bos, 1997; Vohra et al., 2007a). This pathway begins with receptor-induced Pik3 activation. Pik3 converts PIP2 to PIP3. PIP3 in membranes recruits proteins that activate Rac1, a key regulator of lamellipodia formation and part of a positive feedback loop required for migration. Because previous studies demonstrated that blocking Pik3 reduces distal bowel colonization by ENS precursors (Natarajan et al., 2002), we hypothesized that RA deficiency might reduce Pik3 activity or alter localization. We therefore examined the cells that migrated furthest from gut slices with Pik3 and PIP3 immunohistochemistry. Migrating Ret+ cells maintained with GDNF and RA accumulated PIP3 and phospho-Pik3r1, the activated regulatory subunit of Pik3, in lamellipodia. Both PIP3 and pPik3r1 were also abundant in Ret+ cell bodies (Fig. 5A,A′). By contrast, pPik3r1 and PIP3 were not seen at the edge of migrating Ret+ cells cultured with BMS493 (Fig. 5B,B′). Because BMS493 treatment dramatically alters cell shape, however, it was difficult to know if altered pPik3r1 and PIP3 distribution caused, or resulted from, the cell shape changes. We therefore investigated other molecules that could affect lamellipodia formation and cell migration.
Fig. 8.

Signaling pathways controlling ENS precursor migration and lamellipodia formation. (A) This simplified pathway shows that Ret activation by GDNF causes Pik3 activation, generating PIP3 and activating Cdc42 and Rac1 as part of a positive-feedback loop at the leading edge of migrating cells. Rac1 is crucial for lamellipodia formation. Pten reverses Pik3 activity and stops the positive-feedback loop. RA signaling reduces Pten protein abundance in the most actively migrating ENS precursors in vitro and at the leading edge of the wavefront of Ret+ cells organ culture. (B) A model of how cells polarize in response to chemoattractants. In the absence of chemoattractant, signaling proteins might be distributed throughout the cell. In actively migrating cells, Cdc42, Rac1, PIP3 and Pik3 accumulate at the front edge of the cell, inducing cytoskeletal changes and lamellipodia formation. Pten, Rho and Rho kinase (Rock) become restricted to the trailing edge of the cell. High levels of Pten in ENS precursors deprived of Rar signaling disrupts the positive-feedback loop and causes loss of the cell polarity needed for migration.
Fig. 5.

BMS493 alters cell shape, PIP3 and Pik3r1 distribution and Pten abundance. (A-H) E12.5 mid-gut slices were cultured for 16 hours with GDNF (A,A′,C,F), GDNF plus RA (D,G) or GDNF plus BMS493 (B,B′,E,H). (A,A′) Immunohistochemistry for PIP3 or Pik3r1 demonstrates polarization of PIP3 and Pik3r1 in lamellipodia (arrows) of migrating Ret+ cells. (B,B′) In BMS493-treated cells, both PIP3 and Pik3r1 were present but the distribution was not polarized. (C-E) Immunohistochemistry for Ret (green) and Pten (red) demonstrates that Pten levels are low or absent in Ret+ migrating cells cultured with GDNF or GDNF plus RA, but elevated in cells cultured with BMS493. (F,G) Even when Pten is detected, Pten is localized to the trailing edge of migrating cells grown in control media or with added RA. (H) By contrast, Pten is distributed throughout the cytoplasm after culture with BMS493. (I) Analysis of Pten–Ret double-labeling in cells at the outer edge of the migration wavefront (GDNF, n=621 cells; GDNF + RA, n=685; GDNF + BMS493, n=1069; six independent experiments). Scale bars: 25 μm in A-B′,F-H; 100 μm in C-E. *, P<0.0001.
As Pten reverses Pik3 activity by converting PIP3 to PIP2, we hypothesized that Rar antagonism might inhibit migration and lamellipodia formation by increasing Pten. To test this hypothesis, we cultured gut explants for 16 hours with GDNF, GDNF plus RA or GDNF plus BMS493. Remarkably, Pten was much more abundant in migrating Ret+ cells after culture in BMS493 than after culture with GDNF or GDNF plus RA (Fig. 5C-E). In the absence of BMS493, most migrating Ret+ cells were Pten negative (91±1.3%). Even when Pten was present, it was largely excluded from the leading edge of cells (Fig. 5F,G). By contrast, Ret+ cells cultured with BMS493 had abundant Pten (61±5%) and no evidence of polarized Pten accumulation (Fig. 5E,H). These data suggest that RA facilitates ENS precursor migration by reducing Pten accumulation at least in the most actively migrating cells. Furthermore, increased Pten accumulation could account for abnormal cell shape, loss of lamellipodia and reduced migration after treatment with a Rar antagonist. If so, then blocking Pik3 should similarly affect cell shape and migration. Consistent with this hypothesis, treatment of gut slices with Pik3 inhibitor LY294002 significantly reduced ENS precursor migration from explants as previously reported (Natarajan et al., 2002) and reduced the percentage of cells with lamellipodia (22±2%) (Fig. 3H,L,M). Furthermore, LY294002 induced abnormalities in cell shape closely resembling BMS493-treated Ret+ cells. These data support the hypothesis that RA enhances ENS precursor migration by reducing Pten accumulation.
Pten is not expressed in ENS precursors at the migration wavefront in vivo
If regulation of Pten by RA is crucial for normal ENS precursor migration, we might expect low Pten levels in the most actively migrating ENS precursors within the bowel wall. Furthermore, Pten accumulation within ENS precursors should be regulated by Rar. To test this hypothesis, we examined Pten in E11.5 whole gut segments cultured for 48 hours in control media or with added RA or BMS493. These studies demonstrate that Pten protein levels are very low in Ret+ cells at the leading edge of the migration wavefront under control conditions or with RA added, but that BMS493 dramatically increases Pten in Ret+ cells within 200 μm of the wavefront (Fig. 6A-C,E). Interestingly, behind the migration wavefront, Pten was expressed in a subset of Ret+ cells under all conditions, and these Pten expressing cells largely correspond to differentiating TuJ1+ neurons (Fig. 6D). Analysis of Pten in wild-type mice at E11.5 also demonstrated that Pten is largely excluded from Ret+ cells at the migration wavefront (data not shown). Collectively, these data support the hypothesis that Pten abundance is regulated by RA and that reduced Pten facilitates ENS precursor migration.
Fig. 6.

Pten protein levels are low in Ret+ cells at the leading edge of the migration wavefront in organ culture. (A-C″) E11.5 whole guts were cultured for 48 hours in control medium (A), with added RA (B) or with BMS493 (C) before whole-mount immunohistochemistry with Ret (A-C) and Pten (A′-C′) antibodies. Confocal images show the leading edge of the migration wavefront in the colon. (A″-C″) Merged images. Arrows indicate the direction of precursor migration. Ret+ cells at the leading edge of the migration wavefront have very low Pten levels after culture in control media or with RA added. By contrast, Pten is abundant in all Ret+ cells after culture with BMS493, even at the leading edge of the wavefront. (D) Freshly isolated E12.5 mouse hindgut was stained for TuJ1 (D) and Pten (D′). The distal-most immunoreactive bowel is shown. (D″) Merged image. Most TuJ1+ cells are Pten+. Some TuJ1+ cells with faint Pten staining are also detected (arrows). (E) Percentage of Ret+ cells that express Pten within 200 μm of the migration wavefront (n=8 explants and 400 cells under each condition; *, P<0.001). Scale bars: 200 μm in A-C; 50 μm in D.
Pten overexpression reduces GDNF-induced migration of ENS precursors
If RA is important for ENS precursor migration because it reduces Pten, then increased Pten should slow migration. To test this hypothesis, E12.5 cells from murine bowel were infected with lentivirus (FM-Pten) that expresses Venus (a fluorescent protein) and Pten or with control lentivirus (FM) that only produces Venus. Pten production by virus was verified in 293T cells (Fig. 7). Dissociated E12.5 gut cells were infected with virus and embedded in a drop of collagen gel, cultured for 48 hours to allow Pten production, and then surrounded with additional GDNF-containing collagen gel. During an additional 24 hours, ENS precursors migrated into the GDNF-containing gel. Most migrating cells were virus-infected (FM virus: 75±7%; FM-Pten virus: 76±5%) and almost all migrating cells were Ret+. Cells infected with the FM-Pten virus, however, did not migrate as far from the edge of the cell-rich pellet as cells infected with FM virus (Fig. 7E-H). These data suggest that low Pten is required for efficient ENS precursor migration and support the hypothesis that Rar signaling is required for migration of ENS precursors into the distal bowel because RA reduces Pten accumulation in the most rapidly migrating cells.
Fig. 7.

Pten overexpression reduces ENS precursor migration in vitro. (A-B″) FM-Pten efficiently produces Pten. 293T cells that lack significant Pten were infected with lentivirus-expressing Venus (FM virus) (A-A″) or Venus plus Pten (FM-Pten virus) (B-B″). Pten expression in Venus+ cells was evaluated by immunohistochemistry 48 hours after infection (A′,B′). (C-D″) E12.5 dissociated gut cells were infected with FM or FM-Pten, embedded in collagen gel, were cultured for 48 hours and then placed into a larger volume of GDNF-containing collagen gel for 24 hours to encourage migration before Ret immunohistochemistry. Phase contrast (C′) and Venus imaging (C) demonstrate that most cells (∼75%) migrating from the pellet into GDNF-containing gel were Venus+. (D,D′) Essentially all migrating cells were Ret+. (E-F″) FM-Pten-infected cells migrate less rapidly from the cell-gel pellet than FM-infected cells. (G) Diagram shows how cell migration was evaluated. Cells within 100 μm of the cell-rich pellet were too dense to count. Cells in the regions of 100-200 μm and 200-400 μm from the edge of the cell-rich gel pellet were counted separately. (H) Very few FM-Pten-infected cells migrated more than 200 μm from the edge of the cell-rich gel pellet. *, P<0.0001 versus FM. Scale bars: 100 μm.
DISCUSSION
RA signaling is essential for normal ENS development
Both RA excess (Pitera et al., 2001) and reduced RA synthesis (Niederreither et al., 2003) affect ENS development, but the cellular and molecular mechanisms underlying these observations are uncertain. Our studies suggest that RA is part of an essential network of signaling molecules that facilitate ENS precursor migration into the distal bowel. Using Rbp4–/– mice (Quadro et al., 2005; Wendler et al., 2003), we demonstrate that vitamin A deprivation induces distal bowel aganglionosis, but that Rbp4–/– animals rarely develop bowel aganglionosis if maintained on vitamin A-containing food. Ret and Rbp4 mutations also synergistically cause bowel aganglionosis, even when mice are fed retinol. Furthermore, vitamin A deprivation after E7.5 in Rbp–/–;Ret+/– mice causes more extensive aganglionosis than in Rbp4–/–;Ret+/+ animals, demonstrating a new gene-environment interaction causing HSCR-like disease. Together, these data provide compelling evidence that vitamin A is essential for normal ENS development and suggest that retinoid deficiency could increase HSCR penetrance and the extent of aganglionosis, crucial issues for a disease with incomplete penetrance and variable expressivity.
Quantitative analysis of retinoids in Rbp4–/– mice suggests that even mild vitamin A deficiency might increase HSCR penetrance. Previous fetal retinoid measurements demonstrated that E14.5 Rbp4–/– mice have reduced retinol (59% reduction) and retinyl ester (31% reduction) compared with wild-type mice when maintained on vitamin A-sufficient food during gestation (Kim et al., 2008). Our data suggest that, combined with Ret heterozygosity, this modest reduction in fetal retinoids might be enough to cause disease. Depriving Rbp4–/– mice of dietary retinoids at E7.5 led to an additional ∼50% drop in fetal retinoid levels at E12.5. Together, these data suggest that 50-75% reductions in fetal retinoids profoundly influence ENS development. If these findings apply to human populations, then vitamin A deficiency is likely to be a common predisposing factor for HSCR in many regions of the world (West, 2002).
Rar antagonism prevents lamellipodia formation and efficient ENS precursor migration
ENS development requires precursor migration from the vagal and sacral neural crest into and through the developing bowel. Vagal crest-derived cells have a very long migratory route and might therefore be more sensitive to factors that reduce migration efficiency than other cells. Several in vitro models, including fetal gut culture, slice culture, Boyden chamber assays and video microscopy, all demonstrated that blocking RA signaling reduced ENS precursor migration efficiency through the bowel or out of the bowel in response to GDNF. In this regard, Boyden chamber assays provide particularly compelling evidence that ENS precursors migrate toward GDNF and that migration efficiency is RA-dependent.
Rar antagonism also dramatically affects the shape of Ret+ cells migrating onto culture dishes. Normally, most migrating ENS precursors in vitro are polarized with well-defined lamellipodia at the leading edge. By contrast, blocking Rar causes dramatic lamellipodia loss in migrating Ret+ cells and many cells assume unusual shapes consistent with loss of polarity and cytoskeletal dysregulation. We acknowledge that the precise mechanisms required for ENS precursor migration in the three-dimensional environment of the gut wall might be distinct from those used on a two-dimensional tissue culture dish (Doyle et al., 2009), but our consistent finding that reduced Rar signaling in many different assays inhibits ENS precursor migration makes it more probable that common molecular mechanisms underlie these observations. Furthermore, recent studies delineated a model for GDNF-induced control of cytoskeletal dynamics to support neurite growth and migration (Vohra et al., 2007a), providing testable hypotheses to explain our observations.
RA prevents Pten accumulation in migrating ENS precursors to facilitate migration
Pten reverses Pik3 activity by converting PIP3 to PIP2. Pik3 activation at the leading edge of cells is important for efficient migration because it promotes Rac1 activation and lamellipodia formation (Fukuda et al., 2002; Natarajan et al., 2002; Vohra et al., 2007a). In ENS precursors, BMS493-treated cells still have abundant PIP3 and Pik3 immunoreactivity, making it difficult to be convinced that changes in the distribution of these molecules cause the defective cell shape or migration observed after BMS493 treatment. By contrast, Pten levels were much higher in migrating cells maintained in BMS493 than in cells grown with RA. Furthermore, migrating ENS precursors maintained with RA had a striking polarization of Pten protein with exclusion of Pten from the leading edge and accumulation of Pten at the trailing edge of the cell. This polarization of Pten was lost in BMS493-treated cells. These changes in Pten abundance in response to altered Rar signaling provide a reasonable explanation for observed changes in cell shape, polarity and migration efficiency. Furthermore, Pik3 inhibitors cause a similar loss of lamellipodia and altered cell shape. We also show that Pten overexpression reduces ENS precursor migration. Finally, we demonstrate that cells at the migratory wavefront of ENS precursors, a well-defined population of actively migrating cells, have very little Pten, especially when compared with ENS precursors slightly behind the wavefront. Together, these observations suggest that RA is required for efficient ENS precursor migration because Rar signaling reduces Pten accumulation in the most actively migrating cells and that this reduction in Pten permits GDNF-induced Pik3 activity to support lamellipodia formation and cell migration (Fig. 8). Accumulation of Pten in cells behind the migratory wavefront might then be one signal that encourages cells to stop migrating and differentiate. Pten expression might therefore be required for formation of a distributed plexus of neurons and glia along the bowel. This hypothesis is supported by the observation that Pten is expressed in TuJ1+ cells behind the migration wavefront. These observations are consistent with known roles for Pten in the migration of neuronal precursors in the central nervous system (Li et al., 2003; van Diepen and Eickholt, 2008) and in other cells (Kim and Dressler, 2007; Leslie et al., 2007; Liliental et al., 2000; Sanchez et al., 2005; Tamura et al., 1998), supporting the hypothesis that Rar-induced changes in Pten are crucial for normal ENS development.
Several novel aspects of this work should be emphasized. First, effects of RA on Pten are rarely reported. Furthermore, two of the three studies linking RA to Pten show that RA increases Pten accumulation in other cell types (Hisatake et al., 2001; Lee et al., 2006; Lee et al., 2007). Even in the ENS, RA-induced reductions in Pten occur in only subsets of actively migrating cells. Thus, although there are several conserved RARE sequences within 2 kb of the start site for Pten transcription, regulation of Pten protein abundance in ENS precursors is probably complex. Finally, suppressing Pten might be expected to adversely affect ENS precursor migration by increasing PIP3 and enhancing neurite growth (Srinivasan et al., 2005). Our recent work suggests that to counteract this effect, RA also suppresses ENS precursor neurite growth via reduced Smurf1 (Sato and Heuckeroth, 2008). Thus, in the ENS, RA has complex stage-dependent effects that facilitate normal development, but these activities require activation of networks of signaling molecules, controlled protein subcellular localization, and changes in RA responses as cells differentiate. One intriguing possibility is that RA responsiveness of ENS precursors during development might be influenced by Edn3 and Shh, proteins also reported to alter ENS precursor migration, but this hypothesis will require additional investigation (Barlow et al., 2003; Fu et al., 2004; Kapur et al., 1995; Kapur et al., 1993; Kruger et al., 2003; Nagy and Goldstein, 2006; Shin et al., 1999; Sidebotham et al., 2002; Sukegawa et al., 2000; Vohra et al., 2007b).
Conclusions
These studies suggest the intriguing possibility that vitamin A deficiency is a preventable cause of Hirschsprung disease. We now hypothesize that although severe vitamin A deficiency causes many developmental defects, and might contribute to some common anomalies found in children with HSCR (e.g. congenital heart disease), less severe deficiency acts synergistically with genetic mutations to cause penetrant HSCR in individuals who might otherwise have complete colonization of the bowel by ENS precursors if fetal vitamin A supply was optimal. This is similar to the observation that subclinical maternal folate deficiency dramatically increases the risk of neural tube defects (Pitkin, 2007). Furthermore, because RA signaling influences differentiation and proliferation of ENS precursors (Sato and Heuckeroth, 2008a), vitamin A deficiency could contribute not only to HSCR, but also to other intestinal motility disorders.
These observations raise many questions. For example, if vitamin A deficiency is a common cause of HSCR, why has this association not been previously appreciated? One explanation is that although vitamin A deficiency is an important global problem (West, 2002), the epidemiology of HSCR has not been systematically investigated in regions with prevalent vitamin A deficiency. Furthermore, in areas where malnutrition is common, the phenotype of HSCR (abdominal distension, growth failure and early death) might be easily missed, as the diagnosis requires specialized techniques and a high index of suspicion. Given our data and other analyses (Niederreither et al., 2003; Pitera et al., 2001), a systematic study of HSCR epidemiology and maternal vitamin A status now seems appropriate. These data provide new hope that some cases of Hirschsprung disease might be preventable by optimizing maternal nutrition.
Supplementary Material
Acknowledgments
We thank William Blaner for generously sharing Rbp4–/– mice, Chris Zusi for providing BMS493, Bhupinder Vohra, Russell Van Gelder, Louis Muglia and Jonathan Lake for helpful advice, and Sanjay Jain for sharing equipment. This work was supported by NIH/RO1 DK57038, DK6459201, (DDRCC) NIH/P30-DK52574, DK56341, Burroughs Wellcome Fund 1008525, Children's Discovery Institute CH-11-2008-123, P30-DK079333, NCRR C06 RR015502P30 NS057105 and March of Dimes FY02-182. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.040550/-/DC1
References
- Amiel J., Sproat-Emison E., Garcia-Barcelo M., Lantieri F., Burzynski G., Borrego S., Pelet A., Arnold S., Miao X., Griseri P., et al. (2008). Hirschsprung disease, associated syndromes and genetics: a review. J. Med. Genet. 45, 1-14 [DOI] [PubMed] [Google Scholar]
- Araki T., Sasaki Y., Milbrandt J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010-1013 [DOI] [PubMed] [Google Scholar]
- Barlow A., de Graaff E., Pachnis V. (2003). Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron 40, 905-916 [DOI] [PubMed] [Google Scholar]
- Boyden S. (1962). The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 115, 453-466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell V. A., Owens S. E., Chandler R. L., Airey D. C., Bradley K. M., Smith J. R., Southard-Smith E. M. (2004). Interactions between Sox10 and EdnrB modulate penetrance and severity of aganglionosis in the Sox10Dom mouse model of Hirschsprung disease. Hum. Mol. Genet. 13, 2289-2301 [DOI] [PubMed] [Google Scholar]
- Chazaud C., Chambon P., Dolle P. (1999). Retinoic acid is required in the mouse embryo for left-right asymmetry determination and heart morphogenesis. Development 126, 2589-2596 [DOI] [PubMed] [Google Scholar]
- Chazaud C., Dolle P., Rossant J., Mollard R. (2003). Retinoic acid signaling regulates murine bronchial tubule formation. Mech. Dev. 120, 691-700 [DOI] [PubMed] [Google Scholar]
- Clagett-Dame M., DeLuca H. F. (2002). The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 22, 347-381 [DOI] [PubMed] [Google Scholar]
- Doyle A. D., Wang F. W., Matsumoto K., Yamada K. M. (2009). One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol. 184, 481-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H., Crawford P. A., Gorodinsky A., Heuckeroth R. O., Johnson E. M., Jr, Milbrandt J. (2001). RET signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development 128, 3963-3974 [DOI] [PubMed] [Google Scholar]
- Fu M., Lui V. C., Sham M. H., Pachnis V., Tam P. K. (2004). Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J. Cell Biol. 166, 673-684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M., Vohra B. P., Wind D., Heuckeroth R. O. (2006). BMP signaling regulates murine enteric nervous system precursor migration, neurite fasciculation, and patterning via altered Ncam1 polysialic acid addition. Dev. Biol. 299, 137-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T., Kiuchi K., Takahashi M. (2002). Novel mechanism of regulation of Rac activity and lamellipodia formation by RET tyrosine kinase. J. Biol. Chem. 277, 19114-19121 [DOI] [PubMed] [Google Scholar]
- Furness J. B. (2000). Types of neurons in the enteric nervous system. J. Auton. Nerv. Syst. 81, 87-96 [DOI] [PubMed] [Google Scholar]
- Gianino S., Grider J. R., Cresswell J., Enomoto H., Heuckeroth R. O. (2003). GDNF availability determines enteric neuron number by controlling precursor proliferation. Development 130, 2187-2198 [DOI] [PubMed] [Google Scholar]
- Hall A. (2005). Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33, 891-895 [DOI] [PubMed] [Google Scholar]
- Heanue T. A., Pachnis V. (2007). Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies. Nat. Rev. Neurosci. 8, 466-479 [DOI] [PubMed] [Google Scholar]
- Hisatake J., O'Kelly J., Uskokovic M. R., Tomoyasu S., Koeffler H. P. (2001). Novel vitamin D(3) analog, 21-(3-methyl-3-hydroxy-butyl)-19-nor D(3), that modulates cell growth, differentiation, apoptosis, cell cycle, and induction of PTEN in leukemic cells. Blood 97, 2427-2433 [DOI] [PubMed] [Google Scholar]
- Hochgreb T., Linhares V. L., Menezes D. C., Sampaio A. C., Yan C. Y., Cardoso W. V., Rosenthal N., Xavier-Neto J. (2003). A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 130, 5363-5374 [DOI] [PubMed] [Google Scholar]
- Jaffe A. B., Hall A. (2005). Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247-269 [DOI] [PubMed] [Google Scholar]
- Kane M. A., Chen N., Sparks S., Napoli J. L. (2005). Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 388, 363-369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M. A., Folias A. E., Napoli J. L. (2008a). HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal. Biochem. 378, 71-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M. A., Folias A. E., Wang C., Napoli J. L. (2008b). Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem. 80, 1702-1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur R. P., Yost C., Palmiter R. D. (1993). Aggregation chimeras demonstrate that the primary defect responsible for aganglionic megacolon in lethal spotted mice is not neuroblast autonomous. Development 117, 993-999 [DOI] [PubMed] [Google Scholar]
- Kapur R. P., Sweetser D. A., Doggett B., Siebert J. R., Palmiter R. D. (1995). Intercellular signals downstream of endothelin receptor-B mediate colonization of the large intestine by enteric neuroblasts. Development 121, 3787-3795 [DOI] [PubMed] [Google Scholar]
- Kawauchi T., Hoshino M. (2008). Molecular pathways regulating cytoskeletal organization and morphological changes in migrating neurons. Dev. Neurosci. 30, 36-46 [DOI] [PubMed] [Google Scholar]
- Kim D., Dressler G. R. (2007). PTEN modulates GDNF/RET mediated chemotaxis and branching morphogenesis in the developing kidney. Dev. Biol. 307, 290-299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. K., Wassef L., Hamberger L., Piantedosi R., Palczewski K., Blaner W. S., Quadro L. (2008). Retinyl ester formation by lecithin: retinol acyltransferase is a key regulator of retinoid homeostasis in mouse embryogenesis. J. Biol. Chem. 283, 5611-5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger G. M., Mosher J. T., Tsai Y. H., Yeager K. J., Iwashita T., Gariepy C. E., Morrison S. J. (2003). Temporally distinct requirements for endothelin receptor B in the generation and migration of gut neural crest stem cells. Neuron 40, 917-929 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Shin S. Y., Kim S., Choo J., Lee Y. H. (2006). Suppression of PTEN expression during aggregation with retinoic acid in P19 mouse embryonal carcinoma cells. Biochem. Biophys. Res. Commun. 347, 715-722 [DOI] [PubMed] [Google Scholar]
- Lee Y. R., Yu H. N., Noh E. M., Kim J. S., Song E. K., Han M. K., Kim B. S., Lee S. H., Park J. (2007). Peroxisome proliferator-activated receptor gamma and retinoic acid receptor synergistically up-regulate the tumor suppressor PTEN in human promyeloid leukemia cells. Int. J. Hematol. 85, 231-237 [DOI] [PubMed] [Google Scholar]
- Leslie N. R., Yang X., Downes C. P., Weijer C. J. (2007). PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr. Biol. 17, 115-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Liu F., Ross A. H. (2003). PTEN regulation of neural development and CNS stem cells. J. Cell. Biochem. 88, 24-28 [DOI] [PubMed] [Google Scholar]
- Li M., Rossi J. J. (2005). Lentiviral vector delivery of siRNA and shRNA encoding genes into cultured and primary hematopoietic cells. Methods Mol. Biol. 309, 261-272 [DOI] [PubMed] [Google Scholar]
- Liliental J., Moon S. Y., Lesche R., Mamillapalli R., Li D., Zheng Y., Sun H., Wu H. (2000). Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr. Biol. 10, 401-404 [DOI] [PubMed] [Google Scholar]
- Maden M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8, 755-765 [DOI] [PubMed] [Google Scholar]
- McCallion A. S., Stames E., Conlon R. A., Chakravarti A. (2003). Phenotype variation in two-locus mouse models of Hirschsprung disease: tissue-specific interaction between Ret and Ednrb. Proc. Natl. Acad. Sci. USA 100, 1826-1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollard R., Ghyselinck N. B., Wendling O., Chambon P., Manuel M. (2000). Stage-dependent responses of the developing lung to retinoic acid signaling. Int. J. Dev. Biol. 44, 457-462 [PubMed] [Google Scholar]
- Moore T., Holmes P. D. (1971). The production of experimental vitamin A deficiency in rats and mice. Lab. Anim. 5, 239-250 [DOI] [PubMed] [Google Scholar]
- Nagy N., Goldstein A. M. (2006). Endothelin-3 regulates neural crest cell proliferation and differentiation in the hindgut enteric nervous system. Dev. Biol. 293, 203-217 [DOI] [PubMed] [Google Scholar]
- Napoli J. L. (2004). Vitamin A (Retinoids). Encyclopedia of Biological Chemistry, Vol.4 (ed. W. J. Lennarz and M. D. Lane), 354-359 St Louis: Elsevier; [Google Scholar]
- Natarajan D., Marcos-Gutierrez C., Pachnis V., De Graaff E. (2002). Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development 129, 5151-5160 [DOI] [PubMed] [Google Scholar]
- Newgreen D., Young H. M. (2002a). Enteric nervous system: development and developmental disturbances-part 1. Pediatr. Dev. Pathol. 5, 224-247 [DOI] [PubMed] [Google Scholar]
- Newgreen D., Young H. M. (2002b). Enteric nervous system: development and developmental disturbances-part 2. Pediatr. Dev. Pathol. 5, 329-349 [DOI] [PubMed] [Google Scholar]
- Niederreither K., Vermot J., Roux I. L., Schuhbaur B., Chambon P., Dolle P. (2003). The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 130, 2525-2534 [DOI] [PubMed] [Google Scholar]
- Owens S. E., Broman K. W., Wiltshire T., Elmore J. B., Bradley K. M., Smith J. R., Southard-Smith E. M. (2005). Genome-wide linkage identifies novel modifier loci of aganglionosis in the Sox10Dom model of Hirschsprung disease. Hum. Mol. Genet. 14, 1549-1558 [DOI] [PubMed] [Google Scholar]
- Pitera J. E., Smith V. V., Woolf A. S., Milla P. J. (2001). Embryonic gut anomalies in a mouse model of retinoic Acid-induced caudal regression syndrome: delayed gut looping, rudimentary cecum, and anorectal anomalies. Am. J. Pathol. 159, 2321-2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkin R. M. (2007). Folate and neural tube defects. Am. J. Clin. Nutr. 85, 285S-288S [DOI] [PubMed] [Google Scholar]
- Quadro L., Blaner W. S., Salchow D. J., Vogel S., Piantedosi R., Gouras P., Freeman S., Cosma M. P., Colantuoni V., Gottesman M. E. (1999). Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 18, 4633-4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadro L., Hamberger L., Gottesman M. E., Wang F., Colantuoni V., Blaner W. S., Mendelsohn C. L. (2005). Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology 146, 4479-4490 [DOI] [PubMed] [Google Scholar]
- Sanchez T., Thangada S., Wu M. T., Kontos C. D., Wu D., Wu H., Hla T. (2005). PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 102, 4312-4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Heuckeroth R. O. (2008). Retinoic acid regulates murine enteric nervous system precursor proliferation, enhances neuronal precursor differentiation, and reduces neurite growth in vitro. Dev Biol. 320, 185-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. K., Levorse J. M., Ingram R. S., Tilghman S. M. (1999). The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature 402, 496-501 [DOI] [PubMed] [Google Scholar]
- Sidebotham E. L., Woodward M. N., Kenny S. E., Lloyd D. A., Vaillant C. R., Edgar D. H. (2002). Localization and endothelin-3 dependence of stem cells of the enteric nervous system in the embryonic colon. J. Pediatr. Surg. 37, 145-150 [DOI] [PubMed] [Google Scholar]
- Skinner M. (1996). Hirschsprung's Disease. Curr. Probl. Surg. 33, 391-461 [DOI] [PubMed] [Google Scholar]
- Srinivasan S., Anitha M., Mwangi S., Heuckeroth R. O. (2005). Enteric neuroblasts require the phosphatidylinositol 3-kinase/Akt/Forkhead pathway for GDNF-stimulated survival. Mol. Cell. Neurosci. 29, 107-119 [DOI] [PubMed] [Google Scholar]
- Sukegawa A., Narita T., Kameda T., Saitoh K., Nohno T., Iba H., Yasugi S., Fukuda K. (2000). The concentric structure of the developing gut is regulated by Sonic hedgehog derived from endodermal epithelium. Development 127, 1971-1980 [DOI] [PubMed] [Google Scholar]
- Takahashi M. (2001). The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 12, 361-373 [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu J., Matsumoto K., Aota S., Parsons R., Yamada K. M. (1998). Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280, 1614-1617 [DOI] [PubMed] [Google Scholar]
- van Diepen M. T., Eickholt B. J. (2008). Function of PTEN during the formation and maintenance of neuronal circuits in the brain. Dev. Neurosci. 30, 59-64 [DOI] [PubMed] [Google Scholar]
- van Weering D. H., Bos J. L. (1997). Glial cell line-derived neurotrophic factor induces Ret-mediated lamellipodia formation. J. Biol. Chem. 272, 249-254 [DOI] [PubMed] [Google Scholar]
- Vohra B. P., Fu M., Heuckeroth R. O. (2007a). Protein kinase Czeta and glycogen synthase kinase-3beta control neuronal polarity in developing rodent enteric neurons, whereas SMAD specific E3 ubiquitin protein ligase 1 promotes neurite growth but does not influence polarity. J. Neurosci. 27, 9458-9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohra B. P., Planer W., Armon J., Fu M., Jain S., Heuckeroth R. O. (2007b). Reduced endothelin converting enzyme-1 and endothelin-3 mRNA in the developing bowel of male mice may increase expressivity and penetrance of Hirschsprung disease-like distal intestinal aganglionosis. Dev. Dyn. 236, 106-117 [DOI] [PubMed] [Google Scholar]
- Wang F., Herzmark P., Weiner O. D., Srinivasan S., Servant G., Bourne H. R. (2002). Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell. Biol. 4, 513-518 [DOI] [PubMed] [Google Scholar]
- Wedlich-Soldner R., Li R. (2003). Spontaneous cell polarization: undermining determinism. Nat. Cell. Biol. 5, 267-270 [DOI] [PubMed] [Google Scholar]
- Wendler C. C., Schmoldt A., Flentke G. R., Case L. C., Quadro L., Blaner W. S., Lough J., Smith S. M. (2003). Increased fibronectin deposition in embryonic hearts of retinol-binding protein-null mice. Circ. Res. 92, 920-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendling O., Dennefeld C., Chambon P., Manuel M. (2000). Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development 127, 1553-1562 [DOI] [PubMed] [Google Scholar]
- Wendling O., Ghyselinck N. B., Chambon P., Mark M. (2001). Roles of retinoic acid receptors in early embryonic morphogenesis and hindbrain patterning. Development 128, 2031-2038 [DOI] [PubMed] [Google Scholar]
- West K. P., Jr (2002). Extent of vitamin A deficiency among preschool children and women of reproductive age. J. Nutr. 132, 2857S-2866S [DOI] [PubMed] [Google Scholar]
- Wolf G. (1984). Multiple functions of vitamin A. Physiol. Rev. 64, 873-937 [DOI] [PubMed] [Google Scholar]
- Yoshimura T., Arimura N., Kaibuchi K. (2006). Signaling networks in neuronal polarization. J. Neurosci. 26, 10626-10630 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}