

Abstract
14-mer α-helix and a 15-mer β-strand oligopeptides composed of leucine (L) and lysine (K) were used to investigate peptide adsorption and orientation onto well-defined methyl and carboxylic acid terminated self-assembled monolayer (SAM) surfaces with X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). XPS showed both peptides reached monolayer thickness on both SAMs, but significantly higher solution concentrations were required to reach this coverage on the methyl SAMs. This shows that the peptides adsorb more strongly onto the carboxyl-terminated SAMs. The excess oxygen detected by XPS and the H3O+ signal detected by ToF-SIMS for the SAMs with adsorbed peptides indicated that water molecules are associated with the adsorbed peptides, even under ultra-high vacuum conditions. Changes in the amount of L and K fragments detected by ToF-SIMS indicate the β-strand oriented differently on the two SAMs. The L side-chains were preferentially associated with the methyl-terminated SAM and the K side-chains were preferentially associated with the carboxyl SAM. In contrast, little change in the ToF-SIMS K/L ratio was observed for the α-helix peptide absorbed on the two SAMs, indicating ToF-SIMS was not as sensitive to orientation of the α-helix peptide.
Introduction
The interactions between proteins and synthetic surfaces are the focus of much research in the biomedical field. The interest in this topic rests in the fact that denaturation of proteins is often the first step in the thrombogenic cascade or in the foreign-body response, which can lead to formation of blood clots, rejection of the implant, and other adverse effects1,2. Efforts are being made to ascertain the structure and conformation of proteins at surfaces, and to correlate protein surface interactions with the success of the synthetic surface in the body.1,3,4 Developing this correlation is important for biomedical applications such as biomaterials, implantables, drug delivery, and medical equipment that must penetrate the skin barrier. 5
Protein structures are complex and therefore have many independent variables that must be accounted for when designing and interpreting an experiment. A method to reduce these factors is to use peptides as simplified protein models. Model, de novo peptides have been synthesized to mimic many protein structures to investigate the correlation between protein characteristics and their behavior. Examples of this include structural mimics such as synthetic collagen triple-helices6,7, peptides for cell activation and binding domains8–11, self-assembling supramolecular structures12, targeted drug-delivery 13, and behavioral mimics to investigate protein folding14–17 and refolding18.
DeGrado and Lear16 devised short peptides made up of leucine (L) and lysine (K) residues only that would mimic specific secondary structures of larger proteins. Two of these peptides, the 14-mer LK α-helix (LKα14) and the 15-mer LK β-strand (LKβ15), were used in this study to probe the interactions of peptides and proteins with surfaces. LKα14 was designed with a hydrophobic periodicity (HP) of 3.5 and LKβ15 with an HP of 2. When these peptides assume α-helical and β-strand conformations, respectively, the lysine residue side-chains are on one side of the peptide backbone and the leucine side-chains are on the other. This leads to amphiphilic peptides that have a hydrophilic, positively-charged side and a hydrophobic side. Leucine-lysine peptides have been examined in a number of previous studies that include investigation of peptide orientation on hydrophobic and hydrophilic surfaces using sum-frequency generation (SFG)19–21, structural characterization of adsorbed peptides using solid-state nuclear magnetic resonance (ssNMR)22,23, LK peptides as antibacterial agents24, secondary structure of LK peptides at the air/water interface25, peptide chain-length dependence of secondary structure26,27, and infiltration of lipid monolayers by LK peptides28.
Peptides have been characterized in solution and at interfaces using many different techniques. Atomic force microscopy (AFM), circular dichroism (CD), infrared spectroscopy (IR), ellipsometry, and NMR have been used extensively to study secondary structure and structural transitions20,22,23,27,29–36, and compression studies have been used to look at order at interfaces35. These studies have mostly been done in solution at air/water or aqueous/solvent interfaces as opposed to aqueous/solid interfaces which are more relevant for biomaterial applications.
The model surfaces used in this study were self-assembled monolayers of thiols adsorbed onto gold. The sulfur end-group has a high affinity for the gold surface and causes the thiol molecules to orient themselves such that the thiol binds to the gold and the other end of the molecule, the ω-functional group, is displayed at the surface37–40. The thiol molecules form a well-packed structure in which the chains are oriented approximately 30° from the surface normal41. This assembly process allows for versatile, reproducible surfaces. The two surfaces used in this study were hydrophobic, methyl-terminated and hydrophilic, negatively-charged, carboxylic-acid-terminated SAMs.
The techniques used in this study are X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). Not only do these techniques allow for the characterization of peptides adsorbed to solid surfaces, but they also allow for the direct quantification of the adsorbed peptides. XPS in particular gives a quantitative determination of the relative concentrations of atoms on the surface as well as the ability to determine the coordination chemistry of those atoms. ToF-SIMS was then used to investigate the orientation of the adsorbed peptides on the solid surface.
Concurrently, studies of these LK peptides on methyl and carboxyl SAMs were done using sum-frequency generation (SFG) vibrational spectroscopy and near-edge X-ray absorption fine structure (NEXAFS) spectroscopy to determine peptide orientation.42 Phase-sensitive SFG studies showed leucine side-chains interacting with the methyl SAMs and lysine side-chains interacting with the carboxyl SAMs. NEXAFS spectra for the β-strand peptides showed dichroism from preferential orientation of amide bonds in the peptide backbone, indicating that the peptides are lying parallel to the surface with their side-chains interacting with the surface. Together with XPS and ToF-SIMS, these techniques give a detailed view of the adsorption and orientation behavior, and thus the molecular interactions, of the LK peptides on these two SAM surfaces.
Materials and Methods
SAM Preparation
The substrates used in this study were 1 cm × 1 cm pieces cut from a silicon wafer (Silicon Valley Microelectronics, Inc., San Jose), coated with 10-nm Cr and 80-nm Au (99.99%) by electron beam evaporation at pressures below 1×10−6 Torr. The SAMs were prepared by immersing the gold surfaces into 1 mM dodecane thiol (Aldrich) or 0.5 mM 11-mercapto-undecanoic acid thiol (Aldrich) ethanolic (200 proof, Aaper) solutions for approximately 24 hours under nitrogen atmosphere. A lower solution concentration (0.5 mM) was used for assembly of the 11-mercapto-undecanoic acid thiol as it resulted more ordered SAMs (data not shown). The dodecane SAMs were then rinsed copiously with pure ethanol to remove any unbound and oxidized thiol molecules. The undecanoic acid SAMs were sonicated in fresh ethanol for 10 minutes, then rinsed thoroughly with ethanol. Both were then dried under a stream of nitrogen and stored under nitrogen atmosphere until analysis.
Peptide Synthesis
Amino acids used in this study were Fmoc-Leu-OH and Fmoc-Lys(Boc)-OH (Novabiochem). The peptides were synthesized on a PS3 solid-state peptide synthesizer (Rainin) on a Leu-Wang resin (Novabiochem). O-(Benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) (Advanced ChemTech) was used as an activator, and the N-termini of the resin-bound peptides were capped following synthesis by acetylation with about 3 mL of acetic acid (Aldrich). Peptides were cleaved from the resin using a solution of 95% trifluoroacetic acid (Acros), 2.5 % triisopropylsilane (Aldrich) and 2.5 % water according to the procedure from Novabiochem (Fmoc resin cleavage and deprotection) and precipitated in cold tert-butyl methyl ether (Aldrich). The samples were then centrifuged and resuspended 2–3 times in ether to wash the peptide. Centrifuged peptide samples were dried overnight, then resuspended in 5–10 mL water, and dried by overnight lyophilization and their purity was checked using mass spectrometry following the procedures outlined by Long et al.23. The final peptide sequences were Ac-LKLLKKLLKLLKKL-OH for LKα14 and Ac-LKLKLKLKLKLKLKL-OH for LKβ15.
Peptide Adsorption
Peptide adsorption was done by dissolving the peptides at twice the desired concentration in degassed water. A phosphate buffered saline (PBS) 10x solution from Omnipur (EMD), was used to make 1x and 2x PBS solutions (137 mM NaCl, 3 mM KCl, 10 mM phosphate salts for 1x solutions, pH 7.4) that were degassed before use. SAM surfaces were first equilibrated under 1 ml of 2x PBS for 20 minutes at 37 °C before adding 1 ml of the 2x peptide solution. Peptide adsorption was done for 2 hours at 37°C in 24-well plates (non-tissue culture treated plates, Falcon). Samples were never placed in adjacent cells and only near each other if they were at the same concentration. Only a maximum of 4 wells were used on each plate to avoid intermixing. The adsorption was stopped by solution displacement (15 mL buffer added to each 4 mL well), and followed by sequential one-minute rinses in stagnant 1x PBS buffer, stirred buffer, and 3 times in stirred water. Samples were allowed to air-dry and then stored under nitrogen until analysis. Air drying was found to yield more reproducible adsorption isotherms than drying the samples in a stream of nitrogen (data not shown). No difference in the quantity of oxidized or unbound thiol was observed between the two drying methods (data not shown).
X-ray Photoelectron Spectroscopy
XPS is a quantitative technique that measures the elemental composition of the outermost ~100 Å of a sample. Measurements were performed on a Kratos AXIS Ultra DLD instrument using a monochromatic Al Kα X-ray source and a 90° take-off angle in the hybrid mode. The take-off angle is defined as the angle between the sample surface and the axis of the XPS analyzer lens. Compositional survey and detailed scans (N 1s, O 1s, S 2p) were acquired using an analyzer pass energy of 80 eV. Three spots on two or more replicates of each sample were analyzed. Reported compositional data were averages of values determined at each spot on several samples and error bars represent the standard deviation for those average values. XPS data analysis was performed with the Vision Processing data reduction software.
Time-of-Flight Secondary Ion Mass Spectrometry
ToF-SIMS is a semi-quantitative surface analysis tool that can detect the mass fragments present in the top ~20 Å of the surface in the static mode. ToF-SIMS data was acquired with an ION-TOF TOF.SIMS 5–100 instrument using the Bi3+ cluster-ion source to obtain higher fragment yields and improved mass resolution. Mass resolutions (m/Δm) were above 6700 at m/z = 27 for all positive secondary ion mass spectra. Data analysis was performed using the IonSpec data reduction software. The m/z 84 peak (C5H10N+) was chosen as the characteristic peak for lysine (K) and the m/z of 86 peak (C5H12N+) was chosen for leucine (L). These fragments represent the immonium ion fragments from each amino acid.43,44 It should be noted that some of the 84 peak intensity is from leucine (leucine immonium ion with a loss of 2H). The relative ratios of these two peaks were assumed to be proportional to ratios of lysine to leucine at the surface of the samples. Each data point represents the average of the ratios from several spectra on two or more samples. The error bars represent ± one standard deviation. The ToF-SIMS data for adsorbed LKβ15 were also processed using principal component analysis (PCA)45,46 to determine overall trends. The peak set for PCA was chosen by compiling a list of the prominent nitrogen containing peaks from leucine and lysine amino-acids43 (see Table S2 in the supplementary material). Only nitrogen containing peaks were included in the PCA peak set to ensure all peaks were peptide-specific.
Molecular Modeling
Model structures of the LKα14 and LK β15 peptides were constructed and simulated using the CHARMM (version c34b2) suite of molecular simulation tools 47 and parameter libraries (version 27).48 Starting with a terminal amino acid residue with geometry based on parameter library values, additional residues of each structure were added while specifying the appropriate dihedral angles between residues (−57 phi, −47 psi for the LKα14 and −140 phi, +135psi for the LKβ15). Once built, the conformations of these starting structures were relaxed, in the absence of explicit water models, through 500 steps of an energy minimization using the steepest descent algorithm. After this initial minimization, each peptide structure was added to an equilibrated 1 atm, 298 K cubic box of TIP3P water models49 where displaced water molecules were removed from the box. Na+ and Cl− ions sufficient to both neutralize the peptide and approximate a 140 mM saline environment were added to each simulated system by replacing water models with ions as needed. At this point, each entire simulated system underwent a steepest descent energy minimization of 1,000 steps for the water models to adjust to the presence of the ions and a peptide. For both the 0.1 ns equilibration and 1 ns production dynamics simulations, calculation of electrostatic interactions was accomplished using the particle-mesh Ewald method50. The simulations were conducted in the canonical (NVT) ensemble using the VV2 integrator (an implementation of the velocity-Verlet algorithm51). The Nosé-Hoover method52 was used for temperature control, with a target temperature of 298 K and a thermostat time constant of 0.1 ps. Hydrogen bond lengths were held fixed at parameter library values using the SHAKE algorithm,53 which enabled a 2.0 fs time step to be used during the MD simulations.
Results and Discussion
XPS Adsorption Isotherms
The amount of LKα14 and LKβ15 peptide adsorption onto the methyl and carboxylic-acid SAM surfaces was measured using XPS to quantify the nitrogen surface concentration and gold attenuation from the peptide overlayer. Figures 1 and 2 show how the ratio of N atomic percent to Au atomic percent (N/Au ratio) varied with LKα14 and LKβ15 peptide solution concentration for the two SAM surfaces. The amount of nitrogen detected is higher than what would typically be expected for a monolayer of a ~2 kDa peptide due to the high concentration of lysines in the LK peptides (each lysine side chain contains an amine group). Complete XPS data for the adsorption isotherms to methyl and carboxyl SAMs can be found in Tables 1–4. These tables contain the measured XPS composition of the organic overlayers (SAM and adsorbed peptide without the Au signal, renormalized to 100 atomic %) and measured XPS concentration of the substrate (atomic % Au) including the organic overlayer signals. The last column gives the N/Au ratio values shown in Figures 1 and 2. The organic overlayer signals were renormalized to 100 atomic % for comparison with calculated compositions shown at the bottom of Tables 1–4. In Table 5 the theoretical peptide compositions are compared to the XPS measured compositions for thick films of acetylated peptide powders. XPS detected 8–10 % fluorine in thick layers of the LK peptide powders, likely arising from the presence of residual trifluoroacetic acid (TFA) used in cleavage of the peptides from the resin. The chemical formula for TFA is C2HF3O2, and so to remove the TFA contribution from the XPS compositions of the peptides, ½ of the F % was subtracted from the C and O %, and then the C, O and N values were rescaled to 100 %. It is important to note that no fluorine was detected on the adsorbed peptide samples. The presence of TFA in the bulk peptide powders was supported by the presence of a CF3 peak at 292.4 eV in the high-resolution XPS C1s spectra that accounted for about 3.7 % of the total C content, and by the presence of high intensity F−, CF2−, CF3−, and CF3CO2− peaks in the negative ToF-SIMS data.
Figure 1.

LKα14 peptide adsorption isotherms on a) methyl (C12) and b) carboxylic-acid (C10COOH) terminated SAM surfaces. It can be seen that LKα14 is more strongly attracted to the carboxyl-terminated surface than the methyl-terminated SAM. The data in b were fit to a Langmuir isotherm described by Equation 1 and Table 7. Error bars represent the standard deviation for each data point.
Figure 2.

LKβ15 peptide adsorption isotherms on a) methyl (C12) and b) carboxylic-acid (C 10COOH) terminated SAM surfaces. The isotherms are similar to those in Figure 1 and show LK β15 interacts more strongly with the carboxyl-terminated surface than the methyl-terminated SAM. The data in b were fit to a Langmuir isotherm described by Equation 1 and Table 7. Error bars represent the standard deviation for each data point, and are present for each point although some are too small to be visible.
Table 1.
XPS results for LKα14 adsorption onto methyl SAMs. The C, O, N, and S atomic percents were renormalized to 100 atomic % to show the composition of the organic overlayer (SAM plus peptide). The Au atomic % before renormalization and the N/Au ratio show the attenuation of the gold signal by the organic overlayer.
| Peptide Adsorption Concentration (mg/mL) | C atomic %* | O atomic %* | N atomic %* | S atomic %* | Au atomic % | N/Au ratio |
|---|---|---|---|---|---|---|
| 0.1 | 78.9 (2.3) | 15.1 (0.9) | 4.1 (0.4) | 1.9 (0.1) | 48.3 (1.4) | 0.043 (0.005) |
| 0.2 | 83.0 (10.4) | 10.3 (3.6) | 3.8 (1.5) | 2.1 (0.4) | 45.6 (5.3) | 0.050 (0.024) |
| 0.3 | 78.5 (10.2) | 12.2 (2.4) | 7.0 (2.1) | 1.6 (0.3) | 35.7 (7.3) | 0.14 (0.07) |
| 0.4 | 77.3 (2.7) | 12.6 (2.6) | 8.2 (1.7) | 1.4 (0.3) | 26.9 (3.9) | 0.23 (0.08) |
| 0.5 | 75.1 (7.3) | 14.4 (1.6) | 8.5 (1.6) | 1.4 (0.3) | 29.3 (6.7) | 0.22 (0.08) |
| 1 | 74.1 (3.7) | 14.7 (1.0) | 9.6 (0.7) | 1.1 (0.2) | 21.1 (3.4) | 0.37 (0.07) |
| Calculated Values for Monolayer | 83.1 | 6.5 | 8.4 | 2.0 | 34.3 | 0.16 |
| Calculated Values for Bilayer | 78.3 | 8.9 | 11.6 | 1.1 | 22.6 | 0.40 |
Renormalized to 100% without the Au signal.
Table 4.
XPS results for LKβ15 adsorption onto carboxyl SAMs. The C, O, N, and S atomic percents were renormalized to 100 atomic % to show the composition of the organic overlayer (SAM plus peptide). The Au atomic % before renormalization and the N/Au ratio show the attenuation of the gold signal by the organic overlayer.
| Peptide Adsorption Concentration mg/mL | C atomic %* | O atomic %* | N atomic %* | S atomic %* | Au atomic % | N/Au ratio |
|---|---|---|---|---|---|---|
| 0.0005 | 80.7 (1.0) | 13.6 (0.5) | 3.4 (0.7) | 2.4 (0.3) | 37.7 (3.7) | 0.057 (0.016) |
| 0.001 | 77.6 (1.2) | 14.3 (0.9) | 6.4 (1.0) | 1.7 (0.5) | 34.2 (1.4) | 0.12 (0.02) |
| 0.003 | 74.7 (0.3) | 13.7 (0.2) | 10.5 (0.2) | 1.0 (0.1) | 21.9 (0.6) | 0.38 (0.02) |
| 0.005 | 75.3 (0.7) | 13.3 (0.3) | 10.2 (0.4) | 1.3 (0.1) | 22.2 (0.8) | 0.36 (0.03) |
| 0.01 | 74.3 (0.2) | 13.5 (0.1) | 11.2 (0.2) | 1.0 (0.0) | 19.1 (0.2) | 0.47 (0.01) |
| 0.05 | 74.1 (0.5) | 13.8 (0.3) | 11.0 (0.3) | 1.0 (0.1) | 19.3 (0.9) | 0.46 (0.03) |
| 0.3 | 71.3 (0.5) | 15.1 (0.4) | 13.1 (0.1) | 0.5 (0.1) | 8.5 (0.9) | 1.43 (0.16) |
| Calculated Values for Monolayer | 76.2 | 13.7 | 8.2 | 2.0 | 30.0 | 0.19 |
| Calculated Values for Bilayer | 74.6 | 12.7 | 11.7 | 1.1 | 19.7 | 0.48 |
Renormalized to 100% without the Au signal.
Table 5.
XPS composition data for acetylated peptide powder samples after removal of fluorine, carbon and oxygen contributions from the residual TFA. Numbers in brackets are the results before renormalization. The expected theoretical compositions for the acetylated peptides are shown in italics.
| Peptide | C (atomic %) | O (atomic %) | N (atomic %) | F (atomic %) |
|---|---|---|---|---|
| LKα14 | 72.6 (63.8 ± 0.4) 71.4 |
11.4 (14.9 ± 0.4) 12.6 |
16.0 (12.8 ± 0.7) 16.0 |
(8.5 ± 0.1) |
| LKβ15 | 72.2 (61.2 ± 0.6) 71.1 |
11.3 (15.7 ± 0.3) 12.5 |
16.5 (12.3 ± 0.6) 16.4 |
(10.8 ± 0.1) |
The peptide-surface interaction differed significantly between the two SAM surfaces. To obtain similar peptide adsorption amounts on both SAM surfaces, several orders of magnitude higher peptide solution concentrations were required for the methyl-terminated SAM compared to the carboxyl SAM. This was the case for both the LKα14 and LKβ15 peptides, indicating the affinities of the peptides to the methyl and carboxylic acid SAMs were significantly different. These differences would be expected if the amine groups on the lysine side chains interact electrostatically with the carboxylic acid SAMs and the methyl groups on the leucine side chains interact hydrophobically with the methyl SAMs. At pH 7.4, approximately 50% of the carboxyl groups are predicted to be negatively charged,54 and the peptides should be strongly positively charged since the pKa of lysine is around 10.5.55,56 Although the Debye length for the ionic strength of the 1x PBS solution was a few angstroms57, this data indicates that the positively-charged amine groups of the lysine residues were still attracted to the negatively-charged carboxylic acid surface. This suggests that electrostatic screening at physiological ionic strength did not negate charge-charge interactions at the solid-liquid interface.
The adsorption isotherms for the carboxyl SAMs appeared to follow a Langmuir isotherm trend. Accordingly, a Langmuir-type curve, described by Equation 1, was fit to the data for LKα14 and LKβ15 adsorptions to carboxyl SAMs, excluding the last data point in each dataset. However, it is not a true Langmuir isotherm since it is described as a function of the initial peptide solution concentration, rather than the final peptide solution concentration.
| [1] |
The fit parameters for Equation 1 can be seen in Table 7, where A is the amount adsorbed (expressed here in N/Au ratios), K′ is a constant, Amax is the maximum amount adsorbed, and c is the initial peptide solution concentration in mg/mL. The data for adsorption to methyl SAMs were not fit to any curve due to the high standard deviations in the data. From the fit data in Table 7, it is apparent that K′ is the same order of magnitude for both peptides, suggesting similar adsorption kinetics.
Table 7.
Fit parameters for Langmuir isotherms describing LK peptide adsorption onto the carboxyl SAMs.
| Peptide | Amax (N/Au ratio) | K′ (mL/mg) |
|---|---|---|
| LKα14 | 0.32 ± 0.02 | 1.1 ± 0.2 × 103 |
| LKβ15 | 0.52 ± 0.5 | 0.5 ± 0.2 × 103 |
The difference in peptide-surface interactions was also seen by the higher standard deviations observed on the methyl surface as opposed to the carboxyl surface. The data in Figures 1 and 2 are presented such that the error bars represent the N/Au ratio standard deviation at each concentration. It can be seen that on the methyl surface that the peptide concentration was highly variable, suggesting a patchy surface coverage. Even from spot to spot on one sample the N concentration could vary by more than one atomic percent. Possible explanations for the high variability of peptides adsorbed onto the methyl SAMs include adsorption of bundled peptides, a nucleation-site effect where once the initial peptide has adsorbed it increases the probability of other peptides binding in that region, or adsorption at defects in the methyl SAM. The adsorption of bundled peptides is most likely to occur at the highest solution concentrations (see below for further discussion of peptide bundling in solution).
XPS also enabled the comparison of atomic concentrations on the surface with predicted values. The oxygen/nitrogen ratio for the LK peptide should be 0.7, and so, on a methyl SAM, one would expect to observe this ratio since the methyl SAM itself contains no oxygen. However, the amount of oxygen detected by XPS was always higher than expected. Some of this could be attributed to oxidized sulfur species58, but usually only minor amounts of oxidized sulfur species were detected in the XPS S2p spectra, accounting for ≤1 atomic % of the oxygen concentration. Two other possibilities could account for the excess oxygen detected by XPS. One is the oxidation of the peptide itself, which seems unlikely since they were stored in the freezer and should be stable in solution for the three hours of the peptide adsorption experiment (adsorption plus prep time). The second is water associated either with the methyl SAM or the adsorbed peptide. It should be noted that this interaction between water and the SAM or peptide must have been rather strong to withstand the ultra-high vacuum conditions required for XPS analysis. Recent studies have also indicated the presence of water bound to amine terminated SAMs under ultra-high vacuum conditions59.
As shown in Table 6, a methyl SAM soaked in buffer for 2 hours without any exposure to peptide showed an increase from zero to 2.3 atomic % O in the organic overlayer (i.e., without including the Au substrate). There was no oxidized sulfur detected by XPS on this sample. Therefore, in the absence of the peptide, it appears that some water was associating or infiltrating and disrupting the methyl SAM. SFG studies show gauche defects are introduced into alkyl chains when methyl SAMs are exposed to buffer.42 The disruption of the SAM is further corroborated by the fact that the XPS Au signal increased after buffer exposure, indicating decreased integrity of the thiol monolayer resulting either from desorption of the thiol groups from the gold surface or from an increase in disorder of the thiol chains. It is also corroborated by the appearance of a strong ToF-SIMS H3O+ peak at m/z 19 for buffer-soaked and peptide-adsorbed methyl SAMs. This ToF-SIMS water peak is not present in spectra acquired from the bare methyl SAMs. The SAM-associated water does not completely account for all the oxygen detected by XPS on the methyl SAM with adsorbed peptide since, even with the 2.3 atomic % O subtracted from the XPS data, the O/N ratio on a methyl SAM with LKα14 adsorbed at 1 mg/mL was 1.3. Thus, it appears the presence of adsorbed peptide resulted in additional water bound to the surface. Whether this water was trapped at the peptide-SAM interface or whether it was associated with the peptides themselves is currently being investigated. Infrared spectroscopy data for samples previously analyzed by XPS (data not shown) confirms that there is water present on all samples except bare methyl SAMs, even after exposure to ultra-high vacuum conditions.
Table 6.
XPS determined composition for the methyl and carboxyl SAM. The C, O, N, and S atomic percents were renormalized to 100 atomic % to show the composition of theSAM overlayer. The Au atomic % before renormalization shows the attenuation of the gold signal by the SAM overlayer. The expected theoretical compositions for the SAMs are shown in italics.
| C (atomic %)* | O (atomic %)* | N (atomic %)* | S (atomic %)* | Au raw signal (atomic %) | |
|---|---|---|---|---|---|
| Methyl SAM | 96.5 ± 0.4 92.3 |
n.d.** 0.0 |
n.d. 0.0 |
3.5 ± 0.4 7.7 |
47.9 ± 1.7 |
| Carboxyl SAM | 81.7 ± 0.9 78.6 |
15.5 ± 1.0 14.3 |
n.d. 0.0 |
2.8± 0.1 7.1 |
40.5 ± 0.9 |
| Methyl SAM soaked in PBS buffer | 94.6 ± 0.7 92.3 |
2.3 ± 0.4 0.0 |
n.d. 0.0 |
3.1 ± 0.3 7.7 |
50.3 ± 1.4 |
Renormalized to 100% without the Au signal.
n.d. = not detected
To determine if the adsorbed peptides form more than a single layer on the surface, calculations were done to provide expected compositions for both a full monolayer and a bilayer of peptides adsorbed onto the SAM surfaces. These calculations used experimental XPS SAM compositions acquired at a 90° photoelectron take-off angle in the electrostatic mode to narrow the acceptance angle of the analyzer lens. These compositions are listed in Table 8. Theoretical peptide compositions were used in the calculations instead of XPS experimentally determined composition because of the TFA contamination of the peptide powders. The equation used to calculate the theoretical monolayer and bilayer compositions is shown in Equation 2 and has previously been described by Paynter60. θ is the take-off angle with respect to the surface (90° for these experiments), n(x) is the atomic concentration at depth z in atomic %, λ is the inelastic mean free path, F is the transmission function, and k encompasses the capture cross section and emission efficiency.
Table 8.
XPS determined composition for the methyl and carboxyl SAM acquired in electrostatic mode at a take-off angle of 0°. These values were used in the theoretical calculations of XPS composition for LKα14 and LKβ15 peptide monolayers and bilayers. (Data not renormalized).
| C (atomic %) | O (atomic %) | S (atomic %) | Au (atomic %) | |
|---|---|---|---|---|
| Methyl SAM | 46.6 | 0 | 2.0 | 51.5 |
| Carboxyl SAM | 43.6 | 10.0 | 2.0 | 44.4 |
| [2] |
The maximum thickness of the peptides, as determined by measurements made of conformations resulting from molecular dynamic simulations, was 14 Å. Mean free paths were chosen as 32 Å for C, 30 Å for N, 33 Å for S and Au, and 28 Å for O assuming a peptide and SAM densities of ~1.2 g/cm3.61–63 The calculated compositions for peptide monolayer and bilayers are given at the bottom of Tables 1–4.
From the results in Tables 1–4 is it apparent that the gold signal decreases and the nitrogen signal increases with increasing peptide solution, as expected for increasing amounts of peptide adsorption with increasing peptide solution concentration. Comparison of the experimental N/Au ratios to the predicted values for a peptide monolayer indicates that monolayer coverage is reached around solution concentrations of 0.3 mg/mL for the methyl SAM surfaces and 0.001 to 0.003 for the carboxyl SAMs. At the higher concentration peptide solutions required for saturation coverage on the methyl SAM surface, the peptide solution became somewhat cloudy around 0.3mg/mL for the LKβ15 and around 0.5 mg/mL for the LKα14. This suggests, especially at the highest solution concentrations, that the peptides form clusters or bundles, possibly via hydrophobic interactions among the leucine sides, and begin to precipitate from solution. Therefore, the increase in N concentration and decrease in Au concentration for peptide solutions at the highest solutions concentrations likely is due to peptide bundles depositing onto the surface, rather than individual peptides adsorbing to the surface. Thus, when the composition of the peptide covered methyl SAMs exceed the expected values for a peptide monolayer this is likely due to deposition of peptide bundles. Although the predicted N and Au values for a peptide monolayer on methyl SAMs match well with the 0.1 mg/mL experimental data, the measured oxygen concentration is significantly higher than the predicted oxygen concentration. This is consistent with the presence of water on the peptide-adsorbed methyl surfaces and not on the bare methyl SAMs
For the peptides adsorbed onto the carboxyl SAMs, monolayer coverage occurs below solution concentrations where significant bundling of the peptides is expected to occur. Thus the further increase of the N/Au ratio for peptide solution concentrations between 0.001 and 0.05 mg/mL may not be due to significant deposition of peptide bundles. A possible explanation for this additional N is formation of a partial bilayer on the carboxyl SAMs. The experimental surface compositions for these solutions fall between calculation values for a peptide monolayer and a bilayer. The formation of a bilayer on the carboxyl SAMs and not the methyl SAMs is possible since the interactions of the lysines with the carboxyl surface would leave the leucine side chains exposed to the solution, allowing for the leucines from a second layer of peptides to bind to the first peptide layer via hydrophobic interactions. It is interesting to note that the XPS results suggest by ~0.01 mg/mL solution concentrations a peptide bilayer forms on the carboxyl SAMs, which is well below the solution concentrations required to form a peptide monolayer on the methyl SAMs. This suggests the leucine-leucine hydrophobic interactions between two peptides are stronger than the leucine-methyl hydrophobic interactions between the peptide and the methyl SAM. A possible explanation for this observation is that the branched methyl groups on the leucine side chains have more flexibility and mobility than the tightly packed methyl groups on the alkyl chains in the dense SAM structure. This would allow the peptide leucine side chains more freedom to rearrange and adopt conformations leading to more stable hydrophobic interactions between two peptides compared to those formed between individual peptides and the methyl SAM surface.
The calculated monolayer and peptide compositions should only be considered as approximate guidelines. This is because the exact density of the organic film is unknown, the escape depths are averages for organic films and their accuracy for the peptides on SAMs is unknown, and the data was acquired in the Kratos hybrid mode with a large photoelectron acceptance angle (approximately 40°, significantly greater than the 1° acceptance angle used in the calculations). However, the calculations serve as useful guidelines to show that the peptides form bundled layers on the methyl SAM surfaces and possible partial bilayers on the carboxyl SAM surfaces.
ToF-SIMS Orientation Studies
Secondary-ion mass spectrometry experiments were performed to determine the orientation of LKα14 and LKβ15 on the two SAM surfaces. This was done by comparing the relative ratios of two characteristic fragments, one for K (m/z = 84) and one for L (m/z = 86), as shown in Figure 4. The measured ratio of the m/z = 84 to m/z = 86 intensities (not corrected for differences in secondary ion yields) for the LKα14 powder was 0.98±0.03. The 84/86 intensity ratio for the LKα14 peptide adsorbed onto the methyl SAMs was 1.2±0.2 for surface coverages of 4–5 atomic % N and 0.92±0.02 for surface coverages of 8–9 atomic % N. The 84/86 ratio for LKα14 on the carboxyl SAMs was 1.04±0.04 for surface coverages of 4–5 atomic % N and 0.85±0.03 for surface coverages of 8–9 atomic % N. Thus, at the same peptide surface coverage he 84/86 intensity ratio for the LKα14 was slightly higher on the methyl SAMs compared to the carboxyl SAMs, indicating that the lysines were closer to the outermost surface when the LKα14 peptide was adsorbed onto the methyl SAMs. This is expected if the leucines were interacting with the hydrophobic, methyl SAM.
Figure 4.

Ratio of 84/86 characteristic K/L peaks of a) LKα14 and b) LKβ15 adsorbed at different concentrations onto methyl and carboxylic-acid terminated SAMs plotted against approximate N atomic % in the organic layer (XPS). The ratios indicate that the leucine residues interact with the methyl surface and the lysines interact with the carboxyl SAM for the LKβ15 peptide. The orientation of LKα14 is not discernable using this technique. Error bars represent the standard deviation for each data point, and are present for each point although some are too small to be visible.
However, the difference in K/L intensity ratios for the LKα14 peptides is similar to the standard deviations of the measurements, and appears to be more dependent on peptide surface coverage than on the type of SAM. To explore this further, the 84/86 ratio was examined for LKα14 adsorbed onto the methyl SAMs from a range of solution concentrations. These concentration-dependent data showed an inverse relationship between the LKα14 peptide coverage and the 84/86 intensity ratio, as seen in Figure 5. This trend can be explained by the fact that at lower concentrations, the peptides are largely present as individual molecules in solution, and the methyl groups on the leucines are free to interact with the methyl surface giving the expected higher 84/86 ratios. However, at higher peptide concentrations, the peptides are likely forming bundles in solution, which are then deposited onto methyl SAM surface (see the discussion in the XPS results section). This would lead to lower 84/86 intensity ratios.
Figure 5.
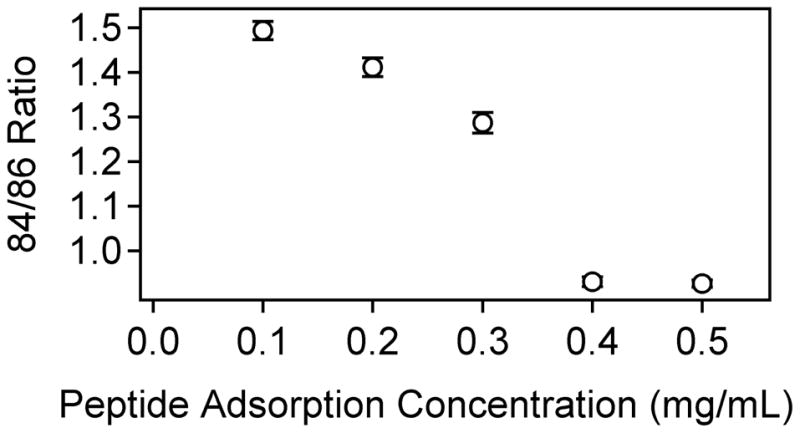
ToF-SIMS data showing the concentration-dependence of the 84/86 ratio for LKα14 on methyl SAMs. Error bars represent the standard deviation for each data point, and are present for each point although some are too small to be visible.
There are two other possibilities why the differences in 84/86 intensity ratios for the LKα14 peptides might be similar on the methyl and carboxyl SAM surfaces. First, ToF-SIMS samples the top 10–20 Å of a surface,64 and the peptide dimensions are only 14 Å in diameter by 22 Å long. Thus, the peptides are the same size as the ToF-SIMS sampling depth. However, it is expected that the residues closer to the outermost surface will have a higher probability of being emitted from the surface than those associated with the SAM surface. The side chains of the LKα14 are arranged such that the leucine side chains are on one side and the lysine side chains on the other. However, the LKα14 structures derived from molecular dynamic simulations in Figure 3 show that the side chains fan out making a polar and an apolar hemisphere, or, more accurately, hemi-cylinder. This greatly reduces the distance between the two side chain types and explains why similar 84/86 intensity ratios would be observed for oriented LKα14 peptides on the two SAM surfaces. SFG results42 showed interactions between the methyl groups of the L side-chains and the methyl SAM and between the amine groups of the K side-chains and the carboxyl SAM for adsorbed LKα14 peptides. For this reason, it is believed that the similarity in 84/86 ratios on the two surfaces is due to the structure of the α-helix.
Figure 3.

Minimized starting conformations of LKα14 and LKβ15 models used for molecular dynamic simulations. All figures are shown with the lysine side-chains pointing up and the leucine side-chains pointing down. Nitrogen atoms are dark blue, carbon atoms are light blue, oxygen atoms are red and hydrogen atoms are grey. Figures a and b show space-fill end-on views of a) LK α14, and b) LKβ15. It can be seen that in contrast to well separated lysine and leucine side chains in the LKβ15 structure, the lysine and leucine side chains fan out and reduce the separation between the lysine and leucine side chains in the LKα14 structure. Figures c and d show ball-and-stick side-views of LKα14 and LKβ15, respectively.
LKβ15 results in Figure 4b, in contrast to the LKα14 results, show significant differences in 84/86 intensity ratios. Although some concentration-dependence is apparent, the 84/86 ratio is significantly greater on the methyl SAM than on the carboxyl SAM. On the methyl SAM, a ratio of 1.81±0.08 was seen for 4–6 atomic % N and 1.7±0.2 for 8–10 atomic % N. On the carboxyl SAM, a ratio of 1.06±0.07 was seen for 4–6 atomic % N and 0.87±0.02 for 8–10 atomic % N. The ratio for the LKβ15 powder was 1.05±0.04. The much greater difference in the 84/86 ratio can be explained by the much greater separation between the leucine and lysine side chains. Whereas LKα14 side chains formed polar and apolar hemi-cylinders, the LKβ15 structures derived from molecular dynamic simulations in Figure 3 show LKβ15 side chains are oriented such that the polar lysine side chains were 180° from the apolar leucine side chains. This provides a much greater distance between the two, and consequently leads to a much more discernable orientation effect using ToF-SIMS. These orientation results are consistent with those obtained using SFG and NEXAFS.42 It should be noted that although the ratio for LKβ15 on the carboxyl SAMs is much lower than that for the methyl SAMs, it is only near monolayer coverages (ca. 8 atomic % N) that the ratio drops significantly below the value for the LKβ15 powder.
Principal component analysis was used to examine the ToF-SIMS data for the LKβ15 peptide adsorbed onto methyl and carboxyl SAMs. The peaks used were major peaks produced by lysine and leucine poly-amino-acids, but since some of these are hydrocarbon peaks or hydrocarbon with oxygen, they also encompass some of the peaks produced by the underlying SAMs. Therefore, any peaks that could be produced by either the SAMs or the LK peptides were eliminated from the PCA peak set so only peptide-specific peaks used (i.e., only nitrogen containing peaks were used for PCA, see Table S2 in the supplementary material for a full list of the peaks used). The scores for PC 2 vs. PC 1 are shown in Figure 6. PC 1 accounts for 69% of the total variance in the data set, and separates samples with and without adsorbed peptide. PC 2 accounts for 24% of the variance and separated the peptide-covered carboxyl SAMs from the other samples. From examining the loadings it appears that the samples with positive scores in PC 2 are those with more peptide and with leucine side-chains closest to the surface, as shown by the positive loadings of peaks 30 (CH4N+, common to all amino-acids) and 86 (C5H12N+, immonium ion for leucine). This would be the case for the peptide-covered carboxyl SAMs as it is expected that the leucines are pointing upwards and the lysines are pointing downwards. Complete tables of scores and loadings can be found in Supplementary Tables S1 and S2. The PCA results confirm that the 84/86 intensity ratio accurately assessed the orientation of the LKβ15 peptides on the methyl and carboxyl surfaces.
Figure 6.
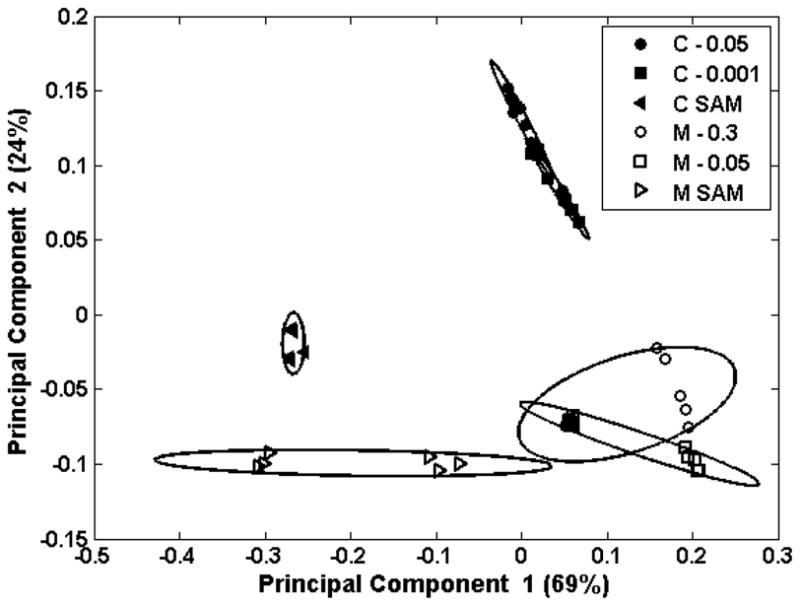

Figure 6a shows the PCA scores plot of PC 2 vs. PC 1 for LKβ15 on carboxyl (C) and methyl (M) SAMs. Numbers in the legend represent the peptide concentration in mg/mL at which the peptides were adsorbed onto the SAMs. PC 1 separates samples with peptide from those without. PC 2 separates the samples with predominantly leucine side-chains nearest the surface from those with other groups close to the surface. Figure 6b shows loadings for PC 1 and Figure 6c shows the loadings for PC 2. L labels the major peaks generated by leucine, K labels major peaks from lysine, and “Both” labels peaks found in both peak lists.
Conclusions
This study investigated the interactions between the amphiphilic LKα14 and LKβ15 peptides and two SAMs, a methyl-terminated and a carboxyl-terminated surface. The XPS results showed that the peptides interact very differently with the two surfaces. Peptides adsorbed at much lower concentrations onto the carboxyl SAMs, indicating a much stronger interaction with the carboxyl SAMs compared to the methyl SAMs. Also, the variability in surface coverage was consistently less on the charged surfaces compared to the hydrophobic surfaces. The XPS results suggest that the peptide lysine side chains were electrostatically bonded to the carboxyl surface while the leucine side chains were bonded to the methyl surface via hydrophobic interactions. ToF-SIMS data comparing the relative lysine/leucine fragment intensity ratios reinforced the XPS results. The K/L intensity ratio was higher for peptides adsorbed onto the methyl surfaces, showing increased lysines at the outermost surface, and lower for the charged surfaces, showing increased leucines at the surface. These data indicate that the leucine side of the LKα14 and LKβ15 peptides interacted with the methyl SAM surface and the lysines interacted with the carboxyl surface. PCA was used to further confirm these results for LKβ15, as the adsorbed LKβ15 peptides showed the greatest difference in the 84/86 intensity ratios for the carboxyl and methyl surfaces. The peptide orientation conclusions drawn from the XPS and ToF-SIMS results are consistent with recent SFG and NEXAFS results.42
XPS and ToF-SIMS results also showed increased oxygen concentrations on the buffer-exposed methyl SAM surfaces, suggesting the presence of bound water interacting with the methyl SAM substrate and also with the adsorbed peptide. Theoretical XPS calculations were consistent with the formation of peptide bundles on the methyl SAM surfaces and possibly partial peptide bilayers on the carboxyl surfaces at higher peptide solutions concentrations. At the highest peptide solution concentrations (> 0.3 mg/mL) it is likely the peptides were bundling in solution and depositing onto the SAM surfaces.
Supplementary Material
Table 2.
XPS results for LKα14 adsorption onto carboxyl SAMs. The C, O, N, and S atomic percents were renormalized to 100 atomic % to show the composition of the organic overlayer (SAM plus peptide). The Au atomic % before renormalization and the N/Au ratio show the attenuation of the gold signal by the organic overlayer.
| Peptide Adsorption Concentration (mg/mL) | C atomic %* | O atomic %* | N atomic %* | S atomic %* | Au atomic % | N/Au ratio |
|---|---|---|---|---|---|---|
| 0.0002 | 82.1 (1.1) | 13.3 (0.2) | 1.9 (0.5) | 2.8 (0.2) | 43.9 (0.9) | 0.024 (0.006) |
| 0.0005 | 79.1 (5.6) | 13.9 (1.2) | 4.9 (1.9) | 2.0 (0.3) | 36.1 (5.2) | 0.094 (0.050) |
| 0.001 | 77.6(0.7) | 12.9(0.5) | 7.9 (0.6) | 1.6 (0.1) | 29.0 (1.2) | 0.19 (0.02) |
| 0.002 | 76.2 (2.1) | 13.7 (0.2) | 8.7 (0.5) | 1.4 (0.1) | 26.9 (1.9) | 0.24 (0.03) |
| 0.005 | 75.1 (2.2) | 14.0 (0.3) | 9.6 (0.5) | 1.3 (0.1) | 26.5 (2.1) | 0.27 (0.03) |
| 0.01 | 75.1 (1.3) | 13.8 (0.4) | 9.8 (0.5) | 1.3 (0.1) | 25.3 (1.4) | 0.29 (0.03) |
| 0.05 | 75.8 (1.2) | 13.3 (0.6) | 9.6 (0.5) | 1.3 (0.3) | 23.9 (0.6) | 0.32 (0.03) |
| 1 | 74.6 (1.1) | 14.2 (0.5) | 10.4 (0.5) | 0.8 (0.1) | 19.1 (1.6) | 0.44 (0.06) |
| Calculated Values for Monolayer | 76.3 | 13.7 | 8.0 | 2.0 | 29.9 | 0.19 |
| Calculated Values for Bilayer | 74.8 | 12.7 | 11.4 | 1.1 | 19.7 | 0.46 |
Renormalized to 100% without the Au signal.
Table 3.
XPS results for LKβ15 adsorption onto methyl SAMs. The C, O, N, and S atomic percents were renormalized to 100 atomic % to show the composition of the organic overlayer (SAM plus peptide). The Au atomic % before renormalization and the N/Au ratio show the attenuation of the gold signal by the organic overlayer.
| Peptide Adsorption Concentration (mg/mL) | C (atomic %)* | O (atomic %)* | N (atomic %)* | S (atomic %)* | Au (atomic %) | N/Au ratio |
|---|---|---|---|---|---|---|
| 0.01 | 89.3 (0.4) | 5.2 (0.5) | 3.0 (0.2) | 2.5 (0.2) | 42.2 (0.7) | 0.041 (0.004) |
| 0.05 | 88.9 (1.3) | 5.7 (1.5) | 3.3 (0.4) | 2.1 (0.5) | 42.1 (1.5) | 0.045 (0.005) |
| 0.1 | 81.9 (3.8) | 10.9 (2.4) | 5.8 (1.9) | 1.4 (0.4) | 37.6 (5.2) | 0.11 (0.07) |
| 0.2 | 80.7 (1.3) | 12.2 (0.7) | 6.2 (0.7) | 0.9 (0.2) | 38.2 (2.3) | 0.10 (0.02) |
| 0.3 | 80.1 (3.3) | 11.1 (1.5) | 7.7 (2.1) | 1.2 (0.3) | 33.1 (7.2) | 0.18 (0.09) |
| 0.4 | 78.4 (2.0) | 14.0 (1.3) | 6.7 (1.7) | 0.9 (0.4) | 31.6 (4.6) | 0.15 (0.07) |
| 0.5 | 75.0 (2.6) | 14.2 (0.7) | 10.2 (2.0) | 0.7 (0.2) | 22.2 (7.0) | 0.42 (0.23) |
| Calculated Values for Monolayer | 82.9 | 6.5 | 8.6 | 2.0 | 34.3 | 0.17 |
| Calculated Values for Bilayer | 78.0 | 8.9 | 12.0 | 1.1 | 22.6 | 0.41 |
Renormalized to 100% without the Au signal.
Acknowledgments
The authors would like to acknowledge the contributions of Nicholas Breen, Gil Goobes, Newton Samuel; and Riki Goobes for help with experiments; and Pat Stayton, Lizzy Mayorga-Szott, Loren Baugh and Scott Curtin for technical discussions. We also acknowledge the support of National Institute of Health grants GM-074511 and NIH EB-002027. This material is based upon work supported under a National Science Foundation Graduate Research Fellowship.
References
- 1.Ratner BD, Bryant SJ. Annual Review of Biomedical Engineering. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 2.Anderson JM. Annual Review of Materials Research. 2001;31:81–110. [Google Scholar]
- 3.Horbett TA, Brash JL. Acs Symposium Series. 1987;343:1–33. [Google Scholar]
- 4.Ratner BD. Advances in Chemistry Series. 1982:9–23. [Google Scholar]
- 5.Latour RA. Biointerphases. 2008;3:FC2–FC12. doi: 10.1116/1.2965132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fields GB, Lauer JL, Dori Y, Forns P, Yu YC, Tirrell M. Biopolymers. 1998;47:143–151. doi: 10.1002/(SICI)1097-0282(1998)47:2<143::AID-BIP3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Gauba V, Hartgerink JD. Journal of the American Chemical Society. 2007;129:8921–8921. doi: 10.1021/ja0683640. [DOI] [PubMed] [Google Scholar]
- 8.Bhadriraju K, Hansen LK. Biomaterials. 2000;21:267–272. doi: 10.1016/s0142-9612(99)00175-1. [DOI] [PubMed] [Google Scholar]
- 9.Belcheva N, Baldwin SP, Saltzman WM. Journal of Biomaterials Science-Polymer Edition. 1998;9:207–226. doi: 10.1163/156856298x00613. [DOI] [PubMed] [Google Scholar]
- 10.Glass J, Blevitt J, Dickerson K, Pierschbacher M, Craig WS. Biochemical Engineering Viii. 1994;745:177–186. doi: 10.1111/j.1749-6632.1994.tb44372.x. [DOI] [PubMed] [Google Scholar]
- 11.Aota S, Nomizu M, Yamada KM. Journal of Biological Chemistry. 1994;269:24756–24761. [PubMed] [Google Scholar]
- 12.Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TCB, Pitkeathly M, Radford SE. Nature. 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 13.Chilkoti A, Dreher MR, Meyer DE. Advanced Drug Delivery Reviews. 2002;54:1093–1111. doi: 10.1016/s0169-409x(02)00060-1. [DOI] [PubMed] [Google Scholar]
- 14.Blondelle SE, Esteve V, Celda B, Pastor MT, Perez-Paya E. Journal of Peptide Research. 2000;56:121–131. doi: 10.1034/j.1399-3011.2000.00744.x. [DOI] [PubMed] [Google Scholar]
- 15.Boon CL, Chakrabartty A. Protein Science. 2000;9:1011–1023. doi: 10.1110/ps.9.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Degrado WF, Lear JD. Journal of the American Chemical Society. 1985;107:7684–7689. [Google Scholar]
- 17.Poon S, Clarke A, Currie G, Schultz C. Bioscience Biotechnology and Biochemistry. 2001;65:1713–1723. doi: 10.1271/bbb.65.1713. [DOI] [PubMed] [Google Scholar]
- 18.Dong H, Hartgerink JD. Biomacromolecules. 2007;8:617–623. doi: 10.1021/bm060871m. [DOI] [PubMed] [Google Scholar]
- 19.Samuel NT. Washington: 2005. [Google Scholar]
- 20.Mermut O, Phillips DC, York RL, McCrea KR, Ward RS, Somorjai GA. Journal of the American Chemical Society. 2006;128:3598–3607. doi: 10.1021/ja056031h. [DOI] [PubMed] [Google Scholar]
- 21.Phillips DC, York RL, Mermut O, McCrea KR, Ward RS, Somorjai GA. Journal of Physical Chemistry C. 2007;111:255–261. [Google Scholar]
- 22.Bower PV, Louie EA, Long JR, Stayton PS, Drobny GP. Langmuir. 2005;21:3002–3007. doi: 10.1021/la040092w. [DOI] [PubMed] [Google Scholar]
- 23.Long JR, Oyler N, Drobny GP, Stayton PS. Journal of the American Chemical Society. 2002;124:6297–6303. doi: 10.1021/ja011624n. [DOI] [PubMed] [Google Scholar]
- 24.Castano S, Desbat B, Laguerre M, Dufourcq J. Biochimica Et Biophysica Acta-Biomembranes. 1999;1416:176–194. doi: 10.1016/s0005-2736(98)00220-x. [DOI] [PubMed] [Google Scholar]
- 25.Maget-Dana R, Lelievre D, Brack A. Biopolymers. 1999;49:415–423. doi: 10.1002/(SICI)1097-0282(19990415)49:5<415::AID-BIP7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Castano S, Desbat B, Cornut I, Meleard P, Dufourcq J. Letters in Peptide Science. 1997;4:195–200. [Google Scholar]
- 27.Dieudonne D, Gericke A, Flach CR, Jiang X, Farid RS, Mendelsohn R. Journal of the American Chemical Society. 1998;120:792–799. [Google Scholar]
- 28.Maget-Dana R, Brack A, Lelievre D. Supramolecular Science. 1997;4:365–368. [Google Scholar]
- 29.Chen HJ, Hsu SL, Tirrell DA, Stidham HD. Langmuir. 1997;13:4775–4778. [Google Scholar]
- 30.Drobny GP, Long JR, Karlsson T, Shaw W, Popham J, Oyler N, Bower P, Stringer J, Gregory D, Mehta M, Stayton PS. Annual Review of Physical Chemistry. 2003;54:531–571. doi: 10.1146/annurev.physchem.54.011002.103903. [DOI] [PubMed] [Google Scholar]
- 31.Kerth A, Erbe A, Dathe M, Blume A. Biophysical Journal. 2004;86:3750–3758. doi: 10.1529/biophysj.103.035964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazo ND, Downing DT. Biochemistry. 1997;36:2559–2565. doi: 10.1021/bi963061b. [DOI] [PubMed] [Google Scholar]
- 33.Powers ET, Kelly JW. Journal of the American Chemical Society. 2001;123:775–776. doi: 10.1021/ja005528i. [DOI] [PubMed] [Google Scholar]
- 34.Wang K, Keasling JD, Muller SJ. International Journal of Biological Macromolecules. 2005;36:232–240. doi: 10.1016/j.ijbiomac.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Yu YC, Berndt P, Tirrell M, Fields GB. Journal of the American Chemical Society. 1996;118:12515–12520. [Google Scholar]
- 36.Cho Y, Ivanisevic A. Langmuir. 2006;22:8670–8674. doi: 10.1021/la060492e. [DOI] [PubMed] [Google Scholar]
- 37.Bain CD, Troughton EB, Tao YT, Evall J, Whitesides GM, Nuzzo RG. Journal of the American Chemical Society. 1989;111:321–335. [Google Scholar]
- 38.Chidsey CED, Loiacono DN. Langmuir. 1990;6:682–691. [Google Scholar]
- 39.Ulman A. An introduction to ultrathin organic films from Langmuir-Blodgett to self-assembly. Academic Press; Boston: 1991. [Google Scholar]
- 40.Ulman A. Characterization of organic thin films. Butterworth-Heinemann; Manning; Boston; Greenwich: 1995. [Google Scholar]
- 41.Grunze M. Physica Scripta. 1993;T49B:711–717. [Google Scholar]
- 42.Weidner T, Apte JS, Gamble LJ, Castner DG. Manuscript in Preparation. [Google Scholar]
- 43.Apte JS, Cheng F, Michel R, Castner DG. Manuscript in Preparation. [Google Scholar]
- 44.Mantus DS, Ratner BD, Carlson BA, Moulder JF. Analytical Chemistry. 1993;65:1431–1438. doi: 10.1021/ac00058a021. [DOI] [PubMed] [Google Scholar]
- 45.Wagner MS, Castner DG. Langmuir. 2001;17:4649–4660. [Google Scholar]
- 46.Wagner MS, Pasche S, Castner DG, Textor M. Analytical Chemistry. 2004;76:1483–1492. doi: 10.1021/ac034873y. [DOI] [PubMed] [Google Scholar]
- 47.Brooks BR, Bruccoleri RE, Olafson BD. Journal of Computational Chemistry. 1983;4:187–217. [Google Scholar]
- 48.Foloppe N, MacKerell AD. Journal of Computational Chemistry. 2000;21:86–104. [Google Scholar]
- 49.Jorgensen W, Jensen C. Journal of Computational Chemistry. 1998;19:1179–1186. [Google Scholar]
- 50.Darden T, York D, Pedersen L. Journal of Chemical Physics. 1993;98:10089–10092. [Google Scholar]
- 51.Swope WC, Andersen HC. Journal of Chemical Physics. 1981;76:637–649. [Google Scholar]
- 52.Evans DJ, Holian BL. Journal of Chemical Physics. 1985;83:4069–4074. [Google Scholar]
- 53.Ryckaert JP, Ciccotti G, Berendsen HJC. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 54.Fears KP, Creager SE, Latour RA. Langmuir. 2008;24:837–843. doi: 10.1021/la701760s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehninger AL, Nelson DL, Cox MM. Lehninger principles of biochemistry. Worth Publishers; New York: 2000. [Google Scholar]
- 56.Meierhenrich U. Amino acids and the asymmetry of life: caught in the act of formation. Springer; Berlin: 2008. [Google Scholar]
- 57.Goldston RJ, Rutherford PH. Introduction to plasma physics. Institute of Physics; 1995. [Google Scholar]
- 58.Castner DG, Hinds K, Grainger DW. Langmuir. 1996;12:5083–5086. [Google Scholar]
- 59.Baio JE, Weidner T, Brison J, Graham DJ, Gamble LJ, Castner DG. Journal of Electron Spectroscopy and Related Phenomena. 2009;172:2–8. doi: 10.1016/j.elspec.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paynter RW. Surface and Interface Analysis. 1981;3:186–187. [Google Scholar]
- 61.Seah WADMP. Surface and Interface Analysis. 1:2–11. [Google Scholar]
- 62.Sato M, Tsukamoto N, Shiratori T, Furusawa T, Suzuki N, Tougaard S. Surface and Interface Analysis. 2006;38:604–609. [Google Scholar]
- 63.Zharnikov M, Frey S, Heister K, Grunze M. Journal of Electron Spectroscopy and Related Phenomena. 2002;124:15–24. [Google Scholar]
- 64.Michel R, Castner DG. Surface and Interface Analysis. 2006;38:1386–1392. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.