

Abstract
PriA, a 3′→5′ superfamily 2 DNA helicase, acts to remodel stalled replication forks and as a specificity factor for origin-independent assembly of a new replisome at the stalled fork. The ability of PriA to initiate replication at stalled forked structures ensures complete genome replication and helps to protect the cell from illegitimate recombination events. This review focuses on the activities of PriA and its role in replication fork assembly and maintaining genomic integrity.
Keywords: DNA replication restart, PriA helicase, recombination, primosome
Over the last decade, it has been well established that replication forks formed at oriC, the bacterial origin of replication, often are unable to complete synthesis of the genome because of encounters with template DNA damage that either stalls or collapses the fork (for reviews, see (12, 45, 68)). Maintenance of cell viability requires that such forks be restarted in a timely fashion and the DNA lesions repaired, demanding a pathway for non-mutagenic, origin-independent restart of replication under normal growth conditions. Because replication forks are limited in range by the frequency of template DNA damage sites, the potential catastrophe of incomplete genome replication may have created selective pressure for the evolution of homologous recombination and replication fork reactivation. However, recombination can be detrimental to cell survival when inappropriate, leading to the loss of genomic information. Therefore, cells require methods to restart replication after fork stalling that do not rely on recombination, but instead restart replication downstream of the damage. In these scenarios, the damage is repaired post-replication, after the forks have been reestablished. As suggested by Cox et al. (12), these systems are an evolutionarily critical step, paving the way for organisms with larger genomes where encounters with damage are more frequent and where restart becomes essential. PriA plays a central role in replication fork reactivation both in the absence of and in conjunction with recombination. It is the interplay of these two activities that preserves cell viability and genomic integrity.
PriA was discovered in Escherichia coli, together with other restart primosomal proteins, PriB, DnaT, PriC, DnaB, and DnaC, because of its requirement in phage ϕX174 DNA replication (74). Primosome assembly in ϕX174 occurs via a series of ordered protein-protein interactions and is initiated by PriA binding to the primosome assembly site (pas) on single-stranded DNA-binding protein (SSB)-coated viral DNA (Fig. 1A) (74). PriA binding to the pas (Fig. 1A(i)) induces interactions with other primosomal proteins. PriB stabilizes the PriA-pas interaction (37, 57), possibly via an interaction with SSB (57), and the PriA-PriB complex then recruits DnaT (Fig. 1A(ii)). PriB is thought to facilitate PriA-DnaT complex formation during primosome assembly because at high concentrations of DnaT, PriB is dispensable (37). Finally, the addition of the replicative DNA helicase, DnaB, from the DnaB-DnaC complex completes the assembly of the preprimosome, which is capable of translocating on the DNA (Fig. 1A(iii)) (56). Complete assembly of a replisome follows via an interaction with the primase, DnaG, and the DNA polymerase III holoenzyme, the cellular replicase (28, 86). The complete primosome (the preprimosome plus primase) can translocate in the 3′→5′ direction via the PriA DNA helicase activity, as well as in the 5′→3′ direction via the DnaB DNA helicase activity (34, 35). PriA-directed replisome loading has been shown to occur both on single-stranded (ss) DNA with a pas sequence (Fig. 1A) or on transcriptionally activated origin sequences in which a stable RNA-DNA hybrid exposes the pas for θ-type DNA replication (63). It was suggested initially that the helicase activity of PriA might function at the replication fork to help form loops in the lagging-strand template during Okazaki fragment synthesis (35), however priA mutants did not exhibit any defect in chromosomal DNA replication (71).
Figure 1.

PriA replisome loading. PriA can load a replisome to various DNA structures. (A) A primosome assembly site on ϕX174 viral DNA. (B) A stalled replication fork where there is a gap in the nascent lagging-strand. Such a structure can form when a leading-strand template block terminates all DNA synthesis. (C) A stalled replication fork where both template strands have been completely copied up to the point of template damage. Similar to (B) above, such a structure may occur when the replisome encounters a leading-strand blockage. Replisome loading on such a structure requires the unwinding of a portion of the nascent lagging-strand DNA to generate a single-stranded region for DnaB binding. (D) A recombinant joint molecule, represented here as a D loop. These structures necessarily occur for restart after the encounter of a template double-strand break and may also be generated under other situations as well. Although the DNA structures are different, the steps in loading the replisome are identical. (i) PriA recognizes the 3′ OH end of the nascent leading-strand DNA at the three-strand junction and (ii) recruits other accessory factors that may be important in fork remodeling (for example, in panel C). Recognition of the pas probably occurs through interaction of PriA with the psuedo-three stand junction formed by the hairpin in the DNA and the adjacent single-stranded DNA. (iii) An interaction between DnaT and DnaC, the helicase loader, allows loading of the replicative helicase, DnaB, forming the preprimosome (composed of PriA, PriB, DnaT, and DnaB). Once loaded, DnaB recruits DnaG via a protein-protein interaction. Synthesis of a primer then attracts the DNA polymerase III holoenzyme and a protein-protein interaction between the τ subunit of the holoenzyme and DnaB cements formation of the replisome. Nascent template 3′ OH ends are denoted by half-arrows.
The priA null phenotype is characterized by the inability to sustain ϕX174 phage infection and a failure to maintain ColE1-based plasmids, which require PriA for replication (33, 53, 61). Conversion of ss ϕX174 phage DNA to the duplex replicative form (RF) also requires PriA in vitro (53). priA-null strains are sensitive to UV irradiation and produce small colonies in which a subset of the population is filamented extensively (33). Although priA-null strains are constitutively induced for the SOS response (61), the expression of PriA is not SOS regulated (74, 80). In addition, priA mutants are defective in both induced stable DNA replication (iSDR) and constitutive stable DNA replication (cSDR) (47). Genetic experiments assessing P1 transduction efficiency indicated that homologous-recombination processes are blocked in priA-null mutants (30). This observation supports the notion that a significant fraction of homologous-recombination events in E. coli affect DNA replication (30) and require PriA function to reload the replisome.
The PriA helicase and primosome assembly activities can be uncoupled by inactivating the ATPase activity of the protein. Unlike priA-null strains, cells carrying the priA300 allele, encoding PriAK230R, a helicase- and ATPase-dead protein that carries a mutation in the Walker P-loop motif (Fig. 2) (87), maintains wild-type growth, viability, UV resistance, recombination proficiency, and cell morphology (71). PriAK230R variants have also been shown to be 2- to 3-fold more active than the wild-type in promoting primosome-dependent DNA replication during synthesis of the complementary strand of ϕX174 in vitro (87). The helicase activity is therefore dispensable for cell viability in an otherwise wild-type background, and the absence of the PriA primosome assembly activity is the most likely factor underlying the priA-null phenotypes.
Figure 2.

Amino acid motifs of E. coli PriA. PriA is divided into two major domains, a DNA-binding domain and a DNA helicase domain. The DNA helicase domain consists of an ATP-binding domain composed of a Walker A box, Walker B box, and SAT motif, and a cysteine-rich region that contains two Zn-finger motifs. Amino acid substitutions in the Walker A box, K230R, yielding a helicase-dead mutant protein, and in the metal binding domain, C479Y, representing the priA300 and priA301 mutations, are shown.
priA-null strains rapidly gain suppressor mutations in dnaC, which encodes a protein that forms a complex with DnaB and is important for loading of the helicase during replisome assembly. Mutations in dnaC, such as dnaC809 and dnaC810 (which are different alleles of the same amino acid substitution (65)), can suppress defects in both priA and priB priC-null strains (65, 69, 71), presumably by bypassing the need for the primosomal proteins in replication restart (85). This suggests that the priA defect occurs at the loading of the replisome. There are three pathways for restart in vivo: PriA-PriB, PriA-PriC, and PriC-Rep. These pathways differ in their recognition of stalled forked structures (Fig. 1B-C, Fig. 3i) (22) and therefore seem to be distinct mechanisms for replisome reloading. Either the priA300 or priA301 (encoding PriA C479Y, which is mutated in the cysteine-rich region of priA and is thought to be important for zinc-binding, helicase activity, and or protein-protein interactions (Fig. 2) (37, 88)) mutations, in combination with a priB null mutation, yield a priA null-like phenotype (70). Therefore, these PriA mutants require PriB for function and it has been suggested that they are deficient specifically in the PriA-PriC pathway (70). Coordination between the pathways may rely on the type of template damage.
Figure 3.
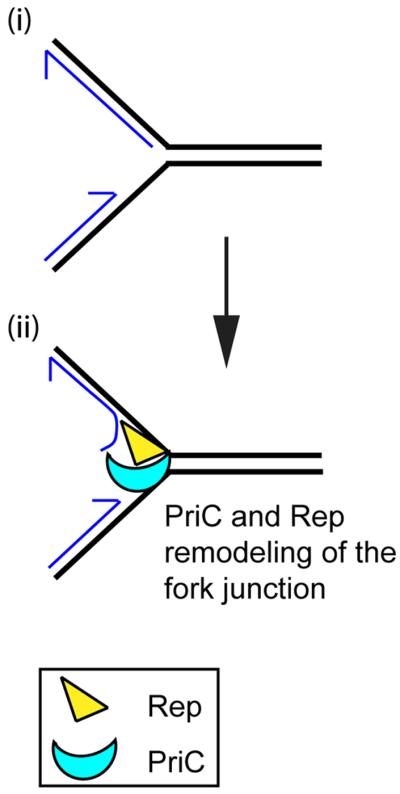
Remodeling of stalled replication forks during replisome loading by the PriC pathway. The preferred DNA structure (i) for the PriC pathway is likely formed when the replisome encounters a leading-strand template blockage and the leading- and lagging-strand polymerases of the replicase uncouple. This results in a gap in the nascent leading strand at the stalled fork with the 5′ of the nascent lagging strand being either right at or close to the fork junction. Displacement of the nascent lagging-strand to allow DnaB loading to the lagging-strand template is most likely accomplished by PriC directing Rep to the proper DNA strand (ii).
PriA in Replication Fork Assembly
PriA has two major roles in replication restart. The first is as a 3′→5′ DNA helicase that remodels stalled fork structures to generate a single-stranded region on the lagging-strand template for DnaB loading (Fig. 1C(ii)). The second is to orchestrate origin-independent replisome loading at the stalled fork (Fig. 1). As suggested by Heller and Marians (21), it is the processed DNA structures generated that are targeted by the restart proteins. PriA-directed assembly of a replisome on a DNA structure involved in restart was first demonstrated on model D-loop oligonucleotide substrates (36) and D loop-containing template DNA (38). Subsequently, two independent DnaB loading systems have been described biochemically using substrates and templates designed to model stalled fork structures: a PriA, PriB, DnaT pathway and a PriC pathway (22). These two systems are differentially affected by gaps in the nascent leading strand and are additive in their activities (Fig. 1B and Fig. 3i) (22). The PriA, PriB, DnaT pathway also can load a replisome to D loops (which are structurally similar to the stalled fork shown in Fig. 1B) on recombinant joint molecules (Fig. 1D) (38, 84). The PriC-pathway is capable of using either the PriA helicase or the Rep helicase to remove the nascent lagging strand in order to generate a single-stranded region on the lagging-strand template for loading of DnaB (Fig. 1C and Fig. 3). These pathways have been described as PriC-Rep and PriA-PriC (23). The specificity factor, PriC, was shown to interact with Rep and therefore coordinates the action of two DNA helicases, Rep and DnaB, for replication restart (23). Interestingly, Rep has also been shown to clear tightly bound proteins from the path of the replisome in vitro and to interact with DnaB (McGlynn, P., personal communication), suggesting a complicated series of interactions for this protein in the replisome and at stalled replication forks.
Using the PriA, PriC, and DnaC810 restart systems, it was demonstrated that DnaG can prime de novo on the leading-strand template, leaving a gap opposite the lesion (20). The variant DnaC810, which suppresses the priA phenotype in vivo, can bypass the requirements for PriA, PriB, PriC, and DnaT in vitro (38), implying that the loading of DnaB is the major requirement for complete replisome assembly and the role of PriA in the cell seems to be loading the replisome at stalled fork structures and restarting replication downstream of the damage. However, a majority of cells can replicate the chromosome without leaving a single gap in the absence of recombination (43). Gaps that are formed are probably filled in by DNA polymerase I, but gap filling does not require either RecQ or RecJ or any of the translesion DNA synthesis (TLS) polymerases (43). This suggests that the gaps left behind during restart by PriA may be either too short or too transient to be detected. Alternatively, repair may be coupled to replication restart, ensuring that all gaps are filled as the replisome is reloaded.
Although the PriAK230R Walker A mutant (Fig. 2) fully restores viability of the priA strain in vivo and is capable of primosome loading in vitro, the viability of a mutant of the DNA polymerase III subunit, holD, is impaired (16). Flores et al. (16) suggest that this defective Ψ subunit of the replicase promotes replication fork reversal at stalled forks. These observations suggest a role for the helicase activity of PriA during replication restart under these circumstances. Interestingly the dnaC809,820 double mutation can fully suppress the holD priA phenotype (16). One presumes this observation indicates that the DnaC809,820 protein is loading DnaB in these situations, however, there is no cognate biochemical analysis of DnaC809,820. In our hands, this protein is essentially inactive in replication restart (Xu and Marians, unpublished data), suggesting that the correct substrate has yet to be found in vitro. However, the DnaC809,820 protein is active in the replication of plasmids containing oriC in vitro (Gabbai and Marians, unpublished data).
PriA Structure and Function Relationships
PriA is a 3′→5′ DNA helicase of the SF2 superfamily of helicases (17). The protein is composed of three functionally significant domains: an N-terminal DNA binding domain, a DNA helicase domain, and a zinc finger motif embedded within the helicase domain (Fig. 2) (79). As a specificity factor, PriA recognizes the 3′-OH end of the nascent leading strand at DNA junctions (Fig. 1i) and the PriA 3′ terminus-binding pocket plays an important role in detecting stalled replication forks (54). The 3′OH-end of the nascent leading strand at the branch point of an arrested fork (Fig. 1B-D) inhibits unwinding of the parental duplex, thereby restricting PriA to the loading site and allowing assembly of a primosome for replication restart (77). PriA proteins mutated in the 3′ terminus binding pocket have low ATPase activity and a decreased affinity for D-loops and arrested replication forks (54). Unlike the helicase dead PriA variant, these variants could not restore growth, morphology, and SDR to priA-null strains when the genes encoding them were provided in trans (54).
PriA recognition of specific DNA structures is controlled by an aspartate residue in the 3′-terminal nucleotide-binding pocket (72). Although these X-ray crystallographic studies were performed with ssDNA, models show that the binding does not prevent base pairing (72) and therefore allows PriA to recognize a forked structure where the nascent leading strand is at the fork template junction (Fig. 1B-D). This mode of recognition by the N-terminal domain of PriA directs proper positioning of the helicase domain on unreplicated double stranded DNA (dsDNA), which in turn allows primosome assembly and restart (72). Additionally, PriA is unable to bind or unwind Holliday junctions because it requires a ssDNA component to support the minimal binding requirement (50). Therefore, for recognition by PriA, Holliday junctions need to be modified.
The 181 amino acid N-terminal domain of PriA can bind to D-loops in the absence of the remainder of the protein, but has lost specificity and will also bind to bubble DNA structures (79). This observation suggests that the C-terminal domain of PriA is important for specificity and stabilized DNA binding (79). In the absence of the DNA-binding domain, the helicase domain of PriA has low levels of DNA-independent ATPase activity, probably because of its higher binding affinity for ATP (11). Thus, both the DNA-binding domain and the helicase domain are important for conferring specificity to PriA.
It has also been noted that in DNA footprinting analyses, PriA protects two separate areas of arrested fork structures with a 3′-end at the template junction (78). The amino acid sequence of PriA suggested that it may contain two nucleotide-binding sites (60). Recent studies provide evidence for two sites that differ in their affinities for nucleotide (41). It has been suggested that the interplay between these two sites controls the affinity of the helicase for DNA with the modulation of PriA structure by ATP hydrolysis being important for the ability of PriA to recognize and specifically bind substrates such as arrested replication forks (41). Furthermore, two modes of PriA stable DNA binding have been identified. The first reflects the 3′ → 5′ DNA helicase activity when bound to a 3′ ssDNA extension from duplex DNA. The second is the D-loop binding activity that suggests PriA binds at the bend of a 3-strand junction (Fig. 1B-D) (60). The interplay between the helicase activity and binding of forked substrates appears to dictate whether PriA will initiate replisome loading or template unwinding.
PriA Interaction with Other Proteins
Replisome loading at a fork during restart is effected by PriA binding followed by its interaction with other restart proteins. Once one PriA molecule is bound to a pas sequence it prevents access of other PriA molecules (2). On substrates with a 3′ overhang, the unwinding activity of an isolated helicase domain is inhibited by the presence of full length PriAK230R and, albeit to a lesser extent, by the isolated DNA-binding domain of PriA (11). This ensures that only a single primosome loads at the pas site and allows efficient replisome loading by blocking access of other PriA molecules. Furthermore, the helicase dead mutant, PriAK230R, is 20-fold more efficient than wild-type PriA at inhibiting DnaB-independent elongation of the invading strand in a D loop (84). This inhibition is probably a result of PriA binding directly to the D-loop and preventing polymerase access to the 3′-OH of the invading strand, but also provides the specificity necessary for eventual replisome loading in a coordinated fashion.
The interaction of PriA with other primosomal proteins and DNA substrates seems to be modulated by PriB. The surface of PriB contains binding sites for PriA, ssDNA, and DnaT. The physical interaction of PriB with other proteins is DNA dependent and promotes ternary complex formation and DnaB loading (Fig. 1, steps (ii)-(iii)) (40). PriB interacts with the helicase domain of PriA (40), which is only accessible when PriA binds to ssDNA. A conformational change in PriA allows interaction with PriB (40).
The inherent processivity of PriA for DNA unwinding is less than 500 bp (26). PriB may act as a processivity factor for PriA helicase by interacting simultaneously with ssDNA and PriA (8). PriA helicase activity stimulation by PriB is specific, neither PriC nor DnaT stimulate PriA and Rep is not stimulated by PriB (8). However, PriA stimulation is not DNA structure specific; PriB can increase the apparent processivity of PriA on both branched and unbranched DNA structures (8). PriC is capable of stimulating Rep helicase activity, but not that of PriA (23).
Unlike many DNA helicases, PriA can bind to specific DNA structures, such as a D-loop or pas, in the presence of SSB (50, 80). SSB binding restrains the polarity of helicase action on a synthetic forked substrate by inhibiting unwinding of the unreplicated duplex, but allows translocation on the lagging-strand template and unwinding of the nascent lagging strand (Fig. 1C) (26). This regulation by SSB is essential for confining primosome assembly to the lagging-strand template and preventing complete degradation of the fork through the unwinding of unreplicated duplex. PriA helicase activity at branched DNA structures is stimulated by a direct interaction between the 15 C-terminal amino acids of SSB and PriA (9). This stimulation is species specific and particular to PriA, since Rep is not activated by SSB (9). Stimulation may also be a result of SSB stabilizing the unwound ssDNA products; however, SSB mutants that disrupt PriA-SSB interactions also inhibit PriA catalyzed unwinding (9).
PriA and RecG
The interaction between PriA and RecG is complex. RecG helicase activity displays similarities to that of PriA, although, unlike PriA, it can also unwind a Holliday junction and reverse a stalled fork structure (50, 51). Initial findings indicated that both RecG and PriA can bind to and unwind the invading strand from a D loop (50) and suppressors of recombination and DNA repair defects in recG mutant strains arose in priA, probably inactivating its DNA helicase activity (1). These observations led to the suggestion that PriA helicase activity might unwind a D-loop structure generated by RecA that would normally be transformed into a Holliday junction by the RecG helicase (1, 50). Subsequent demonstration that both PriA and RecG specifically unwind the nascent lagging strand from model stalled fork structures (23, 52) and the demonstration that the variants encoded by recG suppressor alleles of priA (srgA1 and srgA2) have similar binding affinities for DNA structures as the wild type, but are severely compromised in their ability to unwind the lagging strand at a fork lacking a leading strand, even though they can unwind the lagging-strand from a fork containing both a leading and lagging strand, focused attention to events at stalled replication forks (18). These observations, coupled with the ability of RecG to reverse a stalled replication fork where leading- and lagging-strand synthesis may have uncoupled because of a leading-strand template block (i.e., the 5′ end of the nascent lagging strand was near the fork junction as in Fig. 1C), suggested that RecG typically would reverse these stalled forks, allowing repair of the lesion, and that subsequent to un-reversal, PriA was required for restart. These events could be dependent on RecBCD for trimming of the paired nascent DNA in the reversed fork, but would be independent of RuvAB. However, in the absence of RecG, the 3′→5′ helicase activity of PriA unwound the nascent lagging-strand allowing replisome assembly on a fork that was still blocked, creating toxic intermediates. Thus suppression of the recG phenotype is through inactivation of the PriA helicase activity.
An alternative model suggested that RecG and PriA may act in concert to stabilize arrested replication forks by binding simultaneously to stalled fork structures. RecG helicase then reverses the fork, generating a structure that PriA can capture specifically at the 3′-end of the leading strand (77). This event would ultimately allow loading of the replisome at the correct location (in the absence of damage or once the damage was repaired) and prevent parental duplex unwinding by PriA.
Recent observations, however, reinforce the idea of toxic intermediates generated in recG mutants irradiated with UV. Rudolph et al. (64) demonstrated that such cultures experience extensive filamentation and DnaA-independent DNA replication, even after all the UV lesions are repaired, possibly accounting for the ability of these strains to “recover” and grow on nutrient agar plates. This extensive DNA replication fits the description of iSDR and is suppressed by the priA300 mutation (which is required for iSDR, see below). In addition, priA300 decreases the cell division delay observed after UV irradiation in a recG-null strain, arguing that the extensive iSDR is the toxic event. The authors suggest that induction of SDR can lead to collisions of iSDR forks with nascent DNA at stalled fork structures, leading to displacement of the nascent DNA and generation of 3′ flaps that are targets for PriA-directed replisome assembly, further amplifying the effect. RecG presumably normally helps eliminate these 3′ flaps.
PriA and Recombination
Stalled forks that are not remodeled properly to generate replication restart substrates can also be targets for promiscuous recombination. A recA-null allele improves the growth of a priA300 rep double mutant and confers viability on their ruv derivatives, implying that in the absence of the helicase activities of Rep and PriA, stalled forks become substrates for recombination that can be lethal to the cell if the recombination intermediates are not resolved by RuvABC (43). Nevertheless, the priA300 rep strain is viable (65), suggesting that another helicase, perhaps RecG, may compensate for the loss of helicase activity necessary for remodeling stalled forks to generate the appropriate structure for restart. In the priA300 rep strain, RecFOR, a protein complex implicated in loading RecA on exposed ssDNA, provokes recombination and induces the SOS response (43). A recF-null mutation is synthetically lethal with a priA-null strain (66). However, a recF-null mutation can be combined with a dnaC809 priA-null strain (66). Whereas the DnaC809 variant can suppress the priA-null phenotype, it does not restore a UV inducible SOS response, which is attenuated in recF strains (66). This suggests that PriA and RecF have overlapping roles in cell viability and UV-inducible SOS expression, and dnaC809 can only suppress the priA phenotype.
The extreme filamentation observed in priA cells is dramatically reduced in a priA recA or priA recB double mutant (49), suggesting a detrimental effect of recombination in the absence of the PriA pathway for replication restart. Furthermore, recombination dependent DNA replication is partially impaired in the ATPase-deficient mutants of PriA (48), validating a helicase requirement. The PriA suppressor mutation, dnaC809, can rescue synthetic lethality between priA and either ftsK, recG, or ruvC (49). This implies that the processing of intermediates in recombination pathways via RuvC or RecG is detrimental in the absence of PriA and only the primosome loading activity of the latter protein is required.
RdgC, a protein that binds double-stranded and single-stranded DNA in vitro (55), inhibits RecA promoted DNA strand exchange activity by competing with RecA for DNA binding sites and counteracting DinI stabilization of RecA filaments (13). Therefore, in the absence of RdgC, RecA can allow recombination at unfavorable sites, decreasing genomic stability. PriA is essential in strains lacking RdgC and synthetic lethality of the priA and rdgC mutations can be suppressed by either the dnaC212 or dnaC809 mutation (55). However, these strains are highly filamentous, exhibit slow growth in rich media, are chronically induced for SOS, and are sensitive to UV and other DNA-damaging agents (55), suggesting that the suppressor mutations cannot completely compensate for the priA-null phenotype when RdgC is absent. The priA rdgC strain rapidly accumulates suppressors that inactivate RecF, RecO, and RecR, but mutations also arise in RpoB and SSB (55). In the absence of PriA and RdgC, RecFOR loading of RecA for homologous recombination can be detrimental to the cell. Because RdgC forms stable complexes with both ssDNA and dsDNA, it may function in nucleoid organization as a scaffold or modulator for repair and restart machinery at sites of fork stalling. However, the DNA structures acted on by RdgC and any direct interactions between it and the replication and restart machinery remains to be determined.
Double-strand breaks (DSB) in the genome must be repaired through homologous recombination. Eykelenboom et al (14) analyzed DSB repair in vivo by generating a DSB using the hairpin DNA-cleaving activity of the SbcCD nuclease. They determined that the breaks generated are two ended, that cleavage is dependent on replication, and that repair of the DSB required recA, recBCD, ruvABC, recG, and priA, but not recFOR (14). PriA loading of the replication machinery here helps generate a substrate for reconnection of the broken genome.
PriA is also implicated in the genome restriction and modification defense mechanism of the E. coli host that distinguishes between foreign and host genetic material (3). Alleviation of this defense system is thought to occur when DNA damage activates mechanisms that produce unmethylated target sequences, usually generated by homologous recombination, within the bacterial chromosome (44). Interestingly, sfiA priA strains are reduced in their ability to activate UV-induced restriction alleviation, but the activity is almost completely restored by either the priA300 or dnaC809 mutations (24). The requirement for PriA in UV-induced restriction alleviation may reflect loading of DnaB onto structures formed by recombination.
PriA in Stable DNA Replication
In E. coli, recombination-dependent DNA replication of the genome via iSDR and cSDR can play a role in replication restart. Initiation of iSDR occurs as part of the SOS response and requires RecA, RecB, and RecC, but protein synthesis and transcription is dispensable (42). Formation of D-loops by RecA and RecBC allows DNA replication initiation during iSDR (5, 42). On the other hand, transcription is essential for cSDR, but protein synthesis is dispensable. The cSDR pathway is active in strains mutated in rnhA, which encodes RNase HI, the major ribonuclease H activity in the cell (29), and normally repressed origins of DNA replication, oriKs, are activated (reviewed in (4)). These characteristics led to the R-loop model, proposing that transcripts generated during transcription could be used as sites of DNA replication initiation (83). Both iSDR and cSDR require PriA for initiation of replication at either a D-loop or R-loop structure respectively (Fig. 1D) (4).
In iSDR, PriA appears to be involved after D-loop formation, but prior to initiation of DNA replication (47). PriA Walker A and Walker B mutants (Fig. 2) exhibit decreased levels of iSDR, suggesting that the DNA helicase activity of PriA is required for iSDR (80). As described above, this activity is not necessary for resistance to UV damage or normal growth, indicating that D-loops may require processing by the PriA helicase to allow replisome loading. Furthermore, suppression of the extreme filamentation phenotype of a recG-null strain is effected by priA300 (64), implying that promiscuous iSDR can be detrimental in strains lacking RecG-helicase processing.
In cSDR, the absence of priC but not priB is tolerated, and in a dnaC809 strain, priC is required (67). This suggests that PriC is not essential for loading at a cSDR R-loop, but PriA requires PriB for loading. However, dnaC809 may affect the loading of PriA, possibly by competing for binding sites, and therefore PriC is required to allow primosome formation.
Given that priA-null strains show no major defect in DNA replication and that PriA helicase activity is not required for suppression of the priA-null phenotypes, but is necessary for iSDR and cSDR, the major role for the PriA helicase activity in iSDR and cSDR is likely to be in remodeling of forks for replication restart.
PriA Activity During Mu Phage DNA Replication
PriA plays an important role in initiation of replication on forked DNA intermediates generated by the transposase during bacteriophage Mu transposition (25, 27, 59). Mu phage does not encode its own replication machinery and therefore sequesters the E. coli replication restart machinery to initiate replication during the transition from the transpososome to the replisome (59). This transition involves progressive protein-protein interactions that promote the initiation of Mu DNA replication by the host machinery while the forked ends that were generated remain protected. In vitro, the transition is dependent on both PriA helicase activity and PriC, whereas in vivo, Mu plating efficiency is not affected by either a priB or a priC deletion mutation, suggesting that Mu replication can proceed by either the PriA-PriB or PriA-PriC pathway (59).
IF2, the host translation initiation factor, was isolated as a specificity factor during Mu DNA replication when it was identified as the protein responsible for removing inhibitors and promoting PriA binding (58). The requirement for the PriA helicase activity during Mu DNA replication possibly reflects either displacement of IF2 or remodeling of the Mu fork probably by unwinding of the lagging-strand arm for DnaB loading (Fig. 1C) (58). This is supported by the fact that the helicase dead PriA allele, priA300, is partially defective in the ability to support Mu DNA replication in vivo (25). During nutritional stress, such as Mu infection, IF2 has been shown to stimulate the PriA-PriC restart pathway, suggesting that IF2 is important for specificity in replication restart reactions even in the host cell (58).
PriA Activity in Other Organisms
PriA is found in many bacterial species. In the gram-negative Neisseria meningitidis, priA expression is upregulated when the bacterium infects HeLa cells (76). priA-null strains of N. meningitidis show reduced viability during late logarithmic phase, a growth defect under both aerobic growth (with nitrate) and anaerobic growth, and high sensitivity to oxidative and nitrosative stress (76), demonstrating the requirement for PriA during growth inside the host cell. These strains were as competent as wild type with respect to HeLa cell invasion, but priA mutants could not survive and were found to co-localize with the lysosome (76).
In gram-positive bacteria of low GC content, such as Bacillus subtilis, the PriA pathway requires the activity of the proteins DnaD, DnaB (not to be confused with the replicative E. coli DNA helicase) and DnaI. PriA acts as the specificity factor to recruit and assemble the primosome in B. subtilis, as it does in E. coli (46). DnaB and DnaI are then recruited to coordinate the loading of the replicative helicase DnaC (82), although there is conflicting data suggesting that DnaI alone is the main helicase loader (75). As in E. coli, priA-null strains in B. subtilis rapidly gain suppressor mutations in the helicase loader DnaB; dnaB75 is a gain of function mutation that results in more efficient loading of DnaC because of a stronger affinity to ssDNA (6, 7). DnaD seems to play a similar role to PriB because it has ssDNA binding activity (46) and interacts with both DnaB and PriA in solution (7). It has also been suggested that in B. subtilis, SSB acts as a platform to link the DNA repair toolbox at active replication forks (32). As in E. coli, the B. subtilis SSB C-terminal domain interacts with PriA and, based on studies of GFP-PriA in vivo, PriA is thought to be anchored in the cell in the vicinity of the two active chromosomal replication forks (32).
In eukaryotes, there is now reasonable data suggesting that replication restart pathways exist. While there is no obvious PriA-type activity, various helicases (Sgs1, Srs2, Rrm3, and Fbh1) have roles in remodeling DNA to either promote or to prevent illegitimate recombination events by Rad51, the eukaryotic homolog of RecA (15, 62, 73). Srs2 was also shown to negatively regulate recombination by disrupting Rad51 filaments formed on ssDNA (31, 81).
Based on recent experiments in fission yeast, Callegari and Kelly (10) demonstrated that after UV irradiation, cell cycle checkpoints are not activated in the cycle at which they were irradiated. Instead, they undergo a single checkpoint response after carrying lesions through S phase and replicate the bulk of the DNA before a delay occurs (10). Furthermore, DNA replication is required for the second-cycle delay, implying that replication must be occurring on damaged DNA (10). Therefore, in order to complete chromosome synthesis in the first cell cycle after UV irradiation, the cell must bypass the damage, suggesting the existence of mechanisms to restart replication.
Evidence for replication restart has mounted in S. cerevisiae, where strains defective in nucleotide excision repair showed uncoupling of the leading- and lagging-strand DNA polymerases, yielding long ssDNA stretches that accumulated on the leading-strand side of the fork (39). Smaller internal gaps were also detected along replicated duplexes on both sides of the fork (39), suggesting that damage bypass occurred. Surprisingly, in the absence of TLS activity, fork progression was unaffected but more internal gaps were observed in proximity to elongation points (39), implying that damage bypass and gap filling occurred during post-replication repair. A similar phenotype was also seen in recombination mutants and in all cases, bulk DNA replication was not affected (39). Overall, this study suggests that unrepaired UV damage specifically blocks leading-strand synthesis in vivo, resulting in uncoupled leading- and lagging-strand DNA synthesis and ssDNA regions left randomly behind the fork are not immediately sealed (39).
In embryonic stem cell lines of Mus musculus, Mus81, the catalytic component of a eukaryotic structure-specific endonuclease that preferentially cleaves branched DNA substrates (reminiscent of replication and recombination intermediates), was shown to be involved in the formation of DSBs in response to an inhibition of replication (19). At stalled replication forks, DSBs are useful mechanisms in stimulating the recovery of replication at non-origin locations. Moreover, recovery of stalled replication forks was attenuated in Mus81−/− cell lines, suggesting that Mus81 can potentially convert detrimental replication-associated DNA structures into intermediates for DNA repair (19).
Thus, replication restart seems likely to occur in eukaryotes and may be a major pathway for maintenance of genomic integrity. The exact mechanism is still unclear, and further studies will be required to clarify the picture.
Acknowledgments
We would like to thank Brenda Perez-Cheeks and Ram Madabhushi for thoughtful discussions and careful reading of this manuscript. Studies from the authors’ laboratory were supported by NIH grant GM34557.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Al-Deib AA, Mahdi AA, Lloyd RG. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J Bacteriol. 1996;178:6782–9. doi: 10.1128/jb.178.23.6782-6789.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen GC, Jr., Kornberg A. Assembly of the primosome of DNA replication in Escherichia coli. J Biol Chem. 1993;268:19204–9. [PubMed] [Google Scholar]
- 3.Arber W. Host-controlled variation. In: Hershey AD, editor. The Bacteriophage Lambda. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1971. pp. 83–96. [Google Scholar]
- 4.Asai T, Kogoma T. D-loops and R-loops: alternative mechanisms for the initiation of chromosome replication in Escherichia coli. J Bacteriol. 1994;176:1807–12. doi: 10.1128/jb.176.7.1807-1812.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asai T, Sommer S, Bailone A, Kogoma T. Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. EMBO J. 1993;12:3287–95. doi: 10.1002/j.1460-2075.1993.tb05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruand C, Farache M, McGovern S, Ehrlich SD, Polard P. DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol Microbiol. 2001;42:245–55. doi: 10.1046/j.1365-2958.2001.02631.x. [DOI] [PubMed] [Google Scholar]
- 7.Bruand C, Velten M, McGovern S, Marsin S, Serena C, Ehrlich SD, Polard P. Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Mol Microbiol. 2005;55:1138–50. doi: 10.1111/j.1365-2958.2004.04451.x. [DOI] [PubMed] [Google Scholar]
- 8.Cadman CJ, Lopper M, Moon PB, Keck JL, McGlynn P. PriB stimulates PriA helicase via an interaction with single-stranded DNA. J Biol Chem. 2005;280:39693–700. doi: 10.1074/jbc.M508521200. [DOI] [PubMed] [Google Scholar]
- 9.Cadman CJ, McGlynn P. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 2004;32:6378–87. doi: 10.1093/nar/gkh980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callegari AJ, Kelly TJ. UV irradiation induces a postreplication DNA damage checkpoint. Proc Natl Acad Sci U S A. 2006;103:15877–82. doi: 10.1073/pnas.0607343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen HW, North SH, Nakai H. Properties of the PriA helicase domain and its role in binding PriA to specific DNA structures. J Biol Chem. 2004;279:38503–12. doi: 10.1074/jbc.M404769200. [DOI] [PubMed] [Google Scholar]
- 12.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 13.Drees JC, Chitteni-Pattu S, McCaslin DR, Inman RB, Cox MM. Inhibition of RecA protein function by the RdgC protein from Escherichia coli. J Biol Chem. 2006;281:4708–17. doi: 10.1074/jbc.M513592200. [DOI] [PubMed] [Google Scholar]
- 14.Eykelenboom JK, Blackwood JK, Okely E, Leach DR. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell. 2008;29:644–51. doi: 10.1016/j.molcel.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 15.Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A. 2002;99:16887–92. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flores MJ, Ehrlich SD, Michel B. Primosome assembly requirement for replication restart in the Escherichia coli holDG10 replication mutant. Mol Microbiol. 2002;44:783–92. doi: 10.1046/j.1365-2958.2002.02913.x. [DOI] [PubMed] [Google Scholar]
- 17.Gorbalenya AE, Koonin EV. Helicases: amino acid sequences comparisons and structure-function relationships. Curr Opin Struct Biol. 1993;3:419–429. [Google Scholar]
- 18.Gregg AV, McGlynn P, Jaktaji RP, Lloyd RG. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol Cell. 2002;9:241–51. doi: 10.1016/s1097-2765(02)00455-0. [DOI] [PubMed] [Google Scholar]
- 19.Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 20.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439:557–62. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 21.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7:932–43. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 22.Heller RC, Marians KJ. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol Cell. 2005;17:733–43. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 23.Heller RC, Marians KJ. Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J Biol Chem. 2005;280:34143–51. doi: 10.1074/jbc.M507224200. [DOI] [PubMed] [Google Scholar]
- 24.Ivancic-Bacce I, Vlasic I, Cogelja-Cajo G, Brcic-Kostic K, Salaj-Smic E. Roles of PriA protein and double-strand DNA break repair functions in UV-induced restriction alleviation in Escherichia coli. Genetics. 2006;174:2137–49. doi: 10.1534/genetics.106.063750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones JM, Nakai H. Duplex opening by primosome protein PriA for replisome assembly on a recombination intermediate. J Mol Biol. 1999;289:503–16. doi: 10.1006/jmbi.1999.2783. [DOI] [PubMed] [Google Scholar]
- 26.Jones JM, Nakai H. Escherichia coli PriA helicase: fork binding orients the helicase to unwind the lagging strand side of arrested replication forks. J Mol Biol. 2001;312:935–47. doi: 10.1006/jmbi.2001.4930. [DOI] [PubMed] [Google Scholar]
- 27.Jones JM, Nakai H. The phiX174-type primosome promotes replisome assembly at the site of recombination in bacteriophage Mu transposition. Embo J. 1997;16:6886–95. doi: 10.1093/emboj/16.22.6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim S, Dallmann HG, McHenry CS, Marians KJ. Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell. 1996;84:643–50. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 29.Kogoma T. RNase H-defective mutants of Escherichia coli. J Bacteriol. 1986;166:361–3. doi: 10.1128/jb.166.2.361-363.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kogoma T, Cadwell GW, Barnard KG, Asai T. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J Bacteriol. 1996;178:1258–64. doi: 10.1128/jb.178.5.1258-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–9. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- 32.Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. Embo J. 2007;26:4239–51. doi: 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee EH, Kornberg A. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n’ protein. Proc Natl Acad Sci U S A. 1991;88:3029–32. doi: 10.1073/pnas.88.8.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee MS, Marians KJ. Escherichia coli replication factor Y, a component of the primosome, can act as a DNA helicase. Proc Natl Acad Sci U S A. 1987;84:8345–9. doi: 10.1073/pnas.84.23.8345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MS, Marians KJ. The Escherichia coli primosome can translocate actively in either direction along a DNA strand. J Biol Chem. 1989;264:14531–42. [PubMed] [Google Scholar]
- 36.Liu J, Marians KJ. PriA-directed assembly of a primosome on D loop DNA. J Biol Chem. 1999;274:25033–41. doi: 10.1074/jbc.274.35.25033. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Nurse P, Marians KJ. The ordered assembly of the phiX174-type primosome. III. PriB facilitates complex formation between PriA and DnaT. J Biol Chem. 1996;271:15656–61. doi: 10.1074/jbc.271.26.15656. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Xu L, Sandler SJ, Marians KJ. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc Natl Acad Sci U S A. 1999;96:3552–5. doi: 10.1073/pnas.96.7.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 40.Lopper M, Boonsombat R, Sandler SJ, Keck JL. A hand-off mechanism for primosome assembly in replication restart. Mol Cell. 2007;26:781–93. doi: 10.1016/j.molcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucius AL, Jezewska MJ, Bujalowski W. Allosteric interactions between the nucleotide-binding sites and the ssDNA-binding site in the PriA helicase-ssDNA complex. 3. Biochemistry. 2006;45:7237–55. doi: 10.1021/bi0518287. [DOI] [PubMed] [Google Scholar]
- 42.Magee TR, Kogoma T. Requirement of RecBC enzyme and an elevated level of activated RecA for induced stable DNA replication in Escherichia coli. J Bacteriol. 1990;172:1834–9. doi: 10.1128/jb.172.4.1834-1839.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahdi AA, Buckman C, Harris L, Lloyd RG. Rep and PriA helicase activities prevent RecA from provoking unnecessary recombination during replication fork repair. Genes Dev. 2006;20:2135–47. doi: 10.1101/gad.382306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makovets S, Doronina VA, Murray NE. Regulation of endonuclease activity by proteolysis prevents breakage of unmodified bacterial chromosomes by type I restriction enzymes. Proc Natl Acad Sci U S A. 1999;96:9757–62. doi: 10.1073/pnas.96.17.9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marians KJ. Replication and recombination intersect. Curr Opin Genet Dev. 2000;10:151–6. doi: 10.1016/s0959-437x(00)00059-9. [DOI] [PubMed] [Google Scholar]
- 46.Marsin S, McGovern S, Ehrlich SD, Bruand C, Polard P. Early steps of Bacillus subtilis primosome assembly. J Biol Chem. 2001;276:45818–25. doi: 10.1074/jbc.M101996200. [DOI] [PubMed] [Google Scholar]
- 47.Masai H, Asai T, Kubota Y, Arai K, Kogoma T. Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. Embo J. 1994;13:5338–45. doi: 10.1002/j.1460-2075.1994.tb06868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masai H, Deneke J, Furui Y, Tanaka T, Arai KI. Escherichia coli and Bacillus subtilis PriA proteins essential for recombination-dependent DNA replication: involvement of ATPase/helicase activity of PriA for inducible stable DNA replication. Biochimie. 1999;81:847–57. doi: 10.1016/s0300-9084(99)00211-4. [DOI] [PubMed] [Google Scholar]
- 49.McCool JD, Sandler SJ. Effects of mutations involving cell division, recombination, and chromosome dimer resolution on a priA2::kan mutant. Proc Natl Acad Sci U S A. 2001;98:8203–10. doi: 10.1073/pnas.121007698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGlynn P, Al-Deib AA, Liu J, Marians KJ, Lloyd RG. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol. 1997;270:212–21. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- 51.McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 52.McGlynn P, Lloyd RG. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci U S A. 2001;98:8227–34. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minden JS, Marians KJ. Replication of pBR322 DNA in vitro with purified proteins. Requirement for topoisomerase I in the maintenance of template specificity. J Biol Chem. 1985;260:9316–25. [PubMed] [Google Scholar]
- 54.Mizukoshi T, Tanaka T, Arai K, Kohda D, Masai H. A critical role of the 3′ terminus of nascent DNA chains in recognition of stalled replication forks. J Biol Chem. 2003;278:42234–9. doi: 10.1074/jbc.C300285200. [DOI] [PubMed] [Google Scholar]
- 55.Moore T, McGlynn P, Ngo HP, Sharples GJ, Lloyd RG. The RdgC protein of Escherichia coli binds DNA and counters a toxic effect of RecFOR in strains lacking the replication restart protein PriA. Embo J. 2003;22:735–45. doi: 10.1093/emboj/cdg048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ng JY, Marians KJ. The ordered assembly of the phiX174-type primosome. I. Isolation and identification of intermediate protein-DNA complexes. J Biol Chem. 1996;271:15642–8. doi: 10.1074/jbc.271.26.15642. [DOI] [PubMed] [Google Scholar]
- 57.Ng JY, Marians KJ. The ordered assembly of the phiX174-type primosome. II. Preservation of primosome composition from assembly through replication. J Biol Chem. 1996;271:15649–55. doi: 10.1074/jbc.271.26.15649. [DOI] [PubMed] [Google Scholar]
- 58.North SH, Kirtland SE, Nakai H. Translation factor IF2 at the interface of transposition and replication by the PriA-PriC pathway. Mol Microbiol. 2007;66:1566–78. doi: 10.1111/j.1365-2958.2007.06022.x. [DOI] [PubMed] [Google Scholar]
- 59.North SH, Nakai H. Host factors that promote transpososome disassembly and the PriA-PriC pathway for restart primosome assembly. Mol Microbiol. 2005;56:1601–16. doi: 10.1111/j.1365-2958.2005.04639.x. [DOI] [PubMed] [Google Scholar]
- 60.Nurse P, Liu J, Marians KJ. Two modes of PriA binding to DNA. J Biol Chem. 1999;274:25026–32. doi: 10.1074/jbc.274.35.25026. [DOI] [PubMed] [Google Scholar]
- 61.Nurse P, Zavitz KH, Marians KJ. Inactivation of the Escherichia coli priA DNA replication protein induces the SOS response. J Bacteriol. 1991;173:6686–93. doi: 10.1128/jb.173.21.6686-6693.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osman F, Dixon J, Barr AR, Whitby MC. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol Cell Biol. 2005;25:8084–96. doi: 10.1128/MCB.25.18.8084-8096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parada CA, Marians KJ. Transcriptional activation of pBR322 DNA can lead to duplex DNA unwinding catalyzed by the Escherichia coli preprimosome. J Biol Chem. 1989;264:15120–9. [PubMed] [Google Scholar]
- 64.Rudolph CJ, Upton AL, Harris L, Lloyd RG. Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol. 2009;73:352–66. doi: 10.1111/j.1365-2958.2009.06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sandler SJ. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics. 2000;155:487–97. doi: 10.1093/genetics/155.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sandler SJ. Overlapping functions for recF and priA in cell viability and UV-inducible SOS expression are distinguished by dnaC809 in Escherichia coli K-12. Mol Microbiol. 1996;19:871–80. doi: 10.1046/j.1365-2958.1996.429959.x. [DOI] [PubMed] [Google Scholar]
- 67.Sandler SJ. Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics. 2005;169:1799–806. doi: 10.1534/genetics.104.036962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sandler SJ, Marians KJ. Role of PriA in replication fork reactivation in Escherichia coli. J Bacteriol. 2000;182:9–13. doi: 10.1128/jb.182.1.9-13.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sandler SJ, Marians KJ, Zavitz KH, Coutu J, Parent MA, Clark AJ. dnaC mutations suppress defects in DNA replication- and recombination-associated functions in priB and priC double mutants in Escherichia coli K-12. Mol Microbiol. 1999;34:91–101. doi: 10.1046/j.1365-2958.1999.01576.x. [DOI] [PubMed] [Google Scholar]
- 70.Sandler SJ, McCool JD, Do TT, Johansen RU. PriA mutations that affect PriA-PriC function during replication restart. Mol Microbiol. 2001;41:697–704. doi: 10.1046/j.1365-2958.2001.02547.x. [DOI] [PubMed] [Google Scholar]
- 71.Sandler SJ, Samra HS, Clark AJ. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics. 1996;143:5–13. doi: 10.1093/genetics/143.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sasaki K, Ose T, Okamoto N, Maenaka K, Tanaka T, Masai H, Saito M, Shirai T, Kohda D. Structural basis of the 3′-end recognition of a leading strand in stalled replication forks by PriA. Embo J. 2007;26:2584–93. doi: 10.1038/sj.emboj.7601697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmidt KH, Kolodner RD. Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol Cell Biol. 2004;24:3213–26. doi: 10.1128/MCB.24.8.3213-3226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shlomai J, Kornberg A. A prepriming DNA replication enzyme of Escherichia coli. II. Actions of protein n’: a sequence-specific, DNA-dependent ATPase. J Biol Chem. 1980;255:6794–8. [PubMed] [Google Scholar]
- 75.Soultanas P. A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Res. 2002;30:966–74. doi: 10.1093/nar/30.4.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tala A, De Stefano M, Bucci C, Alifano P. Reverse transcriptase-PCR differential display analysis of meningococcal transcripts during infection of human cells: up-regulation of priA and its role in intracellular replication. BMC Microbiol. 2008;8:131. doi: 10.1186/1471-2180-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tanaka T, Masai H. Stabilization of a stalled replication fork by concerted actions of two helicases. J Biol Chem. 2006;281:3484–93. doi: 10.1074/jbc.M510979200. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka T, Mizukoshi T, Sasaki K, Kohda D, Masai H. Escherichia coli PriA protein, two modes of DNA binding and activation of ATP hydrolysis. J Biol Chem. 2007;282:19917–27. doi: 10.1074/jbc.M701848200. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka T, Mizukoshi T, Taniyama C, Kohda D, Arai K, Masai H. DNA binding of PriA protein requires cooperation of the N-terminal D-loop/arrested-fork binding and C-terminal helicase domains. J Biol Chem. 2002;277:38062–71. doi: 10.1074/jbc.M204397200. [DOI] [PubMed] [Google Scholar]
- 80.Tanaka T, Taniyama C, Arai K, Masai H. ATPase/helicase motif mutants of Escherichia coli PriA protein essential for recombination-dependent DNA replication. Genes Cells. 2003;8:251–61. doi: 10.1046/j.1365-2443.2003.00630.x. [DOI] [PubMed] [Google Scholar]
- 81.Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–12. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 82.Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P, Polard P. A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol Cell. 2003;11:1009–20. doi: 10.1016/s1097-2765(03)00130-8. [DOI] [PubMed] [Google Scholar]
- 83.von Meyenburg K, Boye E, Skarstad K, Koppes L, Kogoma T. Mode of initiation of constitutive stable DNA replication in RNase H-defective mutants of Escherichia coli K-12. J Bacteriol. 1987;169:2650–8. doi: 10.1128/jb.169.6.2650-2658.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu L, Marians KJ. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell. 2003;11:817–26. doi: 10.1016/s1097-2765(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 85.Xu L, Marians KJ. Purification and characterization of DnaC810, a primosomal protein capable of bypassing PriA function. J Biol Chem. 2000;275:8196–205. doi: 10.1074/jbc.275.11.8196. [DOI] [PubMed] [Google Scholar]
- 86.Yuzhakov A, Turner J, O’Donnell M. Replisome assembly reveals the basis for asymmetric function in leading and lagging strand replication. Cell. 1996;86:877–86. doi: 10.1016/s0092-8674(00)80163-4. [DOI] [PubMed] [Google Scholar]
- 87.Zavitz KH, Marians KJ. ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J Biol Chem. 1992;267:6933–40. [PubMed] [Google Scholar]
- 88.Zavitz KH, Marians KJ. Helicase-deficient cysteine to glycine substitution mutants of Escherichia coli replication protein PriA retain single-stranded DNA-dependent ATPase activity. Zn2+ stimulation of mutant PriA helicase and primosome assembly activities. J Biol Chem. 1993;268:4337–46. [PubMed] [Google Scholar]