

Abstract
Polyesters with free functional groups allow facile modifications with biomolecules, which can lead to versatile biomaterials that afford controlled interactions with cells and tissues. Efficient synthesis of functionalizable polyesters is still a challenge that greatly limits the availability and widespread applications of biofunctionalized synthetic polymers. Here we report a simple route to prepare a functionalizable polyester, poly(sebacoyl diglyceride) (PSeD) bearing free hydroxyl groups. The key synthetic step is an epoxide ring-opening polymerization, instead of the traditional polycondensation, that produces poly(glycerol sebacate) (PGS) [1]. PSeD has a more defined structure with mostly linear backbone, more free hydroxyl groups, higher molecular weight, and lower polydispersity than PGS. Crosslinking PSeD with sebacic acid yields a polymer five times tougher and more elastic than cured PGS. PSeD exhibits good cytocompatibility in vitro. Furthermore, functionalization by glycine proceeds with high efficiency. This versatile synthetic platform can offer a large family of biodegradable, functionalized polymers with tunable physiochemical and biological properties useful for a wide range of biomedical applications.
1. Introduction
Aliphatic polyesters such as polylactide form an important class of biomaterial with controllable degradation and excellent biocompatibility [2]. There have been extensive investigations on these biomaterials [3] and many polyesters such as polyglycolide, polylactide, and other polyhydroxyalkanoates have been widely used as resorbable sutures, drug delivery matrices, and tissue-engineering scaffolds [4-7]. Most of these polyesters are, however, semi-crystalline, hydrophobic, and lack free functional group(s) for further modification by biomolecules [5, 8-10]. One way to address these drawbacks is the introduction of functional groups. The presence of functionalizable groups such as hydroxyl and carboxylate in polyesters can efficiently increase their hydrophilicity and degradability, plus modulate their mechanical, thermal, chemical, and biologic properties [8, 9, 11]. Furthermore, these functional groups provide access to versatile routes to conjugate a variety of bioactive molecules such as peptides [12], saccharides [13], and biotin [14]. This can lead to novel biomaterials with diverse bioactivities. The reaction of pendant groups can further convert functionalizable linear polyesters to branched and network polymers, providing even more means to modulate their properties. Therefore, the properties of functionalized polyesters can be effectively tailored to meet a variety of demands in biomedical applications.
The state of the art in the synthesis of functionalizable polyesters has been well presented in several recent reviews [8-10, 15, 16]. As mentioned in these reviews, chemical preparation of functionalizable polyesters is difficult because it usually involves a complex multistep synthetic route with a low overall yield. Two general strategies are used: one is pre-functionalization via polymerization of functionalized monomer [17, 18]; the other is post-polymerization functionalization via modification of non-functionalized polyester [19, 20]. The former usually requires complicated monomer preparation, and tedious protection and deprotection of functional groups. The latter often has to be performed under strict conditions and sometimes it is hard to control the final chemical structure. Moreover, both approaches are often associated with degradation in the transformation after polymerization [19, 21].
In light of this, efficient synthesis of linear polyesters bearing suitable functionalities is highly valuable. We designed the title polymer according to three criteria. (1) Functionality: we chose the versatile hydroxyl group as the pendant functional group in the polymer. Hydroxyl group is one of the best understood organic functional groups, and many mild reactions are available for further modifications with biomolecules such as peptides [12, 22]. To minimize side reactions in subsequent biofunctionalization, hydroxyl group was designed to be the only functional group susceptible to further chemical modification. (2) Biocompatibility: this is required for the eventual application of any biomaterial. Thus, the resultant polymer and degradation products must be well tolerated by the cells and tissues. (3) Availability: this is important for widespread usage of the materials. Therefore, the principal polymer should be prepared from low-cost reagents by simple synthetic transformations in relatively fewer steps.
Poly(sebacoyl diglyceride) (PSeD) (Fig. 1) satisfies these design criteria. PSeD can be viewed as a linear analog of poly(glycerol sebacate) (PGS) [1]. PGS is synthesized directly by polycondensation of glycerol and sebacic acid and has a mixed structure: some of its secondary hydroxyl groups remain free and some are converted to ester groups that link two polymer chains. The distinct synthesis method of PSeD by an epoxide ring opening polymerization leads to a well defined polymer structure with free hydroxyl groups, which is important for control of subsequent functionalization with bioactive molecules [12]. This versatile synthetic platform is applicable to other diglycidyl esters and diacids, which will lead to biodegradable and biofunctionalizable polymers with a variety of physiochemical properties.
Fig. 1.

The newly designed synthetic strategy of PSeD leads to a biodegradable polymer that retains the biocompatibility of PGS but with a more defined structure that is advantageous for subsequent functionalization.
The in vitro and in vivo biocompatibilities of PGS have been reported by multiple research groups and it is widely used in soft tissue engineering [1, 23-29]. PSeD will likely have the same biocompatibility of PGS due to its structural relationship with PGS. To the best of our knowledge, common routes to synthesize polyesters by either polycondensation or ring-opening polymerization of lactones are not effective in the synthesis of linear polyesters with free functional groups without protection and deprotection steps. Glycerol-based polyesters prepared using direct chemical (Scheme 1) or lipase-catalyzed polycondensation are usually compromised by premature crosslinking, large polydispersity index (PDI), or low molecular weight [1, 30, 31]. We recently reported the application of epoxide ring-opening polymerization between biomolecules with primary amines and diglycidyl esters that led to polymers with free -OH groups [32, 33]. We reasoned that a similar polymerization between a diacid and a diglycidyl ester would produce a useful PSeD polyester (Fig. 1). In this synthetic strategy, glycidyl group serves as both the active polymerization functionality and the precursor of the glyceryl moiety. Thus, the ring-opening polymerization would form the polyester backbone and the pendant hydroxyl groups in one step; as an added advantage, the reaction would not produce small molecule byproducts.
2. Materials and Methods
2.1. Materials and Equipments
Sebacoyl chloride (TCI America, 90%) and glycidol (Acros, 96%), were distilled under reduced pressure. Triethylamine (Alfa Aesar, 99%) was dried by anhydrous NaOH and distilled. Anhydrous solvents toluene, dioxane, and N,N-dimethylformamide (DMF), were purchased from EMD. Ethyl ether, ethyl acetate, and acetone were purchased from PHARMCO-AAPER. All solvents were used without further purification. Sebacic acid (Alfa Aesar, 98%) was recrystallized three times from 95% ethanol and dried under vacuum. Tetrabutylammonium bromide (Acros, 99+%), maleic anhydride (Alfa Aesar, 99%), Boc-Gly-OH (Peptides International), N,N′-Dicyclohexylcarbodiimide (DCC) (Alfa Aesar, 99%), and dimethylaminopyridine (DMAP) (99%, Avocado Research Chemicals Ltd) were used without further purification.
All compound characterization was carried out at ambient temperature. The molecular weight was determined via gel permeation chromatography (GPC) on a Viscotek GPCmax VE2001 system. For PSeD, the measurement was performed on a Viscotek organic I-MBMMW-3078 column and a VE3580 differential refractive index using DMF as the eluent. Polystyrene (Varian EasiVial PS-M, Part No. 2010-0301) was used for calibration. For PSeD glycinate and maleic monoester, the measurement was performed on a PSS SDV 1000Å column and a Viscotek 270 dual detector (differential refractive index and right angle light scattering) using THF as the eluent. Polystyrene (American polymer standards PS170K) was used for calibration. 1H NMR spectra were recorded on a Varian Mercury-400 NMR in deuterated chloroform solution with tetramethylsilane as the internal chemical reference unless otherwise specially noted. 13C and 2D NMR spectra were recorded on a Bruker Avance 600 NMR. Fourier transformed infrared (FTIR) spectra were recorded on a Thermo Nicolet IR-100 spectrometer via sample films coated on NaCl crystal windows unless noted otherwise. Differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) were performed on Seiko DSC 220C and TG/DTA 320 instruments, respectively, at a heating rate of 10 °C/min under a nitrogen atmosphere.
2.2. Synthesis of PSeD
Preparation of monomer diglycidyl sebacate
Sebacoyl chloride (11.37 g, 47.6 mmol) was added dropwise to a solution of glycidol (7.8 ml, 116.9 mmol) and triethylamine (31.5 ml, 224.1 mmol) in 180 ml of anhydrous toluene cooled in an ice/ethanol bath (−15 °C) under a nitrogen atmosphere over 30 min. The reaction mixture was stirred for another 6 h before it was filtered and concentrated. The residue was purified by flash chromatography on silica gel (3:1 hexane:ethyl acetate) to afford diglycidyl sebacate (10.4 g, 70% yield) as a white solid. 1H NMR (400 MHz, CDCl3, Me4Si) δ 4.42 (dd, J = 12.6, 3.0 Hz, 2H), 3.91 (dd, J = 11.8, 6.2 Hz, 2H), 3.19-3.23 (m, 2H), 2.85 (t, J = 4.4 Hz, 2H), 2.65 (dd, J = 5.2, 2.4 Hz, 2H), 2.35 (t, J = 7.8 Hz, 4H), 1.60-1.65 (m, 4H), 1.25-1.31 (m, 8H).
Synthesis of PSeD
Diglycidyl sebacate, an equimolar amount of sebacic acid, and a catalytic amount of tetrabutylammonium bromide were mixed and dissolved in anhydrous DMF or dioxane in a Schlenk flask in a glove box filled with nitrogen. The flask was sealed, transferred out of the glove box, and connected to a Schlenk line. The flask was heated and its contents were stirred under a nitrogen atmosphere. The reaction mixture was purified via dilution in ethyl acetate, precipitation in ethyl ether, and dried under a vacuum at ambient temperature overnight. (See Table 1 for variations of the reaction parameters)
Table 1.
Optimization of polymerization between diglycidyl sebacate and sebacic acid.
| Entry | nBu4NBr | Solvent | Temperature | Time | Mn | PDI | Yielda |
|---|---|---|---|---|---|---|---|
| 1 | none | DMF | 90 °C | 50 h | 11.5 kDa | 6.5 | 60% |
| 2 | 3.4 mol % | Dioxane | 100 °C | 114 hb | Crosslinked | – | 75% |
| 3 | 1.4 mol % | Dioxane | 80 °C | 160 hb | 12.7 kDa | 5.9 | 78% |
| 4 | 0.1 mol % | Dioxane | 95 °C | 174 h | 16.6 kDa | 2.5 | 90% |
Unoptimized.
The final reaction mixture formed a resin and could not be stirred.
2.3. Functionalization of PSeD using the -OH groups
PSeD glycinate
PSeD (118.9 mg, 0.460 mmol), Boc-Gly-OH (88.7 mg, 0.506 mmol), DCC (189.9 mg, 0.921 mmol), and DMAP (2.81 mg, 0.023 mmol) were dissolved in anhydrous dichloromethane (8 ml). The mixture was stirred at room temperature under a nitrogen atmosphere for 18 h, before it was filtered and the filtrate was concentrated. The raw product was redissolved in acetone, precipitated using ethyl ether and washed with deionized water. The precipitate was dried to furnish PSeD glycinate (155 mg) as a colorless solid.
PSeD maleic monoester
PSeD (262.3 mg, 1.015 mmol) and maleic anhydride (199.1 mg, 2.031 mmol) were dissolved in 0.46 ml of anhydrous DMF in a Biotage 0.5-2 ml microwave vial and sealed in a glove box filled with nitrogen. The vial was transferred out of the glove box and reacted at 110 °C for 30 min using a Biotage Initiator 2.5 microwave reactor. The reaction mixture was precipitated in H2O (18 Mohm-cm) and dried under vacuum at ambient temperature overnight to obtain the maleic monoester of PSeD (292.9 mg) as a transparent solid.
2.4. Crosslinking of PSeD and the mechanical properties of the resultant elastomer
A mixture of PSeD and sebacic acid (1.13 wt.%) was melted and poured into an aluminum mold coated with hyaluronic acid (mold release). The mixture was heated at 120 °C for 20 h to remove all the bubbles, then subjected to vacuum (1.1 Torr) at 120 °C for another 21 h to yield crosslinked PSeD elastomer.
Tensile strength testing was conducted on an MTS insight mechanical analyzer equipped with a 50 Newton load cell according to ASTM standard D142-06a. Three dog bone-shaped samples (D142-06a die A design scaled by 1/4, 14.75 mm × 3mm × 1.3 mm, length × width × thickness) were tested and averaged. Deflection speed was kept at 125 mm/min. In the simple tensile strength test, the sample was elongated to failure. In the cyclic tensile strength test, the sample was pulled to 50%, then allowed to recover to 1% before immediately stretched to 50% five times.
2.5. Evaluation of in vitro biocompatibility of PSeD
The wells of a tissue culture-treated polystyrene (TCPS) 24-well plate were coated with 80 μl of 1 g/l PSeD solution in MeOH (0.2 μm filtered). The plate was dried under vacuum overnight after evaporation of the solvent in air. Both the TCPS plate and the polymer-coated plate were sterilized by UV light for 30 min, then washed with 1 ml phosphate buffered saline three times and with 1 ml culture medium once with gentle shaking (20 rpm). Each well was seeded with 50,000 nonimmortalized smooth muscle cells (passage 12) isolated from the carotid arteries of 4-year-old male baboons (Papio cyncephalus) in 1 ml of MCDB 131 medium supplemented with 10% FBS, 1% l-glutamine, and 20 μg/ml gentamycin. The cells were incubated at 37 °C with 5% CO2. Medium was exchanged every 2 days. The number of metabolically active cells was determined colorimetrically by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay according to a previously published protocol [34] via absorption at 570 nm measured with a Molecular Devices SpectraMax M2/M2e Microplate Reader. Statistical analysis was performed using a two-tailed Student's t test. A p value of_± 0.05 was considered statistically significant. All values are reported as mean ± standard deviation. Both polymer wells and the control were imaged on day 3 using an inverted phase contrast microscope [Nikon TE-2000U (Melville, NY) equipped with a 4 MP Diagnostics Spot Flex digital camera (Sterling Heights, MI)].
3. Results
3.1. Synthesis of PSeD
The first step of the synthesis was the preparation of the monomer - diglycidyl sebacate. Due to the high reactivity of the epoxide groups, our preliminary attempts, including Tl(OAc)3-catalyzed transesterification, DCC/DMAP-mediated condensation, and the reaction between sodium sebacate and epichlorohydrin [35], led to complex reaction mixture and very low yields, and were therefore considered to be failures. Later, we prepared diglycidyl sebacate by epoxidation of diallyl sebacate, which was prepared via esterification of allyl alcohol and sebacic acid [33]. The yield was only approximately 60%, however, despite attempts to vary conditions such as reaction temperature, reaction time, and reactant ratio. Furthermore, excessive amounts of the oxidant m-chloroperoxybenzoic acid had to be used to accomplish the epoxidation. m-Chloroperoxybenzoic acid and its byproduct m-chlorobenzoic acid were difficult to remove completely and often led to lower purified yield (50%) in relatively large scale (2.6 g) reactions. Therefore, we explored the esterification of sebacoyl chloride by glycidol in toluene as a way to synthesize diglycidyl sebacate (Fig. 2) [36]. We found that the side reaction, ring-opening of the epoxide moiety by HCl, could be efficiently prevented using excessive triethylamine (5×) as an acceptor of HCl and lowering the reaction temperature to −15 °C. No ring-opened byproducts were observed in the 1H NMR spectra of the reaction mixture. The purification was convenient because the byproduct, triethylamine hydrochloride, was only sparingly soluble in toluene and could be removed by a simple filtration. Under optimized conditions, diglycidyl sebacate was produced at 70% purified yield on a 10-g scale.
Fig. 2.

Direct esterification under optimized conditions produced the monomer diglycidyl sebacate in one step with simpler procedure and higher yield than the previous two-step route.[33]
The second step of the synthesis was the epoxide ring-opening polymerization between diglycidyl sebacate and sebacic acid to yield PSeD. To minimize side reactions, we performed this reaction at relatively low temperature and, therefore, we chose solution polymerization instead of bulk polymerization because the melting point of sebacic acid is 132 °C. Diglycidyl sebacate and sebacic acid were first polymerized in DMF without catalyst at 90 °C to produce PSeD (Table 1, entry 1). The resultant polymer had a moderate molecular weight (Mn = 11.5 kDa) and a high PDI (6.5). Quaternary ammonium salts have been reported to be efficient catalysts for addition reactions of epoxy compounds,[37] so we expected that they would be useful in our polymerization. When equimolar amount of diglycidyl sebacate and sebacic acid reacted in the presence of 3.4% nBu4NBr at 100 °C in dioxane (Table 1, entry 2), however, the catalyst induced gelation and the resultant polymer did not dissolve in common solvents such as CH2Cl2, THF, acetone, ethanol, and DMF. This indicated that the polymer was crosslinked. Reducing the nBu4NBr concentration and lowering the reaction temperature successfully prevented crosslinking of the polymer (Table 1, entry 3); however, the resultant polymer had a high PDI (5.9). A more significant reduction in catalyst concentration and a slight elevation of reaction temperature [0.1% molar ratio nBu4NBr at 95 °C (Table 1, entry 4)] yielded PSeD with an increased molecular weight (Mn = 16.6 kDa), a narrower polydispersity (PDI = 2.5), and an excellent isolated yield (90%).
3.2. Structural Characterization of PSeD
The polymer was characterized by standard analytical methods. Its structure was analyzed by FT-NMR and FTIR spectroscopy. In the 1H NMR spectrum (Fig. 3A), the three signals marked ‘a’ at δ 1.31, 1.62 and 2.35 ppm were ascribed to the sebacoyl moiety in the polymer backbone. The broad peak marked ‘b’ at δ 2.89 ppm indicated the presence of a hydroxyl group (-OH). The signal at chemical shift 3.55-4.54 ppm marked ‘c, d’ corresponded to the CH protons of the glyceryl moiety. Comparison of the integrations of the various protons suggested that each polymer repeating unit consisted of one -OH group. Moreover, it revealed that essentially all the epoxide functionalities had been consumed because no proton signals associated with epoxy groups (δ around 3.0 ppm, please see the 1H NMR data of diglycidyl sebacate in the experimental section) were observed. The 13C NMR spectrum of PSeD (Fig. 3B) further confirmed its structure. The three signals marked ‘a’ at δ 24.78, 28.95 and 34.05 ppm were attributed to the sebacoyl moiety in the backbone of the polymer. The signals at chemical shifts 61.31 and 72.08 ppm marked ‘c, d’ were attributed to the carbons of glyceryl moiety. The signal marked ‘b’ at δ 173.94 ppm corresponded to carbon of the ester group.
Fig. 3.
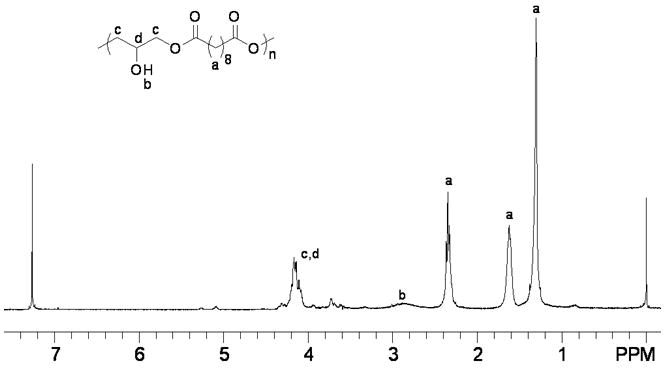
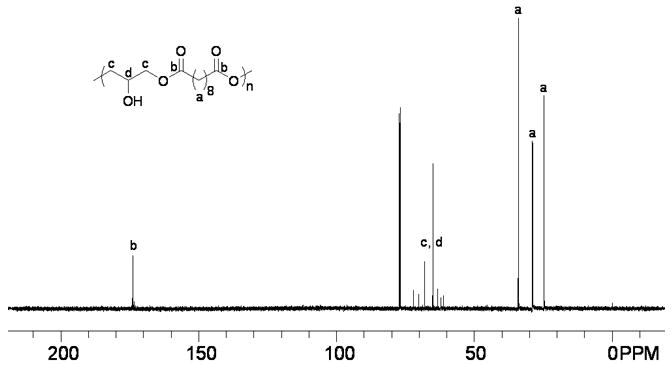
(A) The 1H NMR spectrum of PSeD is consistent with the proposed structure with glyceryl moiety at δ from 3.55-4.54 ppm, sebacoyl moiety at δ 1.31, 1.62, and 2.35 ppm, and -OH group at δ 2.89 ppm. (B) The 13C NMR spectrum of PSeD corroborates well with the 1H NMR. The resonance of the glyceryl moiety occurs at δ 61.31-72.08 ppm, the sebacoyl moiety at δ 24.78, 28.95, and 34.05 ppm, and the ester group at δ 173.94 ppm.
To examine the linearity of PSeD and the relative amount of free -OH groups, 13C DEPT 135 (distortionless enhancement by polarization transfer) (Fig. 4A) and 1H-13C HETCOR (heteronuclear correlation) NMR experiments (Fig. 4B) were performed. In the 13C DEPT 135 NMR spectrum (Fig. 4A), the glyceryl CH2 groups (c) appeared as negative peaks and the CH (d) appeared as positive peaks. When glyceryl OH groups were converted to esters, their signals shifted downfield (d′) because of the electron withdrawing effect of the carbonyl group. The intensity of d is much higher than that of d′, indicating most of the -OH groups remain free. The 1H-13C HETCOR NMR spectrum (Fig. 4B) revealed that most of the proton signals of the glyceryl moiety appeared around δ 4.00 ppm (strong peaks marked with an *). The weak proton signal at 5.09 ppm (#) was associated with the carbon at 72.06 ppm (d′), a negative peak in the DEPT 135 NMR spectrum indicating a glyceryl CH group. Furthermore, the downfield chemical shift of the # peak suggested that it was an ester CH group (d′). The higher intensity of the d signal vs. d′ again indicated the linear diglyceride structure of PSeD and the fact that most of the hydroxyl groups are free. A ratio of the free glyceryl OH groups vs. glyceryl ester group was estimated using 1H NMR. We set the proton integration of sebacoyl moiety (δ 1.31, 1.62 and 2.35 ppm) as 16 protons and the integration of the CH group (d+d′) in glyceryl moiety as 1 proton (Fig. 4C), which corresponded to one PSeD repeating unit. The resulting integration of 0.1 for the glyceryl ester moieties (δ5.09 to 5.28 ppm, d′, Fig. 3C), representing the branched PSeD, corresponded to approximately 10%. In summary, about 90% of the glyceryl hydroxyl groups in PSeD are free, that is, PSeD is approximately 90% linear, 10% branched. A similar analysis of PGS revealed that only 45% of the hydroxyl groups are free (see supplementary data Fig. S1).
Fig. 4.
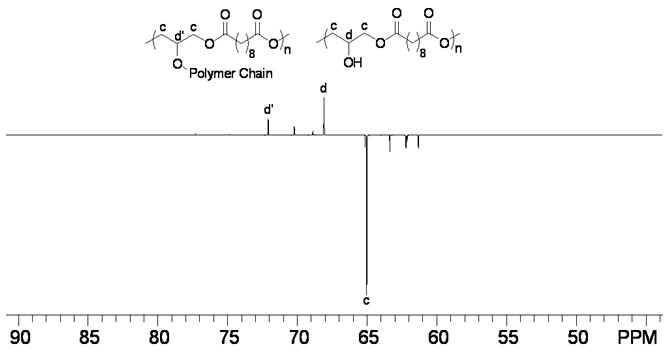
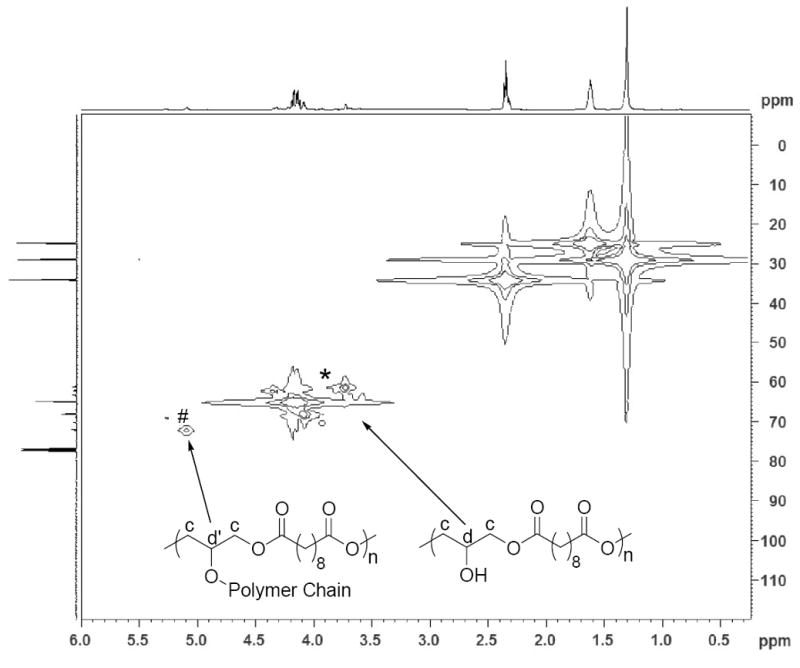
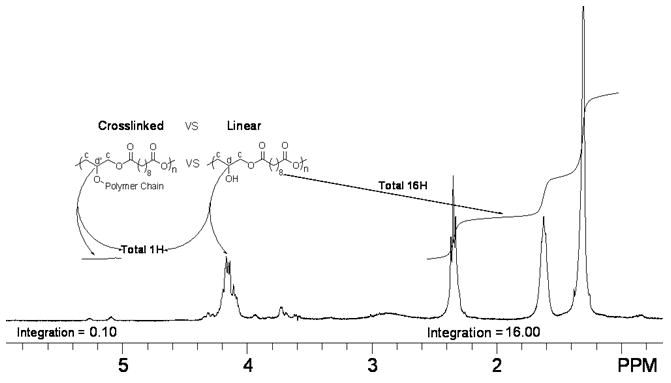
(A) 13C DEPT 135 NMR spectrum of PSeD was used to distinguish the different carbons of PSeD. The positive peaks and negative peaks correspond to CH and CH2 groups, respectively. (B) The 1H-13C HETCOR NMR spectrum revealed the correlation of protons and carbons in PSeD. The strong peaks ‘*’ represented most NMR signals of the glyceryl moiety, which displayed at the normal chemical shift region of a diglyceride and indicated a mostly linear structure of PSeD. The weak signal ‘#’ revealed that the proton at 5.09 and 5.28 ppm was attached to the carbon at 72.06 ppm, which corresponded to the CH group of the glyceryl unit in the ester form (d′). (C) According to the relative chemical shifts and the results of 13C DEPT 135 and 1H-13C HETCOR NMR spectra, the protons from 5.09-5.28 ppm were deduced to be the secondary hydrogen d′, which corresponds to the triglyceride, branched part of PSeD. Thus, the relative integration (0.1) of protons from 5.09 to 5.28 ppm revealed the low crosslinking of PSeD (approximately 10%).
The characteristic functional groups of PSeD were corroborated by FTIR investigation (Fig. 5). The ester bond in the backbone and the presence of hydroxyl groups on the side chain showed a sharp, intense C=O stretch at 1740 cm−1 and a broad, intense O-H stretch at 3461 cm−1, respectively.
Fig. 5.

The FTIR spectrum of PSeD displayed the signal of expected functionalities: O-H bonds at 3461 cm−1and ester C=O bonds at 1740 cm−1.
3.3. Physical properties of PSeD and PGS prepolymer
In order to compare PSeD with PGS prepolymer synthesized by direct polycondensation of glycerol and sebacic acid, we prepared PGS prepolymer as previously reported [38] and characterized it under identical conditions as PSeD. The results are summarized in Table 2. The polymers' thermal properties were determined by DSC and TGA. Similar to PGS, the glass transition temperature (Tg) of PSeD was very low (< −100 °C), which suggested that, after curing, it would be an elastomer at ambient temperature. PSeD, however, showed higher melting (Tm) and crystallization (Tc) temperatures than PGS prepolymer. This was consistent with a linear structure for PSeD, different from the branched structure of PGS. Furthermore, the TGA experiment revealed that PSeD had a high decomposition temperature (Td) at 382.9 °C and therefore, it could be used in a wide temperature range.
Table 2.
Comparison of the properties of PSeD and PGS prepolymer.
| Compound | Mn (kDa) | PDI | Crosslinking (%)a | Tm (°C) | Tc (°C) | Tg (°C) | Td (°C) |
|---|---|---|---|---|---|---|---|
| PSeD | 16.6 | 2.5 | 10 | 26.4, 52.3 | 1.2 | Noneb | 382.9 |
| PGS | 9.0 | 9.3 | 55 | -3.4, 15.0, 33.3 | −12.0 | Noneb | c |
According to the analysis of NMR spectra. For PSeD please see Figure 4C. For PGS please see Figure S1 in supporting information.
No glass transition temperature was observed above −100 °C.
Not measured.
PSeD was designed to be a versatile intermediate for further functionalization, so we tested its solubility in various solvents at ambient conditions. PSeD was soluble in most common organic solvents except those with low polarities (Table 3), which made it easy to be cast into various shapes and ready for further functionalization. It also indicated that the polymer had few crosslinks [31, 39]. The polymer was insoluble in water, making it a candidate for a tissue engineering scaffold and various implantation applications. Moreover, PSeD can be stored for more than one year without significant change in solubility. In contrast, PGS became insoluble after strorage under the same conditions for 3 months, likely due to crosslinking of the polymer. This improved property makes PSeD more amenable to practical use.
Table 3.
Solubility of PSeD in various solvents.
| Solventa | Hexane | Et2O | CH2Cl2 | THF | CHCl3 | Ethyl acetate |
| Solubilityb | − | − | + | + | + | + |
| Solventa | Dioxane | Acetone | MeOH | EtOH | DMF | H2O |
| Solubilityb | + | + | + | + | + | − |
Ranked according to the polarity.
insoluble; +: soluble
3.4. Functionalization of PSeD
To demonstrate the potential ability to use the -OH groups of PSeD for biofunctionalization, we investigated the coupling reaction between PSeD and glycine, a representative for biomolecules. PSeD readily reacted with N-tert-butoxylcarbonyl (Boc)-protected glycine in the presence of excess DCC and a catalytic amount of DMAP at room temperature to yield PSeD-glycinate (Fig. 6, Mn = 25.1 kDa, PDI = 1.4) with around 90% glycination (according to the relative integrations in 1HNMR spectrum; see supplementary data Fig. S2A).
Fig. 6.

A model reaction for the conjugation of biomolecules to PSeD.
To further explore the potential of PSeD for derivatization and to illustrate the versatility of PSeD, we reacted PSeD with excess maleic anhydride at 110 °C in DMF by microwave-assisted esterification (Fig. 7). The reaction was performed for 30 min and produced PSeD-maleic monoester (Mn = 20.3 kDa, PDI = 2.0) with around 80% esterification (according to the relative integrations in 1HNMR spectrum; see supplementary data Fig. S3A).
Fig. 7.

A model reaction for further derivatization of PSeD. Maleic monoester derivative of PSeD was readily synthesized by microwave-assisted esterification, which yielded a polymer with free α, β-unsaturated carboxylic groups and provided more approaches to further functionalization.
3.5. Curing PSeD and the mechanical properties of the resultant elastomer
To keep crosslinking low and elasticity high for the final network, PSeD was cured with a small amount sebacic acid (1.1% by weight) under moderate vacuum at 120 °C, Tensile tests of the cured PSeD revealed a stress-strain curve characteristic of an elastomeric and tough material (Table 4, Fig. 8A). The Young's modulus of the original linear region was found to be 1.57 ± 0.48 MPa and the ultimate tensile stress was found to be 1.83 ± 0.06 MPa. The elongation at break was up to 409 ± 29 %. Moreover, cyclic tensile testing was performed with 50% strain for 5 cycles (Fig. 8B). Only limited hysteresis was observed.
Table 4.
Mechanical properties of cured PSeD and PGS.
| Elastomer | Young's modulus (MPa) | Tensile strength (MPa) | Elongation (%) |
|---|---|---|---|
| PSeD | 1.57 ± 0.48 | 1.83 ± 0.06 | 409 ± 29 |
| PGSa | 0.282 ± 0.025 | 0.5 | 267 ± 59.4 |
Representative data of PGS is shown [1]. PGS and PSeD were cured via different mechanisms; the crosslinking density were both low, but unlikely to be identical.
Fig. 8.


Typical tensile stress vs. strain curve of cured PSeD elastomer. (A) Simple tensile test to failure. (B) Cyclic tensile test at 50% for 5 cycles. Note the reversibility of the deformation after the first cycle.
3.6. Biocompatibility of PSeD
The biocompatibility of PSeD was tested by examining the metabolic activity of nonimmortalized baboon smooth muscle cells seeded onto PSeD-coated 24-well plates. TCPS was used as a control. The cells showed similar long spindle morphology on PSeD and TCPS (Fig. 9A and B). Identical numbers of cells were seeded on all surfaces. From day 1 to day 3, MTT assays revealed a significantly higher number of metabolically active cells (P < 0.05) on PSeD surfaces than on TCPS surfaces (Fig. 9C). On day 4, cells approached confluence on both PSeD and TCPS surfaces and there was no difference in cell number statistically. This demonstrated that PSeD was at least as cytocompatible as TCPS in vitro.
Fig. 9.



Baboon smooth muscle cells cultured on PSeD surface exhibited normal morphology, similar to that on TCPS. Representative phase contrast photomicrographs of baboon smooth muscle cells at day 3 after seeding in PSeD-coated wells (A) and TCPS wells (B) (magnification 100×, scale bar = 300 μm). (C) MTT assay of baboon smooth muscle cells in PSeD wells and TCPS wells showed that there were more metabolically active cells on PSeD than on TCPS in the first three days. Data represent mean ± SD. * Statistical significance (p < 0.05). Normalized values shown.
4. Discussion
Functionalized polyesters are promising new biomaterials because they add tunable biodegradability, physical, chemical, and biological properties to established polyesters such as polylactide and polyglycolide. The free functional groups allow facile modifications with biomolecules that lead to versatile biomaterials with controlled interactions with cells and tissues. Efficient synthesis of functionalizable polyesters is, however, still a major challenge that greatly limits the availability and widespread applications of biofunctionalized synthetic polyesters. Here we have presented a simple strategy to prepare functionalizable polyester with free hydroxyl groups. The epoxide ring-opening polymerization between a dicarboxylic acid and the corresponding diglycidyl ester produces the target functionalizable polyester in one step without the need for protection and deprotection (Fig. 1, Table 1). The synthetic strategy is versatile and can be used with other diacids and diepoxides to yield various linear functionalizable polyesters. Compared to other current approaches to functionalized polyester [8-10, 15, 16], it is simpler and more efficient.
PSeD exhibits several unique advantages over PGS prepolymer synthesized by polycondensation of glycerol and sebacic acid (Table 2): better defined structure with more free -OH groups and higher linearity (10% vs. 55% branching); higher molecular weight (16.6 kD vs. 9.0 kD); narrower polydispersity (2.5 vs. 9.3); and longer shelf time (>1 year vs. 3 months). In addition, the direct polycondensation for PGS requires higher temperature (120 °C) and vacuum (40 mTorr) to remove H2O and accelerate the reaction [1]. PSeD synthesis proceeds at lower temperature (95 °C) with no need for vacuum to remove small molecule byproducts (Fig. 1).
The more defined structure of PSeD provides a suitable platform for further biofunctionalization. We selected glycine as the representative for biomolecules. The DCC coupling reaction ran smoothly and produced PSeD glycinate in high yield. Since many biomolecules, such as amino acids, peptides, proteins, glycans, and biotin contain free carboxylates, the reaction described here (Fig. 6) would serve as an effective bioconjugation route for PSeD. In addition, microwave-assisted esterification with maleic anhydride provides PSeD maleic monoester with good purity and yield (Fig. 7). This is a much more efficient synthetic route than a similar coupling reaction reported between hydroxylated polyester and succinic anhydride (reaction time: 30 min vs. 10 days) [12]. The free α, β-unsaturated carboxylic acid groups in PSeD-maleic monoester can easily react with a biomolecule through esterification, amidation, Michael addition, and photo-crosslinking [12, 40-42]. This further enriches the variety of chemistry that can be performed to functionalize PSeD to obtained biomaterials with unique mechanical and biological properties.
For applications in tissue engineering, biomaterials that mimic the mechanical aspect of native extracellular matrix by transducing mechanical stimuli to cells and tissues may be advantageous [43-46]. Since many soft tissues such as blood vessels and lungs are elastic, several biodegradable elastomers have been developed [1, 47-56]. Among them, crosslinked elastomers made from aliphatic polyesters have received much attention recently [24, 49, 51, 56]. They show good biocompatibility and biodegradability. Their mechanical properties are in the range of soft tissues and they primarily degrade by surface erosion, retaining their structural integrity and mechanical stability during degradation in vivo [57]. Most crosslinked polyester elastomers are typically synthesized by random polycondensation [1, 49-53]. This method produces prepolymers with relatively low molecular weight, high polydispersity, undefined branched structure, and are prone to premature crosslinking. This compromises the mechanical properties of the elastomer. Various approaches from several different laboratories have been used to improve the mechanical properties of PGS; these include modulating monomer feeding ratios, curing conditions, and crosslinking reagents [24, 50, 58-60], and all of these relied on polycondensation as the synthetic method. Consequently, the resultant polymers have mechanical properties very similar to the original PGS [1]. We reasoned that polymeric networks produced by crosslinking linear functionalized polyester with higher molecular weight and narrower polydispersity would provide more elastic materials than their counterparts obtained by direct polycondensation. Accordingly, we investigated the crosslinking of PSeD with sebacic acid to produce a biodegradable elastomer that would be structurally related to PGS. The motivation was to maintain the good biocompatibility of PGS while improving its mechanical properties. As expected, sebacic-acid cured PSeD demonstrated a much tougher (approximately 5 times greater area under curve) and more elastic properties than cured PGS (Table 4, Fig. 8): 5 times higher Young's modulus; > 3 times higher tensile strength; and greater maximum strain at break. It therefore resembles bovine ligament elastin mechanically (tensile strength and Young's modulus of 2 MPa and 1.1 MPa, respectively [61]). The strain at break was higher than that of most soft tissues (for examples, arteries and veins can be elongated up to 260% [62]). To the best of our knowledge, the elongation at break of the cured PSeD is one of the highest among crosslinked polyester elastomers used in biomedical applications. In addition, the cyclic tensile test revealed the elastomer's ability to recover from deformations (Fig. 8B), indicating that it is suitable for applications in a mechanically dynamic environment. Moreover, the mechanical properties of PSeD can be further modulated via controlling curing conditions and selecting different types and amount of crosslinking reagents (data will be reported elsewhere).
The in vitro cytocompatibility of PSeD was evaluated by monitoring metabolic activities of nonimmortalized baboon smooth muscle cells seeded on PSeD surfaces. Examination of the morphology of cells and the number of metabolically active cells on PSeD film suggested that cells on PSeD surfaces proliferate at least as well as those on TCPS surfaces (Fig. 9). This suggests that PSeD may be suitable for many biomedical applications including tissue engineering, wound healing, and drug delivery. We are currently investigating the in vivo biocompatibility of PSeD.
PSeD and PGS exhibit several Tm values and one distinct Tc in their thermograms. This could be caused by melt-recrystallization during the DSC heating cycle. The polymer was first heated from room temperature to 100 °C, then cooled to -100 °C before being heated again. Cooling of the polymer might yield a less thermodynamically stable form. Upon heating the crystalline domains melted and the molten polymers recrystallized into a more stable form that subsequently melted. Because the two melting transitions were adjacent to each other, a distinct Tc was not observed. We are investigating possible deconvolution of the transitions using advanced software to further characterize the thermal behavior of the polymer.
5. Conclusions
A functionalizable polyester bearing free hydroxyl groups was designed and successfully synthesized in two simple steps from commercially available starting materials. The synthetic route is practical, featuring easily synthesized monomers, mild polymerization conditions, and no protection and deprotection steps. The usefulness of this approach has been demonstrated by the tougher and more elastic nature of the cured polymer as compared to the first generation PGS, and the ready functionalization with glycine. The enhanced mechanical properties and easy derivatizations of PSeD were enabled by virtue of its more defined polymer structure than that of PGS. Initial evaluation using nonimmortalized cells showed PSeD to be cytocompatible in vitro. The synthetic strategy is general and can be used to synthesize other glycerol-based linear polyesters. We are currently investigating modification of PSeD with oligopeptides, optimizing the curing conditions, and expanding the application of this synthetic platform to other diacids. We expect this new design platform will lead to a diverse family of biodegradable and bioactive polymers with versatile mechanical, physical, chemical, and biological properties tailored for a wide range of biomedical applications.
Supplementary Material
Acknowledgments
The authors are grateful for the financial support from NHLBI R01HL 089658.
Footnotes
Functionalizable polymer is defined as a polymer with functional groups that readily react with biomolecules and functionalized biomaterial as one already modified with biomolecules.
Supplementary Data: The 1H NMR spectrum of PGS, the 1H NMR and FTIR spectra of PSeD maleic monoester, the 1H NMR, 13C NMR, and FTIR spectra of PSeD glycinate are available as supplementary data in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang YD, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. 2002;20(6):602–6. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 2.Albertsson AC, Varma IK. Degradable Aliphatic Polyesters. Berlin: Springer-Verlag Berlin; 2002. Aliphatic polyesters: Synthesis, properties and applications; pp. 1–40. [Google Scholar]
- 3.Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32(89):762–98. [Google Scholar]
- 4.Yang SF, Leong KF, Du ZH, Chua CK. The design of scaffolds for use in tissue engineering. Part 1. Traditional factors. Tissue Eng. 2001;7(6):679–89. doi: 10.1089/107632701753337645. [DOI] [PubMed] [Google Scholar]
- 5.Lavik E, Langer R. Tissue engineering: current state and perspectives. Appl Microbiol Biotechnol. 2004;65(1):1–8. doi: 10.1007/s00253-004-1580-z. [DOI] [PubMed] [Google Scholar]
- 6.Philip S, Keshavarz T, Roy I. Polyhydroxyalkanoates: biodegradable polymers with a range of applications. J Chem Technol Biotechnol. 2007;82(3):233–47. [Google Scholar]
- 7.Chen GQ, Wu Q. The application of polyhydroxyalkanoates as tissue engineering materials. Biomaterials. 2005;26(33):6565–78. doi: 10.1016/j.biomaterials.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 8.Lecomte P, Riva R, Schmeits S, Rieger J, Van Butsele K, Jerome C, et al. New prospects for the grafting of functional groups onto aliphatic polyesters. Ring-opening polymerization of alpha- or gamma-substituted epsilon-caprolactone followed by chemical derivatization of the substituents. Macromol Symp. 2006;240:157–65. [Google Scholar]
- 9.Parrish B, Emrick T. Strategies in Aliphatic Polyester Synthesis for Biomaterial and Drug Delivery Applications. ACS Symp Ser. 2006;939:248–66. Degradable. [Google Scholar]
- 10.Williams CK. Synthesis of functionalized biodegradable polyesters. Chem Soc Rev. 2007;36(10):1573–80. doi: 10.1039/b614342n. [DOI] [PubMed] [Google Scholar]
- 11.Vert M. Aliphatic polyesters: Great degradable polymers that cannot do everything. Biomacromolecules. 2005;6(2):538–46. doi: 10.1021/bm0494702. [DOI] [PubMed] [Google Scholar]
- 12.Noga DE, Petrie TA, Kumar A, Weck M, Garcia AJ, Collard DM. Synthesis and modification of functional poly(lactide) copolymers: Toward biofunctional materials. Biomacromolecules. 2008;9(7):2056–62. doi: 10.1021/bm800292z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Studer P, Larras V, Riess G. Amino end-functionalized poly(ethylene oxide)-block-poly(methylidene malonate 2.1.2) block copolymers: synthesis, characterization, and chemical modification for targeting purposes. Eur Polym J. 2008;44(6):1714–21. [Google Scholar]
- 14.Kwon GS, Okano T. Polymeric micelles as new drug carriers. Adv Drug Del Rev. 1996;21(2):107–16. [Google Scholar]
- 15.Taniguchi I, Kuhlman WA, Mayes AM, Griffith LG. Functional modification of biodegradable polyesters through a chemoselective approach: application to biomaterial surfaces. Polym Int. 2006;55(12):1385–97. [Google Scholar]
- 16.Jerome C, Lecomte P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv Drug Del Rev. 2008;60(9):1056–76. doi: 10.1016/j.addr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Gerhardt WW, Noga DE, Hardcastle KI, Garcia AJ, Collard DM, Weck M. Functional lactide monomers: Methodology and polymerization. Biomacromolecules. 2006;7(6):1735–42. doi: 10.1021/bm060024j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leemhuis M, van Nostrum CF, Kruijtzer JAW, Zhong ZY, ten Breteler MR, Dijkstra PJ, et al. Functionalized poly(alpha-hydroxy acid)s via ring-opening polymerization: Toward hydrophilic polyesters with pendant hydroxyl groups. Macromolecules. 2006;39(10):3500–8. [Google Scholar]
- 19.Hao QH, Yang J, Li QB, Li Y, Jia L, Fang Q, et al. New facile approach to novel water-soluble aliphatic poly(butylene tartarate)s bearing reactive hydroxyl pendant groups. Biomacromolecules. 2005;6(6):3474–80. doi: 10.1021/bm050317x. [DOI] [PubMed] [Google Scholar]
- 20.Ponsart S, Coudane J, Vert M. A novel route to poly(epsilon-caprolactone)-based copolymers via anionic derivatization. Biomacromolecules. 2000;1(2):275–81. doi: 10.1021/bm005521t. [DOI] [PubMed] [Google Scholar]
- 21.Zhang SP, Yang J, Liu XY, Chang JH, Cao AM. Synthesis and characterization of poly(butylene succinate-co-butylene malate): A new biodegradable copolyester bearing hydroxyl pendant groups. Biomacromolecules. 2003;4(2):437–45. doi: 10.1021/bm0201183. [DOI] [PubMed] [Google Scholar]
- 22.Deng C, Chen XS, Sun J, Lu TC, Wang WS, Jing XB. RGD peptide grafted biodegradable amphiphilic triblock copolymer poly(glutamic acid)-b-poly(L-lactide)-b-poly(glutamic acid): Synthesis and self-assembly. J Polym Sci, Part A: Polym Chem. 2007;45(15):3218–30. [Google Scholar]
- 23.Motlagh D, Yang J, Lui KY, Webb AR, Ameer GA. Hernocompatibility evaluation of poly(glycerol-sebacate) in vitro for vascular tissue engineering. Biomaterials. 2006;27(24):4315–24. doi: 10.1016/j.biomaterials.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Chen QZ, Bismarck A, Hansen U, Junaid S, Tran MQ, Harding SE, et al. Characterisation of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue. Biomaterials. 2008;29(1):47–57. doi: 10.1016/j.biomaterials.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Gao J, Crapo P, Nerern R, Wang YD. Co-expression of elastin and collagen leads to highly compliant engineered blood vessels. J Biomed Mater Res A. 2008;85A(4):1120–8. doi: 10.1002/jbm.a.32028. [DOI] [PubMed] [Google Scholar]
- 26.Neeley WL, Redenti S, Klassen H, Tao S, Desai T, Young MJ, et al. A microfabricated scaffold for retinal progenitor cell grafting. Biomaterials. 2008;29(4):418–26. doi: 10.1016/j.biomaterials.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radisic M, Park H, Chen F, Salazar-Lazzaro JE, Wang YD, Dennis R, et al. Biomirnetic approach to cardiac tissue engineering: Oxygen carriers and channeled scaffolds. Tissue Eng. 2006;12(8):2077–91. doi: 10.1089/ten.2006.12.2077. [DOI] [PubMed] [Google Scholar]
- 28.Sales VL, Engelmayr GC, Johnson JA, Gao J, Wang YD, Sacks MS, et al. Protein precoating of elastomeric tissue-engineering scaffolds increased cellularity, enhanced extracellular matrix protein production, and differentially regulated the phenotypes of circulating endothelial progenitor cells. Circulation. 2007;116(11):I55–I63. doi: 10.1161/CIRCULATIONAHA.106.6806637. [DOI] [PubMed] [Google Scholar]
- 29.Redenti S, Neeley WL, Rompani S, Saigal S, Yang J, Klassen H, et al. Engineering retinal progenitor cell and scrollable poly(glycerol-sebacate) composites for expansion and subretinal transplantation. Biomaterials. 2009;30(20):3405–14. doi: 10.1016/j.biomaterials.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kline BJ, Beckman EJ, Russell AJ. One-step biocatalytic synthesis of linear polyesters with pendant hydroxyl groups. J Am Chem Soc. 1998;120(37):9475–80. [Google Scholar]
- 31.Kumar A, Kulshrestha AS, Gao W, Gross RA. Versatile route to polyol polyesters by lipase catalysis. Macromolecules. 2003;36(22):8219–21. [Google Scholar]
- 32.Gao J, Kim YM, Coe H, Zern B, Sheppard B, Wang YD. A neuroinductive biomaterial based on dopamine. Proc Natl Acad Sci U S A. 2006;103(45):16681–6. doi: 10.1073/pnas.0606237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gumera CB, Wang Y. Modulating neuronal responses by controlled integration of acetylcholine-like functionalities in biomimetic polymers. Adv Mater. 2007;19(24):4404–9. [Google Scholar]
- 34.Gao J, Ensley AE, Nerem RM, Wang YD. Poly(glycerol sebacate) supports the proliferation and phenotypic protein expression of primary baboon vascular cells. J Biomed Mater Res A. 2007;83A(4):1070–5. doi: 10.1002/jbm.a.31434. [DOI] [PubMed] [Google Scholar]
- 35.Maerker G, Carmichael JF, Port WS. Glycidyl esters. I. Method of preparation and study of some reaction variables. J Org Chem. 1961;26:2681–8. [Google Scholar]
- 36.Kester EB, Gaiser CJ, Lazar ME. Glycidyl esters of aliphatic acids. J Org Chem. 1943;8(6):550–6. [Google Scholar]
- 37.Nishikubo T, Kameyama A. Addition-reactions of cyclic ethers with various carbonyl - compounds and their application for polymer synthesis. Prog Polym Sci. 1993;18(5):963–95. [Google Scholar]
- 38.Gao J, Crapo PM, Wang YD. Macroporous elastomeric scaffolds with extensive micropores for soft tissue engineering. Tissue Eng. 2006;12(4):917–25. doi: 10.1089/ten.2006.12.917. [DOI] [PubMed] [Google Scholar]
- 39.Kallinteri P, Higgins S, Hutcheon GA, St Pourcain CB, Garnett MC. Novel functionalized biodegradable polymers for nanoparticle drug delivery systems. Biomacromolecules. 2005;6(4):1885–94. doi: 10.1021/bm049200j. [DOI] [PubMed] [Google Scholar]
- 40.Hersel U, Dahmen C, Kessler H. RGD modified polymers: biomaterials for stimulated cell adhesion and beyond. Biomaterials. 2003;24(24):4385–415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 41.Jiao YP, Cui FZ. Surface modification of polyester biomaterials for tissue engineering. Biomedical Materials. 2007;2(4):R24–R37. doi: 10.1088/1748-6041/2/4/R02. [DOI] [PubMed] [Google Scholar]
- 42.Ifkovits JL, Burdick JA. Review: Photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Eng. 2007;13(10):2369–85. doi: 10.1089/ten.2007.0093. [DOI] [PubMed] [Google Scholar]
- 43.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260(5110):920–6. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 44.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428(6982):487–92. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 45.Zimmermann WH. TISSUE ENGINEERING Polymers flex their muscles. Nature Materials. 2008;7(12):932–3. doi: 10.1038/nmat2328. [DOI] [PubMed] [Google Scholar]
- 46.Dado D, Levenberg S. Cell-scaffold mechanical interplay within engineered tissue. Semin Cell Dev Biol. 2009;20(6):656–64. doi: 10.1016/j.semcdb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Poirier Y, Nawrath C, Somerville C. Production of polyhydroxyalkanoates, a family of biodegradable plastics and elastomers, in bacteria and plants. Bio-Technology. 1995;13(2):142–50. doi: 10.1038/nbt0295-142. [DOI] [PubMed] [Google Scholar]
- 48.Amsden B. Curable, biodegradable elastomers: emerging biomaterials for drug delivery and tissue engineering. Soft Matter. 2007;3(11):1335–48. doi: 10.1039/b707472g. [DOI] [PubMed] [Google Scholar]
- 49.Yang J, Webb AR, Ameer GA. Novel citric acid-based biodegradable elastomers for tissue engineering. Adv Mater. 2004;16(6):511–6. [Google Scholar]
- 50.Nijst CLE, Bruggeman JP, Karp JM, Ferreira L, Zumbuehl A, Bettinger CJ, et al. Synthesis and characterization of photocurable elastomers from poly(glycerol-co-sebacate) Biomacromolecules. 2007;8:3067–73. doi: 10.1021/bm070423u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruggeman JP, Bettinger CJ, Nijst CLE, Kohane DS, Langer R. Biodegradable xylitol-based polymers. Adv Mater. 2008;20(10):1922–7. [Google Scholar]
- 52.Bettinger CJ, Bruggeman JP, Borenstein JT, Langer RS. Amino alcohol-based degradable poly(ester amide) elastomers. Biomaterials. 2008;29(15):2315–25. doi: 10.1016/j.biomaterials.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruggeman JP, de Bruin BJ, Bettinger CJ, Langer R. Biodegradable poly(polyol sebacate) polymers. Biomaterials. 2008;29(36):4726–35. doi: 10.1016/j.biomaterials.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dey J, Xu H, Shen JH, Thevenot P, Gondi SR, Nguyen KT, et al. Development of biodegradable crosslinked urethane-doped polyester elastomers. Biomaterials. 2008;29(35):4637–49. doi: 10.1016/j.biomaterials.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guan JJ, Sacks MS, Beckman EJ, Wagner WR. Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: synthesis, characterization and cytocompatibility. Biomaterials. 2004;25(1):85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- 56.Younes HM, Bravo-Grimaldo E, Amsden BG. Synthesis, characterization and in vitro degradation of a biodegradable elastomer. Biomaterials. 2004;25(22):5261–9. doi: 10.1016/j.biomaterials.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 57.Wang YD, Kim YM, Langer R. In vivo degradation characteristics of poly(glycerol sebacate) J Biomed Mater Res A. 2003;66A(1):192–7. doi: 10.1002/jbm.a.10534. [DOI] [PubMed] [Google Scholar]
- 58.Liu QY, Tian M, Ding T, Shi R, Zhang LQ. Preparation and characterization of a biodegradable polyester elastomer with thermal processing abilities. J Appl Polym Sci. 2005;98(5):2033–41. [Google Scholar]
- 59.Liu QY, Tian M, Shi R, Zhang LQ, Chen DF, Tian W. Structure and properties of thermoplastic poly(glycerol sebacate) elastomers originating from prepolymers with different molecular weights. J Appl Polym Sci. 2007;104(2):1131–7. [Google Scholar]
- 60.Liu QY, Tan TW, Weng JY, Zhang LQ. Study on the control of the compositions and properties of a biodegradable polyester elastomer. Biomedical Materials. 2009;4(2):9. doi: 10.1088/1748-6041/4/2/025015. [DOI] [PubMed] [Google Scholar]
- 61.Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Elastic proteins: biological roles and mechanical properties. Philos Trans R Soc Lond B Biol Sci. 2002;357(1418):121–32. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee MC, Haut RC. Strain rate effects on tensile failure properties of the common carotid-artery and jugular vein of ferrets. J Biomech. 1992;25(8):925–7. doi: 10.1016/0021-9290(92)90232-p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.