

Abstract
The hypoxia-inducible factor (HIF)-1 is critically involved in the cellular adaptation to a decrease in oxygen availability. The influence of HIF-1α for the development of cardiac hypertrophy and cardiac function that occurs in response to sustained pressure overload has been mainly attributed to a challenged cardiac angiogenesis and cardiac hypertrophy up to now. Hif-1α +/+ and Hif-1α +/− mice were studied regarding left ventricular hypertrophy and cardiac function after being subjected to transverse aortic constriction (TAC). After TAC, both Hif-1α +/+ and Hif-1α +/− mice developed left ventricular hypertrophy with increased posterior wall thickness, septum thickness and increased left ventricular weight to a similar extent. No significant difference in cardiac vessel density was observed between Hif-1α +/+ and Hif-1α +/− mice. However, only the Hif-1α +/− mice developed severe heart failure as revealed by a significantly reduced fractional shortening mostly due to increased end-systolic left ventricular diameter. On the single cell level this correlated with reduced myocyte shortenings, decreased intracellular Ca2+-transients and SR-Ca2+ content in myocytes of Hif-1a +/− mice. Thus, HIF-1α can be critically involved in the preservation of cardiac function after chronic pressure overload without affecting cardiac hypertrophy. This effect is mediated via HIF-dependent modulation of cardiac calcium handling and contractility.
Electronic supplementary material
The online version of this article (doi:10.1007/s00424-009-0748-x) contains supplementary material, which is available to authorized users.
Keywords: Hypoxia, Transverse aortic constriction, Cardiac hypertrophy, Hypoxia-inducible factor, Heart failure
Introduction
An intact oxygen homeostasis in the heart is important not only for cardiac development but also for adaptation of the adult myocardium to a decrease in oxygen supply [18, 21]. Hypoxia affects cardiac vessel density and cell fate of cardiomyocytes [10]. Like in other tissues, hypoxia initiates a hypoxia-inducible gene expression programme, including genes regulating angiogenesis, anaerobic glycolysis, etc., in the heart [14]. The hypoxia-inducible factor (HIF)-1 is the transcriptional master regulator for these hypoxia-inducible genes [36]. At the molecular level, hypoxia is sensed by three oxygen-, iron- and 2-oxoglutarate-dependent prolyl-4-hydroxylase domain (PHD) enzymes. The basis for the hypoxia-induced stability and activity of HIF-1 is the oxygen-dependent, PHD-mediated hydroxylation of distinct prolyl residues of the HIF-1α subunit [11, 12, 25]. Hydroxylated HIF-1α is rapidly ubiquitinated by the von Hippel–Lindau E3 ligase complex and degraded in the proteasome [26].
The ventricular geometry of the heart is a major determinant for myocardial oxygen consumption. Accordingly, myocardial oxygen consumption is proportional to ventricular wall tension, which is increased as a consequence of pressure overload [17]. Sustained pressure overload, which occurs for example in chronic arterial hypertension or aortic valve stenosis, initially causes left ventricular hypertrophy and subsequently heart failure. Regarding cardiac pathophysiology, the role of HIF-1α for cardiac adaptation to ischemia has been predominantly analyzed so far [5, 16]. Less is known, however, about the functional involvement of HIF-1α for the adaptation to sustained pressure overload, although increased protein levels of HIF-1α have been observed in heart samples from various models of pathologic cardiac hypertrophy [19].
Homozygous inactivation of the Hif1α gene causes embryonic lethality because of failed cardiac and vascular development, whereas heterozygous Hif-1α +/− mice develop normally. Partial HIF-1α deficiency in Hif-1α+/− mice results in an impaired response to continuous or intermittent systemic hypoxia including pulmonary hypertension [33, 38]. However, under resting conditions Hif-1α+/− mice do not demonstrate an appreciable cardiovascular phenotype. This is in contrast to HIF-1α cardiac-specific knockout mice (MCL2v-Cre x Hif-1α loxP mice), which present a reduction in cardiac vascularity already under resting conditions [31]. Cardiac vessel density is critically involved in the development of cardiac hypertrophy in chronic pressure overload. To gain insight if HIF-1α affects cardiac hypertrophy and function in response to chronic pressure overload independent from cardiac vascularity, Hif-1α+/− mice were used in a model of transverse aortic constriction (TAC). Ca2+ handling is critically involved in mechanical load-dependent cardiac function. Therefore, Hif-1α+/+ and Hif-1α+/− mice were analyzed regarding indicators for cardiac hypertrophy in vivo (posterior wall thickness (PWT), septum thickness (ST), left ventricular weight (LVW) and cardiac angiogenesis), heart failure (left ventricular endsystolic (LVESD) and enddiastolic diameter (LVEDD), fractional shortening (FS)) and myocyte function in vitro (cell shortening and Ca2+-transients) after TAC.
Materials and methods
Animals and surgical intervention
All protocols regarding animal experimentation were approved by the Niedersächsische Landesamt für Verbraucherschutz und Lebensmittelsicherheit (33.42502-105/06). Surgical intervention was performed with littermate mice that were either WT or heterozygous for the Hif-1α mutant allele in which exon 2 has been replaced with a neo R gene. Hif-1α +/− mice were kindly provided by R. Johnson (University of California, USA) and are described in [30]. HIF-1α protein levels in the heart of the Hif-1α +/− mice were reduced by 43% ± 18.7% as determined by immunoblots. Pressure overload was induced by TAC in 12–14-week-old female and 8–10-week-old male mice. Mice were anaesthetized by intraperitoneal injection of a mixture of xylazine and ketamine. The aorta was constricted with polyviolene non-absorbable braided nylon strings (5-0 USP) using blunted 25-gauge (male mice) and 26-gauge (female mice) needles as placeholders that were removed after ligation. After aortic constriction, the chest was closed and mice were allowed to recover from anaesthesia. Sham-operated animals were anaesthetized and handled like the TAC-operated animals with the exception of the aortic constriction. The following number of animals were treated with TAC or sham and underwent follow up analysis by echocardiography for 11 weeks: TAC: male Hif-1α+/+ n = 7, male Hif-1α+/− n = 3, female Hif-1α+/+ n = 5, female Hif-1α+/− n = 10; sham: male Hif-1α+/+ n = 5, male Hif-1α+/− n = 6, female Hif-1α+/+ n = 5, female Hif-1α+/− n = 6.
Echocardiography
Two-dimensional images and M-mode tracings were recorded from the parasternal long axis view at midpapillary level (Vevo 660™, VS-0 M-VE660 version 3.1, Visual Sonics). Heart rate, PWT, ST, LVESD and LVEDD were determined. FS of the left ventricle was defined as the LVEDD minus LVESD divided by the LVEDD. FS was used as marker for cardiac contractile function.
Immunofluorescence analysis
Hearts were fixed in 4% paraformaldehyde for 10–20 min. They were rinsed in phosphate-buffered saline (PBS), transferred to 5% and 15% sucrose in PBS, and embedded in Tissue Freeze Medium (Sakura Finetek Europe, NL). Cryosections of 16 μm thickness were prepared. Non-specific binding of antibodies was blocked by incubation with 1% bovine serum albumin (BSA) for 1 h before incubation with anti-vascular endothelial growth factor receptor (VEGFR)-2-antibodies (rat-anti-mouse; BD Pharmingen, San Diego, USA; 1:100), or monoclonal rat anti-mouse CD31 antibodies (SantaCruz Biotechnology, Santa Cruz, USA; 1:50). The sections were incubated with the primary antibodies for 1 h or at 4 C overnight, respectively. After rinsing, secondary antibodies (Alexa 488- or 555-conjugated goat anti-rat; Molecular Probes, Leiden, The Netherlands) were applied at 1:200 dilution for VEGFR-2 or 1:400 dilution for CD31 for 1 h. Cell nuclei were counterstained with DAPI. Then, sections were mounted under coverslips with Fluoromount-G (Southern Biotechnology Associates, Birmingham, GB). They were studied with epifluorescent microscope (Axio Imager Z1, Zeiss, Goettingen, Germany). Capillaries were counted in at least five different fields per mouse and quantified as capillaries/mm2.
Western blot
Heart tissue was rapidly homogenized in a buffer containing 4 M Urea, 140 mM Tris (pH 6.8), 1% SDS, 2% NP-40 and protease inhibitors (Roche). Protein concentrations were quantified (Bio-Rad, DC Protein Assay). For immunoblot analysis protein samples were resolved by SDS/PAGE and transferred onto nitrocellulose membranes (Amersham Biosciences) by semi-dry blotting (PeqLab). Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), HIF-1α, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were detected using mouse anti-SERCA (Dianova), rabbit anti-HIF-1α (Novus) and rabbit anti-GAPDH (Cell Signalling) antibodies followed by a goat horseradish peroxidase (HRP)-labelled anti-mouse or anti-rabbit antibody (Santa Cruz Biotechnology). Chemiluminescence detection of HRP was performed by incubation of the membranes with 100 mM Tris/HCl (pH 8.5), 2.65 mM H2O2, 0.45 mM luminol and 0.625 mM coumaric acid for 1 min at room temperature and analysed by imaging with a chemiluminescence camera (LAS3000; Fujifilm).
Isolation of cardiomyocytes, cardiomyocyte shortening and intracellular Ca2+-measurements
Isolation of adult mouse ventricular myocytes was carried out as described previously [23]. Briefly, hearts were excised from mice that were anaesthetized in a gas chamber with isoflurane. Hearts were mounted on a Langendorff-perfusion apparatus driven by gravity and perfused with nominally Ca2+-free Tyrode’s solution containing (in millimolar) NaCl, 115; KCl, 4.7; KH2PO4, 0.6; Na2HPO4; MgSO4, 1.2; NaHCO3, 12; KHCO3, 10; HEPES, 10; taurine, 30; 2,3-butanedionemonoxime, 10; glucose, 5.5 (pH 7.46) for 2–4 min at 37 C. Perfusion was then switched to the same solution containing Liberase blendzyme 1 (Roche) 0.25 mg/ml and Trypsin 0.14 mg/ml with perfusion continuing until the heart became flaccid (7–12 min). Ventricular tissue was removed, dispersed, filtered, and suspensions were rinsed several times. After Ca2+ reintroduction (stepwise increase to 0.8 mM), isolated myocytes were then plated onto superfusion chambers, with the glass bottoms treated with laminin to allow cell adhesion and used for immediate measurements.
Shortening and [Ca]i measurements were performed simultaneously and performed as reported previously [23] using a fluorescence detection system (IonOptix Corp., Milton, MA). Myocytes were loaded with fluo-3 by incubation with 10 μM of the acetoxymethyl ester (AM) form of the dye (Molecular Probes, Eugene, OR) for 20 min at room temperature in darkness. The dye was excited with a wavelength at 480 ± 15 nm using a 75 W xenon arc lamp (Ushio, Japan) on the stage of a Nikon Eclipse TE200-U inverted microscope. Emitted fluorescence was measured using a photomultiplier (at 535 ± 20 nm; IonOptix Corp., Milton, MA). From the raw fluorescence, ΔF/F0 was calculated by dividing through the baseline fluorescence (F0), after subtraction of the background fluorescence. Myocytes were field-stimulated (voltage 25% above threshold) at 1, 2 and 4 Hz and 37 C until steady-state was achieved, and only those cells exhibiting stable steady-state contractions were included in the study. To measure myocyte shortening, cells were simultaneously transilluminated by red light (>650 nm, to avoid interference with fluorescence measurements), and shortening was measured using a sarcomere length detection system (IonOptix Corp., Milton, MA). In case of determining SR Ca2+ content, cells were stimulated with 10 mM caffeine.
Statistical analysis
Data are shown as mean ± SEM. Multiple comparison was performed by two-way ANOVA analysis followed by the Bonferroni test for comparisons of means. In case of comparing the progress of cardiac hypertrophy (PWT and ST) and cardiac function (FS, LVEDD and LVESD) overtime paired t tests were performed. Values of p < 0.05 were considered to be statistically significant.
Results
No difference in angiogenesis between Hif-1α+/+ and Hif-1α+/− mice following sustained pressure overload
The vascular endothelial growth factor is a HIF-1 target gene and represents one of the major growth factors in reference to angiogenesis [24]. The spatio-temporal expression of its tyrosine kinase receptor VEGFR-2 in endothelial cells is indicative of an angiogenetic process. In cardiac-specific HIF-1α knockout mice, an influence of HIF-1α on cardiac vessel density after TAC has been described before [31]. To analyze if a partial deletion of the HIF-1α expression affects cardiac angiogenesis following sustained pressure overload, TAC intervention was performed in Hif-1α+/+ and Hif-1α+/− female and male mice. Subsequently, VEGRF-2 expression in Hif-1α+/+ and Hif-1α+/− mice was analyzed (Fig. 1a). The VEGFR-2 expression in Hif-1α+/+ versus Hif-1α+/− mice did not show any apparent differences, indicating that angiogenesis was not impaired in the heterozygous mice. This result was substantiated by the fact that vessel density as measured by CD31 staining did not differ comparing Hif-1α+/+ and Hif-1α+/− mice (Fig. 1b and c).
Fig. 1.
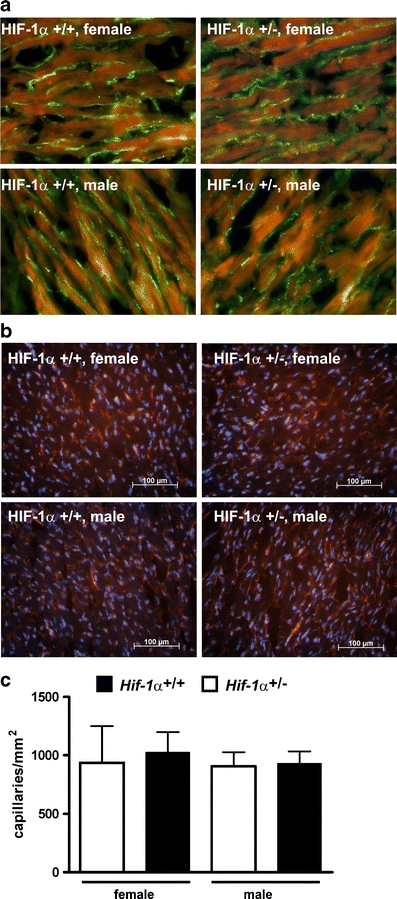
Cardiac VEGF R2 expression and vessel density in mice after transverse aortic constriction (TAC). Sustained pressure overload was induced in female and male Hif-1α+/+ and Hif-1α+/− mice by TAC. Hearts were excised and analyzed for angiogenesis by a VEGF R2 (green FITC-stained VEGF R2; red autofluorescence) and b CD31 (red TRITC-stained CD31; blue DAPI-stained nuclei) immunofluorescence analyses before and 1.5 weeks after TAC treatment. c Capillary density of three female and male HIF-1α +/+ and HIF-1α +/− mice after TAC treatment was determined based on the CD31 staining as shown in b
No difference in left ventricular hypertrophy in Hif-1α+/+ and Hif-1α+/− mice in response to sustained pressure overload
It has recently been hypothesized, that cardiac vessel density and cardiac hypertrophy are mutually related [13, 35]. Hif-1α+/+ and Hif-1α+/− mice developed cardiac hypertrophy to a similar extent after TAC. This finding is in line with the above-demonstrated data, in which we could not detect a difference in cardiac vessel density comparing TAC-treated Hif-1α+/+ with TAC-treated Hif-1α+/− mice. Left ventricular hypertrophy was indicated by significantly increased PWT and ST in TAC-treated female and male mice (Fig. 2a and b, respectively). In sham-operated animals, no significant differences in PWT or ST compared with pre-treatment values were observed (Suppl Fig. 1a and b). Additionally, post-mortem LVW and total heart weights (HW) were determined as indicators for cardiac hypertrophy (Fig. 3). In Hif-1α+/+ and Hif-1α+/− mice increased LVW/HW-ratios after TAC were observed. In line with the data obtained in the echocardiography analyses, there were no significant differences comparing the LVW/HW-ratios of Hif-1α+/+ and Hif-1α+/− mice.
Fig. 2.
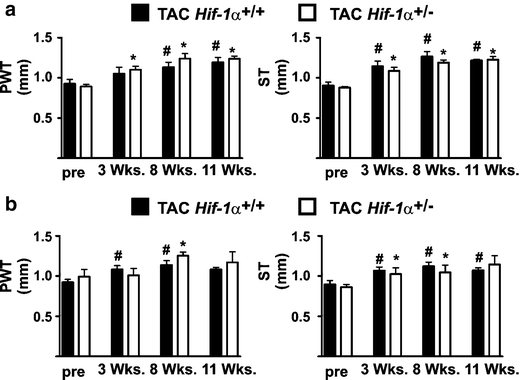
HIF-1α does not affect cardiac hypertrophy after transverse aortic constriction (TAC) in female and male mice. a Sustained pressure overload was induced in female Hif-1α+/+ and Hif-1α+/− mice by TAC. Subsequently, posterior wall thickness (PWT) and septum thickness (ST) were analyzed up to 11 weeks by echocardiography. # p < 0.05 (TAC-treated Hif-1α+/+ versus non-treated/pre Hif-1α+/+ mice), *p < 0.05 (TAC-treated Hif-1α+/− versus non-treated/pre Hif-1α+/− mice). b Sustained pressure overload was induced in male Hif-1α+/+ and Hif-1α+/− mice by TAC. Subsequently, PWT and ST were analyzed up to 11 weeks by echocardiography. # p < 0.05 (TAC-treated Hif-1α+/+ versus non-treated/pre Hif-1α+/+ mice), *p < 0.05 (TAC-treated Hif-1α+/− versus non-treated/pre Hif-1α+/− mice)
Fig. 3.
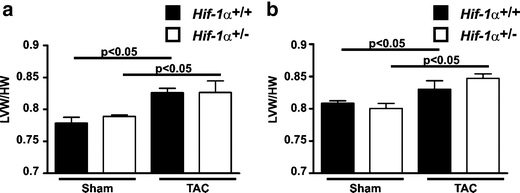
HIF-1α does not affect the left ventricular hypertrophy after transverse aortic constriction (TAC). Male (a) and female (b) Hif-1α+/+ and Hif-1α+/- mice underwent TAC or sham-intervention. Fifteen weeks after the intervention, the hearts were excised and left ventricular weights (LVW) as well as total heart weights (HW) were determined
Impaired cardiac function in Hif-1α+/− mice as a consequence of sustained pressure overload
In contrast to cardiac hypertrophy, there was an impact of HIF-1α on heart function after TAC as indicated by the FS of Hif-1α+/+ and Hif-1α+/− mice (Fig. 4). In Hif-1α+/+ mice, the FS did not significantly change over the course of eleven weeks after TAC compared with pre-treatment values. In Hif-1α+/− mice, however, FS was significantly reduced starting from 8 or 3 weeks after TAC in female (Fig. 4a) and male (Fig. 4b) mice, respectively, while reduced FS was mainly due to a significantly increased LVESD. The TAC-induced heart failure in the Hif-1α +/− mice was further substantiated by the finding that lung weights were significantly increased 15 weeks after TAC intervention in the Hif-1α+/− compared with Hif-1α+/+ mice (Hif-1α+/+ mice: 0.15 g ± 0.008; Hif-1α+/− mice: 0.18 g ± 0.011, p < 0.05). Since cardiac hypertrophy was similar in Hif-1α+/+ and Hif-1α+/− mice, it has to be assumed that the development of heart failure in the Hif-1α+/− mice occurred independent of cardiac hypertrophy. In sham-treated female and male mice, no significant difference in LVEDD, LVESD and FS was observed (Suppl Fig. 2a and b).
Fig. 4.
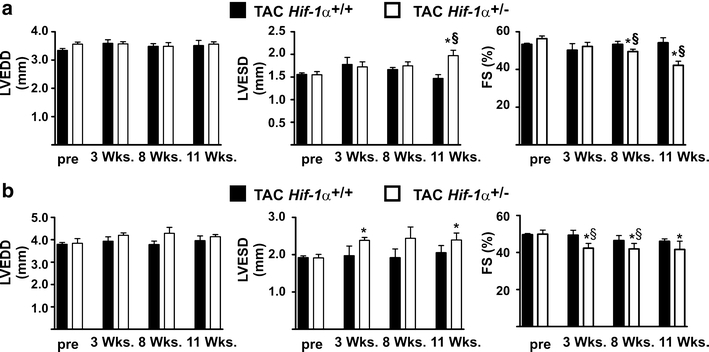
Impaired cardiac function in female and male Hif-1α+/− after transverse aortic constriction (TAC). a Sustained pressure overload was induced in female Hif-1α+/+ and Hif-1α+/− mice by TAC. Subsequently, left ventricular end-diastolic diameter (LVEDD), left ventricular endsystolic diameter (LVESD) and fractional shortening (FS) were analyzed up to 11 weeks by echocardiography. *p < 0.05 (TAC-treated Hif-1α+/− versus non-treated/pre Hif-1α+/− mice), § p < 0.05 (TAC-treated Hif-1α+/+ versus TAC-treated Hif-1α+/− mice). b Sustained pressure overload was induced in male Hif-1α+/+ and Hif-1α+/− mice by TAC. Subsequently, left ventricular end-diastolic diameter (LVEDD), left ventricular endsystolic diameter (LVESD) and fractional shortening (FS) were analyzed up to 11 weeks by echocardiography. *p < 0.05 (TAC-treated Hif-1α+/− versus non-treated/pre Hif-1α+/− mice), § p < 0.05 (TAC-treated Hif-1α+/+ versus TAC-treated Hif-1α+/− mice)
Impaired myocyte contractility and Ca2+ handling in cardiomyocytes of Hif-1α+/− mice
Intact Ca2+ handling is critically important for heart function under resting conditions as well as during adaptation towards sustained pressure overload [20]. Therefore, we determined mechanical performance and cytosolic Ca2+ dynamics of isolated cardiomyocytes. The reduced contractility observed as a consequence of pressure overload in the Hif-1α+/− mice by echocardiography was also detectable at the cellular level. The contractile behaviour of Hif-1α+/− cardiomyocytes, isolated 3 weeks after TAC from male mice, displayed a reduction in myocyte shortening compared with cardiomyocytes isolated from Hif-1α+/+ mice (Fig. 5a). This correlated with abnormalities in Ca2+-transients of the Hif-1α +/− cells. Hif-1α+/− cardiomyocytes isolated from TAC-treated mice demonstrated significantly reduced intracellular Ca2+-transients when stimulated with 1–4 Hz (Fig. 5b). Twitch relaxation and [Ca2+]i decline were significantly impaired and thus slower in cardiomyocytes isolated from TAC- versus sham-treated mice. However, there was no difference comparing cardiomyocytes isolated from Hif-1α+/+ or Hif-1α+/− mice (Fig. 5c and d). During excitation–contraction coupling, the Ca2+ entry via voltage dependent l-type Ca2+ channels triggers Ca2+ release from the sarcoplasmic reticulum (SR) through ryanodine receptors (Ca2+-induced Ca2+ release), which then leads to activation of the myofilaments and contraction of the cell. Alterations in the amplitude of the Ca2+ transients and of the subsequent contraction may result from changes in the SR Ca2+ content. In line with the decreased Ca2+ transients SR Ca2+ content was indeed significantly decreased in cardiomyocytes isolated of TAC-treated versus sham-treated HIF-1α +/− mice, whereas no significant difference was observed in cardiomyocytes isolated of TAC-treated versus sham-treated HIF-1α +/+ mice (Fig. 5e). The SERCA is the major pump, which is responsible for Ca2+ reuptake into the SR. SERCA protein levels, however, did not differ comparing HIF-1α +/+ and HIF-1α +/− mice (Fig. 5f). Taken collectively, HIF-1α+/− mice demonstrated significantly diminished Ca2+ transients and fractional shortenings after TAC treatment most likely due to a decrease in SR Ca2+ content.
Fig. 5.
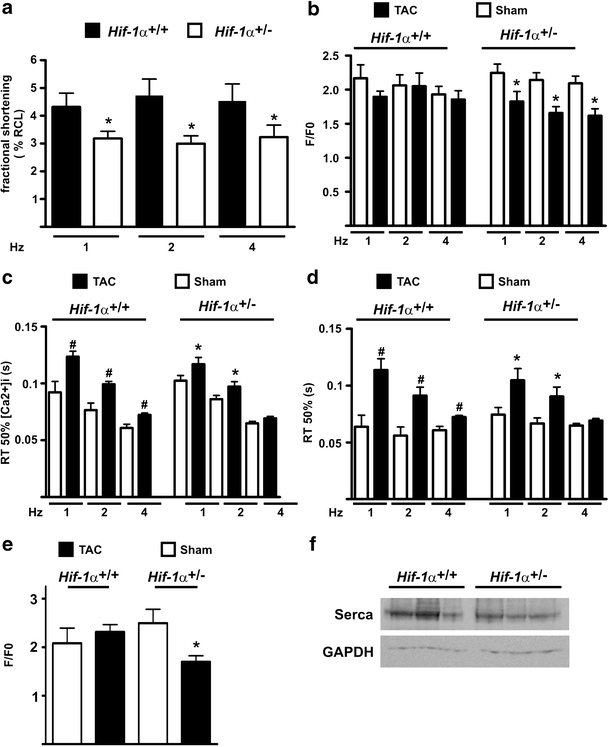
Impaired function of isolated Hif-1α+/- cardiomyocytes after transverse aortic constriction (TAC). Cardiomyocytes were isolated from male Hif-1α+/+ and Hif-1α+/− mice 3 weeks after TAC or sham-intervention. Cells were stimulated with 1–4 Hz and were analyzed for a fractional shortening, which was measured as percent resting cell length (RCL) after TAC treatment (*p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/+ versus TAC-treated Hif-1α+/− mice), b Ca2+ transient amplitude (*p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/− versus sham-treated Hif-1α+/− mice), c RT 50% [Ca2+]i decline (*p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/− versus sham-treated Hif-1α+/− mice, # p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/+ versus sham-treated Hif-1α+/+ mice), d RT 50% twitch relaxation (*p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/− versus sham-treated Hif-1α+/− mice, # p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/+ versus sham-treated Hif-1α+/+ mice) and e SR Ca2+ content measured by caffeine-induced Ca2+ transients (*p < 0.05 cardiomyocytes derived from TAC-treated Hif-1α+/− versus sham-treated Hif-1α+/− mice). f Protein extracts derived from hearts of Hif-1α+/+ and Hif-1α+/− mice were analyzed by immunoblots
Discussion
HIF-1 has been assigned an important role in coordinating gene expression with cardiac tissue oxygen tensions [32]. A decrease in the oxygen availability is apparent during the course of ischemic heart disease. Under these hypoxic conditions HIF-1 plays an important role in the cellular adaptation of cardiomyocytes [22]. Examples of HIF-1-mediated adaptations include increased expression of VEGF to promote angiogenesis, glucose transporter 1 to enhance glucose uptake and glycolytic enzymes to facilitate glucose metabolism. Patients with HIF1A point mutations (HIF1A.2 or HIF1A.50, which result in both cases in HIF-1α proteins with a diminished transactivation activity) develop less coronary artery collaterals compared with patients with the wild type protein [8, 28]. A protective role of HIF-1α in the ischemic adult heart was further demonstrated in Hif-1α+/− mice. Hif-1α+/− animals have a reduction in the HIF-1α protein expression of ∼30–50%. Under resting conditions, they do not present an appreciable cardiovascular phenotype. However, in case of an acute ischemic stimulus they have a complete loss of ischemia pre-conditioning-induced cardioprotection [4]. Using the Hif-1α+/− mice in a TAC model, our data indicate that HIF-1α likewise confers protection of cardiac tissue in case of sustained pressure overload.
Hif-1α+/− mice developed a severe impairment of FS after aortic constriction in sharp contrast to their respective wild type littermates. The PWT and ST after TAC, however, did not differ comparing Hif-1α+/− and Hif-1α+/+ mice. The impact of HIF-1α for cardiac hypertrophy is discussed controversially in the literature. In Tet-inducible cardiac-specific Hif-1α knockout mice mechanical load-induced cardiac hypertrophy is impaired, which correlates with a decreased cardiac vessel density [31]. Huang et al., however, demonstrated a mild hypertrophy with increased wall thickness and heart weight to body weight ratios in resting constitutive cardiac specific Hif-1α knockout mice despite of cardiac hypovascularization [10]. In the present study, we did not observe an impaired TAC-induced cardiac angiogenesis and hypertrophy comparing Hif-1α+/+ and Hif-1α+/− mice. The discrepancies regarding the impact of HIF-1α on cardiac hypertrophy may rely on the onset and extent of the Hif-1α knockout in the different genetic mouse models used.
Regarding the development of heart failure, female and male mice responded differently to the applied pressure overload in our study. Transition to heart failure developed 8–11 weeks after onset of pressure overload in female mice, whereas FS was decreased already 3 weeks after onset of pressure overload in male mice. A gender-dependent difference in the extent of heart failure as a consequence of mechanical load is in line with the literature [37]. Nevertheless, the data obtained in our study have to be analyzed with caution regarding gender effects, since female and male mice differed regarding the extent of the TAC applied and the age at which they were analyzed.
Our results provide insights into the role of HIF-1α in the development of heart failure. Our findings link HIF-1α to an altered SR Ca2+ handling since we found a decreased SR Ca2+ content in the TAC-treated HIF-1α+/− mice. Abnormal SR Ca2+ uptake and release lead to depressed [Ca2+]i, which eventually results in the contractile phenotype in heart failure. Altered Ca2+-handling in the Hif-1α+/− mice thus could be contributing to the observed heart failure in chronic pressure overload.
In our study, we found a decreased SR Ca2+ content in the Hif-1α+/− cardiomyocytes after TAC. In this regard, it is interesting to note that it has been previously reported that HIF-1α modulates Ca2+ signalling during T-cell receptor stimulation in thymocytes [27]. This has been attributed to an increased expression of SERCA in pVHL-depleted thymocytes, which could be reversed by inhibiting HIF-1α stabilization. Up to now, the molecular link between HIF-1α and SERCA expression is not clear. However, it has been excluded that SERCA is a direct transcriptional target of HIF-1 [27]. In our loss of function mouse model, we could not observe a HIF-related difference in SERCA protein levels, when comparing heart samples from Hif-1α+/− and Hif-1α+/+ mice. SR-Ca2+ content is determined by Ca2+ release as well as SR Ca2+ reuptake. The reuptake is mainly due to SERCA function, which is highly regulated by for example post-translational modifications as well as its regulator protein phospholamban. Thus, the decrease in SR Ca2+ content after TAC in the Hif-1α+/- mice may rely on an increased SR Ca2+ release or a diminished SERCA function. Taken together, it is tempting to speculate that HIF-1α plays a more general role in Ca2+ handling, which affects adaptation of cardiomyocytes to various forms of mechanical stress. Nevertheless, further studies are needed, to define the molecular role of HIF-1α for cardiac Ca2+-handling and cardiomyocyte function in detail.
HIF-1α can be stabilized by inhibiting the activity of the HIFα-regulating PHDs independent of the oxygen concentration [3, 6, 15]. Applying inhibitors of the PHDs has been demonstrated to induce tissue protection in case of ischemic diseases in the central nervous [1, 34], renal [2, 7], gastrointestinal [29] and cardiovascular system [9]. Recent reports describing the consequences of inhibiting the activity of PHDs in the heart have proven that HIF-1α is a central component for this protection [5]. These data imply that short-term elevation of HIF-1α levels in response to hypoxia or ischemia drives beneficial adaptive processes. The presented data indicate that HIF-1α has a likewise positive effect on the cardiac function in case of adaptation towards chronic pressure overload. Therefore, subsequent studies will aim to analyze, if stabilizing HIF-1α in the heart via inhibiting PHD activity will positively influence cardiac function in case of increased mechanical load.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(a) Female Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, posterior wall thickness (PWT) and septum thickness (ST) were analyzed up to 11 weeks by echocardiography. (b) Male Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, posterior wall thickness (PWT) and septum thickness (ST) were analyzed up to 11 weeks by echocardiography (DOC 302 kb)
Female Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, left ventricular enddiastolic diameter (LVEDD), left ventricular endsystolic diameter (LVESD) and fractional shortening (FS) were analyzed up to 11 weeks by echocardiography. (b) Male Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, left ventricular enddiastolic diameter (LVEDD), left ventricular endsystolic diameter (LVESD) and fractional shortening (FS) were analyzed up to 11 weeks by echocardiography (DOC 416 kb)
Acknowledgment
This work was supported by the Deutsche Forschungsgemeinschaft (DFG Ka 1269/9-1 to DMK and HK and Klinische Forschergruppe 155 to LSM and KS). We acknowledge the excellent technical help from Sabine Krull.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor-1α increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernhardt WM, et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol. 2006;17:1970–1978. doi: 10.1681/ASN.2005121302. [DOI] [PubMed] [Google Scholar]
- 3.Bernhardt WM, Warnecke C, Willam C, Tanaka T, Wiesener MS, Eckardt KU. Organ protection by hypoxia and hypoxia-inducible factors. Methods Enzymol. 2007;435:221–245. doi: 10.1016/S0076-6879(07)35012-X. [DOI] [PubMed] [Google Scholar]
- 4.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1α. Cardiovasc Res. 2008;77:463–470. doi: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- 5.Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 6.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- 7.Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, Maxwell PH. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19:39–46. doi: 10.1681/ASN.2006090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hlatky MA, et al. Polymorphisms in hypoxia inducible factor 1 and the initial clinical presentation of coronary disease. Am Heart J. 2007;154:1035–1042. doi: 10.1016/j.ahj.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 9.Huang M, et al. Short hairpin RNA interference therapy for ischemic heart disease. Circulation. 2008;118:S226–S233. doi: 10.1161/CIRCULATIONAHA.107.760785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, Young LH, Johnson RS, Giordano FJ. Cardiac myocyte-specific HIF-1α deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. Faseb J. 2004;18:1138–1140. doi: 10.1096/fj.03-1377com. [DOI] [PubMed] [Google Scholar]
- 11.Ivan M, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 12.Jaakkola P, et al. Targeting of HIF-α to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y, et al. Dietary copper supplementation reverses hypertrophic cardiomyopathy induced by chronic pressure overload in mice. J Exp Med. 2007;204:657–666. doi: 10.1084/jem.20061943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Katschinski DM. In vivo functions of the prolyl-4-hydroxylase domain oxygen sensors: direct route to the treatment of anaemia and the protection of ischaemic tissues. Acta Physiol (Oxf) 2009;195:407–414. doi: 10.1111/j.1748-1716.2008.01952.x. [DOI] [PubMed] [Google Scholar]
- 16.Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, Thistlethwaite PA. Hypoxia-inducible factor 1α reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. doi: 10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 17.Kissling G. Mechanical determinants of myocardial oxygen consumption with special reference to external work and efficiency. Cardiovasc Res. 1992;26:886–892. doi: 10.1093/cvr/26.9.886. [DOI] [PubMed] [Google Scholar]
- 18.Krishnan J, Ahuja P, Bodenmann S, Knapik D, Perriard E, Krek W, Perriard JC. Essential role of developmentally activated hypoxia-inducible factor 1α for cardiac morphogenesis and function. Circ Res. 2008;103:1139–1146. doi: 10.1161/01.RES.0000338613.89841.c1. [DOI] [PubMed] [Google Scholar]
- 19.Krishnan J, et al. Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Lehnart SE, Maier LS, Hasenfuss G (2009) Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev. doi:10.1007/s10741-009-9146-x [DOI] [PMC free article] [PubMed]
- 21.Liu H, Fisher SA. Hypoxia-inducible transcription factor-1α triggers an autocrine survival pathway during embryonic cardiac outflow tract remodeling. Circ Res. 2008;102:1331–1339. doi: 10.1161/CIRCRESAHA.107.167858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686–690. doi: 10.1038/cdd.2008.13. [DOI] [PubMed] [Google Scholar]
- 23.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 24.Marti HH. Angiogenesis—a self-adapting principle in hypoxia. Exs. 2005;94:163–180. doi: 10.1007/3-7643-7311-3_12. [DOI] [PubMed] [Google Scholar]
- 25.Masson N, Ratcliffe PJ. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O2 levels. J Cell Sci. 2003;116:3041–3049. doi: 10.1242/jcs.00655. [DOI] [PubMed] [Google Scholar]
- 26.Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 27.Neumann AK, Yang J, Biju MP, Joseph SK, Johnson RS, Haase VH, Freedman BD, Turka LA. Hypoxia inducible factor 1 α regulates T cell receptor signal transduction. Proc Natl Acad Sci U S A. 2005;102:17071–17076. doi: 10.1073/pnas.0506070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Resar JR, et al. Hypoxia-inducible factor 1α polymorphism and coronary collaterals in patients with ischemic heart disease. Chest. 2005;128:787–791. doi: 10.1378/chest.128.2.787. [DOI] [PubMed] [Google Scholar]
- 29.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan HE, Lo J, Johnson RS. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sano M, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 32.Semenza GL. O2-regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF-1. J Appl Physiol. 2004;96:1173–1177. doi: 10.1152/japplphysiol.00770.2003. [DOI] [PubMed] [Google Scholar]
- 33.Semenza GL. Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp Physiol. 2006;91:803–806. doi: 10.1113/expphysiol.2006.033498. [DOI] [PubMed] [Google Scholar]
- 34.Siddiq A, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tirziu D, et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest. 2007;117:3188–3197. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 37.Witt H, et al. Sex-specific pathways in early cardiac response to pressure overload in mice. J Mol Med. 2008;86:1013–1024. doi: 10.1007/s00109-008-0385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu AY, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Female Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, posterior wall thickness (PWT) and septum thickness (ST) were analyzed up to 11 weeks by echocardiography. (b) Male Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, posterior wall thickness (PWT) and septum thickness (ST) were analyzed up to 11 weeks by echocardiography (DOC 302 kb)
Female Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, left ventricular enddiastolic diameter (LVEDD), left ventricular endsystolic diameter (LVESD) and fractional shortening (FS) were analyzed up to 11 weeks by echocardiography. (b) Male Hif-1α+/+ and Hif-1α+/− mice underwent sham-treatment. Subsequently, left ventricular enddiastolic diameter (LVEDD), left ventricular endsystolic diameter (LVESD) and fractional shortening (FS) were analyzed up to 11 weeks by echocardiography (DOC 416 kb)