

Abstract
The alpha-kinase family represents a class of atypical protein kinases that display little sequence similarity to conventional protein kinases. Early studies on myosin heavy chain kinases in Dictyostelium discoideum revealed their unusual propensity to phosphorylate serine and threonine residues in the context of an alpha-helix. Although recent studies show that some members of this family can also phosphorylate residues in non-helical regions, the name alpha-kinase has remained. During evolution, the alpha-kinase domains combined with many different functional subdomains such as von Willebrand factor-like motifs (vWKa) and even cation channels (TRPM6 and TRPM7). As a result, these kinases are implicated in a large variety of cellular processes such as protein translation, Mg2+ homeostasis, intracellular transport, cell migration, adhesion, and proliferation. Here, we review the current state of knowledge on different members of this kinase family and discuss the potential use of alpha-kinases as drug targets in diseases such as cancer.
Keywords: Signal transduction, Protein phosphorylation, Atypical kinases, Alpha-kinase family
Introduction
The reversible phosphorylation of proteins catalyzed by protein kinases and phosphatases is one of the principal regulatory mechanisms of signal transduction. Kinase regulated signaling cascades control a large variety of cellular functions and physiological processes such as transcription, protein translation, DNA replication and repair, cell adhesion, and cell migration as well as cell growth and proliferation. As a consequence, defects in protein phosphorylation are frequently associated with the pathogenesis of human diseases including cancer, diabetes, and inflammatory disorders. Owing to their well-defined catalytic properties, kinases represent one of the most favored groups of drug targets of the pharmaceutical industry. To identify novel specific kinase inhibitors and facilitate translational research into the use of such drugs, a detailed understanding of the regulation and function of kinases and their substrates is essential.
The protein kinase-like superfamily consists of a large collection of proteins sharing functional properties that allow them to catalyze the transfer of phosphate from ATP to a variety of substrates including proteins and lipids [1]. The majority (90%) of protein serine/threonine and tyrosine kinases are classified as conventional protein kinases (CPKs) since they possess a common architecture within the catalytic domain. The remaining protein kinases (about 10%), which display little sequence similarity to CPKs, are referred to as atypical protein kinases (APKs) and include protein kinases related to histidine kinases from bacteria, phosphoinositide 3-kinase related kinases, and alpha-kinases. Phylogenic studies point out that both CPKs and APKs are broadly distributed in prokaryotes and eukaryotes [2]. In addition, the ancient origin of both groups of kinases suggest that APKs developed in parallel with CPKs and do not simply represent modified CPKs. However, a specific subfamily of APKs known as alpha-kinases is exceptional in that it appeared recently in evolution and therefore displays a distribution limited to eukaryotes [2, 3]. Scheeff and Bourne hypothesized that alpha-kinases developed to provide a novel signaling capacity within more complex eukaryotic organisms [2]. Here, we highlight the research performed on alpha-kinases to date and their potential involvement in human disease.
Alpha-kinases, a novel class of protein kinases
Alpha-kinases were first recognized as a novel class within the protein kinase superfamily by Ryazanov and colleagues [4] after earlier publications on the identification and characterization of the Dictyostelium myosin heavy chain kinase A (MHCK-A), B (MHCK-B), and C (MHCK-C) [5–7]. Following the cloning of human, mouse, rat and Caenorhabditis elegans eukaryotic elongation factor-2 kinase (eEF2K, previously known as CaMKIII) [8], sequence analysis revealed a surprising lack of sequence similarity to known protein kinases [4]. However, its catalytic domain was found to be strikingly similar to that of the Dictyostelium MHCKs, suggesting the existence of a new class of protein kinases with a novel catalytic domain structure [8]. The cloning of several alpha-kinases allowed for the rapid identification of other members of this kinase family in various genomes [1, 4] (Fig. 1).
Fig. 1.

Phylogenetic analysis of the alpha-kinase family. A PSI-Blast [152] search was done, starting with the TRPM7 alpha-kinase domain and using three iterations, to detect all sequenced alpha-kinases. For those sequences that were detected with PSI-Blast, but that were not detected as having an alpha-kinase domain by SMART [151], reciprocal PSI-Blast searches were performed to ensure that they were indeed homologous alpha-kinases. From the results, species were included so that the major eukaryotic taxa with alpha-kinases would be represented in the tree. Selected sequences were then aligned using ClustalW2 [153]. A phylogenetic tree was generated using PHYML [154] as implemented in Seaview [155]. Bootstrap values of the major branches are indicated. The dashed line separates vertebrate from invertebrate alpha-kinases. Red branches indicate N-terminal alpha-kinases. Alpha-kinases with a typical subdomain architecture are indicated in the colors that refer to the schematic representations in Table 1
The alpha-kinase family was named after the unique mode of substrate recognition by its initial members, the Dictyostelium heavy chain kinases. In contrast to the target sites of CPKs, which are usually located within loops, β-turns, or irregular structures [9], the substrate residues targeted by MHCKs were found to be located within protein sequences that adopt an alpha-helical conformation [10, 11]. However, recent in vitro data suggest that this is not the consensus as multiple alpha-kinases were found to also target residues in non-helical regions [12, 13]. Currently, alpha-kinases represent a family of atypical protein kinase with a unique catalytic domain architecture homologous to the Dictyostelium discoideum MHCKs. Determinants for substrate recognition remain largely unknown, although the necessity of a basic residue following the phosphoacceptor aminoacid has been shown previously [14].
To date six human alpha-kinases have been identified. In addition to eEF2K, the human genome encodes alpha-kinase 1 (lymphocyte alpha-kinase, LAK or ALPK1), alpha-kinase 2 (heart alpha-kinase, HAK or ALPK2), and alpha-kinase 3 (muscle alpha-kinase, MAK or ALPK3), initially named after the tissue in which they were identified [4]. The remaining two mammalian alpha-kinases, TRPM6 and TRPM7, represent cation channels belonging to the TRP ion channel family.
The alpha-kinases found in man are widely distributed among vertebrates. In contrast to the other human alpha-kinase containing proteins, eEF2K can also be found in invertebrates such as the metazoan Trichoplax adhaerens and in the diatom Thalassiosira pseudonana, indicating that eEF2K represents the oldest alpha-kinase within the vertebrates. According to the phylogenetic tree, eEF2K appears to be most closely related to the Dictyostelium discoideum MHCKs, especially to MHCK-B and MHCK-C, consistent with the unique N-terminal localization of their alpha-kinase domains (Fig. 1; Table 1). The four Dictyostelium MHCKs share many structural and functional features which is supported by their isolated position in the phylogeny [7, 15–19]. The closely related Dictyostelium alpha-kinase 1 (AK1) contains an N-terminal Arf-GAP domain [19] and is, like the MHCKs, specifically expressed by Dictyostelium discoideum, indicating taxon specific gene duplications and recombination. As AK1 has not been functionally characterized, it will not be discussed in depth. In the ciliate Tetrahymena thermophila, gene amplification of alpha-kinases is evident as its genome encodes eight very similar alpha-kinases with an N-terminal von Willebrand factor A motif. Another striking example is the collection of alpha-kinases encoded by the genome of Entamoeba histolytica which includes many different domain architectures, including proteins with SH3 and p53-like domains [20]. This phylogenetic tree contains a number of alpha-kinases which are neither highlighted (Fig. 1) nor discussed in Table 1 as they have no typical subdomain organization and have yet to be characterized. Overall, we postulate that the divergent family of alpha-kinases arose by independent taxon specific gene duplications and recombination with a variety of subdomains.
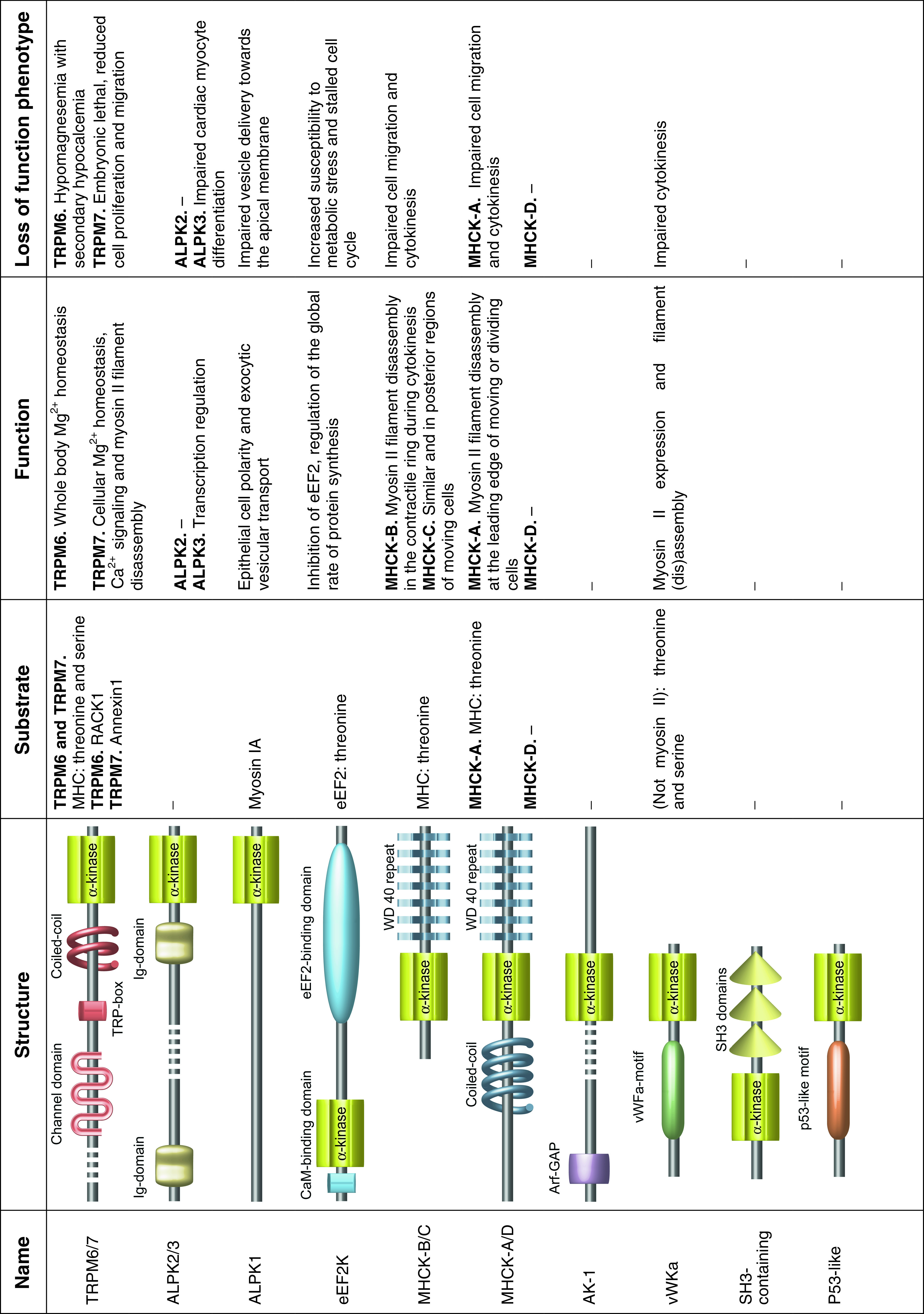
Table 1.
Overview of alpha-kinases. The main characteristics of alpha-kinases and the domain architectures of these proteins. Domains were determined using SMART [151] and published data. The colors of these domains refer to colors used in Fig. 1 to indicate the specific alpha-kinase subtypes. If known, substrate specificity is indicated together with the preference for threonines and/or serines
Elucidation of the crystal structure of the TRPM7 kinase domain by Yamaguchi and colleagues led to the striking observation that, despite a lack of sequence similarity, the alpha-kinase catalytic core has an architecture related to that of CPKs [21]. CPKs and alpha-kinases share a remarkably similar N-terminal lobe that predominantly folds into a curved β-sheet and contains the phosphate binding loop (P-loop). The C-terminal lobe, however, might appear to be more divergent although in both cases it largely consists of alpha-helices and the activation loop can easily be distinguished (Fig 2a). The interlobe cleft serves as binding pocket for ATP and contains well-conserved key catalytic residues, such as invariant lysine- (K1648 in TRPM7) and aspartate- (D1767, D1777 in TRPM7), that are located in similar positions to position the ATP and coordinate Mg2+ ions (Figs. 2b and 3).
Fig. 2.

Alpha-kinases: structure and conservation. a Crystal structure of TRPM7 as determined by Yamaguchi et.al. [21]. N-terminus (N) refers to residue G1579 in the human TRPM7 amino acid sequence. Please note that this residue corresponds to G1577 in the mouse sequence. Indicated are domains that resemble the N-terminal lobe (containing P-loop), C-terminal lobe (containing activation loop) and interlobe cleft of CPKs. Conserved residues within the alpha-kinase family (Fig. 3) are indicated in red. b The well-conserved interlobe cleft functions as ATP binding pocket. The ATP molecule (yellow) is positioned within the binding pocket by multiple residues that are well conserved within the alpha-kinase family (red). The indicated residue identities refer to the human TRPM7 amino acid sequence (Fig. 3). c Typical zinc-finger domain within the C-terminal lobe of alpha-kinases. Residues that bind the zinc molecule (green) are well conserved between the alpha-kinase family members (red). The indicated residue identities refer to the human TRPM7 amino acid sequence (Fig. 3)
Fig. 3.
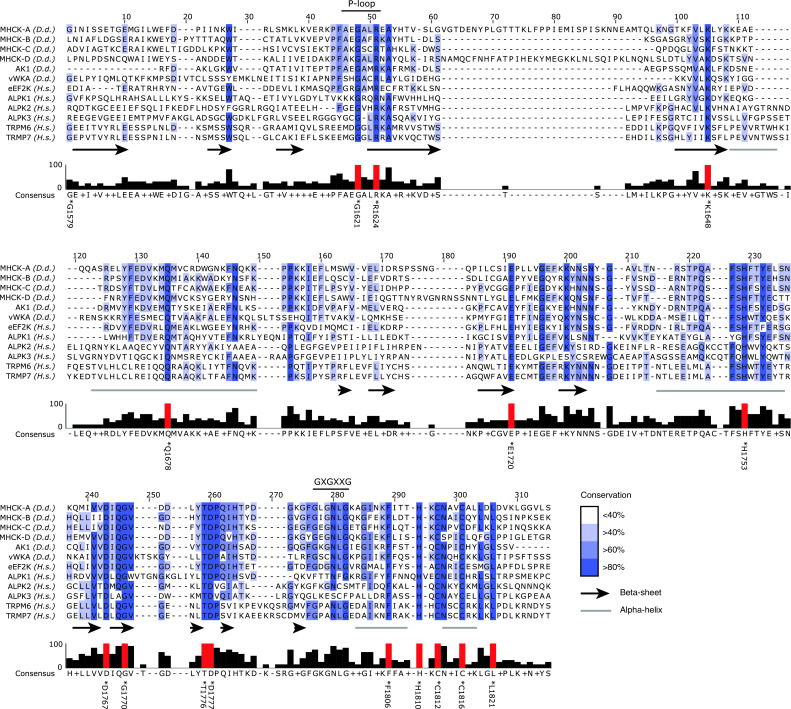
Sequence alignment of Dictyostelium and human alpha-kinases. Dictyostelium and human alpha-kinase amino acids sequences corresponding to the human TRPM7 alpha-kinase domain (G1579-T1830) were aligned using ClustalW2 sequence alignment software [153]. Names and species, Dictyostelium discoideum (Dd), Homo sapiens (Hs) are indicated on the left. Conservation of specific residues within this group of alpha-kinases, corrected for missing sequences, is indicated by shadings of blue and a barchart supporting the alignement (0 not conserved, 100 fully conserved). Red bars represent fully conserved residues and are indicated red in Fig. 2. Sequences representing the P-loop and GXGXXG motif are indicated on top. The indicated residue identities refer to the human TRPM7 amino acid sequence
Although the ATP binding pockets of CPKs and alpha-kinases are structurally very alike, the nature of interactions between the kinase and base and sugar moieties of ATP differ significantly and may also differ within the alpha-kinase family itself [21]. One striking difference is the localization of the conserved GXGXXG motif which, in CPKs, is positioned within the catalytic P-loop and serves to bind phosphates. Although alpha-kinases do contain a similar glycine rich motif, it is not located within the P-loop but in another loop proximal to the C-terminus (Fig. 3). This loop structurally resembles the activation loop in CPKs which is involved in peptide substrate recognition, suggesting a similar function for the glycine rich motif in alpha-kinases [21]. The different modes of substrate recognition between alpha-kinases and CPKs may explain the distinct set of alpha-kinase substrates.
Another major distinction between alpha-kinases and CPKs is the presence of a zinc-finger motif within the C-terminus of alpha-kinases (Figs. 2c and 3). This motif coordinates a zinc ion and is thought to play an important role in stabilizing the tertiary structure of the catalytic core. Indeed, mutation of the conserved cysteine residues (C1812, C1816 in TRPM7) abrogates kinase activity [22, 23]. For an in-depth comparison between the structures of alpha-kinases and CPKs, see review in [3].
Members of the alpha-kinase family
Myosin heavy chain kinases in Dictyostelium
Tight regulation of myosin II induced contractility is essential for processes like cytokinesis and cell migration. Myosin II regulation by light chain phosphorylation has been described in many eukaryotic systems and is well known to regulate activity of the myosin motor domain [24]. A role for myosin II heavy chain phosphorylation in regulating myosin II activity and function was initially established in lower eukaryotes such as Dictyostelium and Acanthamoeba [25–28]. The incorporation of phosphate into the Dictyostelium myosin heavy chain, residues Thr1823 [11], Thr1833 and Thr2029 [10], inhibits its ability to self-assemble into bi-polar filaments, which is required for the generation of contractile forces [26]. Mutagenesis of these residues into non-phosphorylatable alanines (3*Ala) induces overassembly of myosin II proteins, severely affecting polarization, chemotaxis, and cytokinesis [28, 29].
In search of protein kinases responsible for heavy chain phosphorylation in Dictyostelium, MHCK-A was identified in actomyosin precipitations and shown to specifically incorporate phosphate into the alpha-helical myosin heavy chain [15]. Thereby, MHCK-A reduced the filament forming properties of myosin II and inhibited overall myosin II function [15, 26, 28, 30]. Cloning of MHCK-A revealed three separate domains (Table 1). A coiled-coil N-terminal region (~500aa) is involved in the heteromerization of the kinase [31] and required for MHCK-A translocation to actin-rich cell regions such as the cortex. Moreover, it directly interacts with actin filaments in vitro which increases its kinase activity ~40-fold [32, 33]. More recently, actin crosslinking was shown to be an additional feature of this domain [34]. This suggests that, in addition to driving myosin II disassembly, MHKC-A may also affect the actin-myosin cytoskeleton by crosslinking actin filaments. The second domain represents the catalytic domain (~aa550–800), which showed no homology to known eukaryotic protein kinases [5, 6]. Finally, the C-terminus contains a stretch of seven WD-repeats that generally coordinate multi-protein complex assemblies [35]. Steimle and colleagues showed that the MHCK-A WD-repeat domain binds directly to the myosin heavy chain and thereby targets the kinase towards its substrate [36]. Neither the N-terminal coiled-coil domain nor the WD-repeats are required for catalytic activity [6]. However, these domains are essential in substrate phosphorylation which is strongly enhanced by autophosphorylation of the region that links the coiled-coil region to the catalytic core (aa500–550) [6, 37].
Apart from MHKC-A, the Dictyostelium genome encodes three additional myosin heavy chain kinases, MHCK-B, MHCK-C, and MHCK-D [7, 16, 38], with a catalytic domain very similar to that of MHCK-A. Each of these MHCK isoforms contains a C-terminal WD domain, but in contrast to MHCK-A, MHCK-B, and MHCK-C lack the large N-terminal coiled-coil region (Table 1). Although MHCK-D remains functionally uncharacterized to date, it does contain this coiled-coil region and, according to sequence similarity, it appears to be the closest relative of MHCK-A (Fig. 1). MHCKA, B, and C share many biochemical features. Kinase activity is regulated by autophosphorylation [17, 37, 39] and all three specifically target threonine residues within the myosin heavy chain in vitro, which results in myosin II filament disassembly [7, 16–19]. MHCK-A, B, and C triple gene knockouts induce myosin II overassembly and severely affect cytokinesis, resembling the phenotypes observed when MHC phosphorylation was inhibited by substituting the targeted residues with alanine (3*Ala) [19, 28]. Overexpression of MHCK-A, B, and C, on the other hand, also results in impaired cytokinesis by hyperphosphorylation of myosin II inducing myosin II filament disassembly [30, 39, 40].
In spite of these similarities, the spatial regulation of the MHCK isoforms differs significantly, indicating specific functions for each isoform. Both MHCK-A localization and activity appear to be actin dependent [32, 41, 42]. In response to stimulation by chemoattractants such as cAMP, MHCK-A is enriched in the actin-rich leading edge of the cell where it disassembles myosin II filaments [41]. MHCK-A might thus provide a mechanism by which myosin II is locally inactivated, allowing actin based cellular protrusions such as lamellipodia and phagocytic cups to be formed. Hence, MHCK-A plays an important role in maintaining cell polarity, chemotaxis and cell migration [29]. Both MHCK-B and C are enriched in the contractile ring during late cytokinesis, driving myosin II disassembly for efficient cell division. In addition, MHCK-C is enriched in the posterior region of moving cells where it could play a role in the disassembly of the actomyosin cytoskeleton and myosin II recycling required for proper cell body translocation [16, 17, 19, 38]. Notably, in contrast to MHCK-A, MHCK-C distribution fully depends on myosin II [16, 17]. Altogether, these spatial differences indicate specific roles for MHCK isoforms in fine tuning the Dictyostelium actomyosin cytoskeleton.
vWFa-kinase
vWFa-kinase (vWKa) is one of the more recently identified alpha-kinases in Dictyostelium, carrying a kinase catalytic domain (aa398–592 in Dictyostelium) similar to the MHCKs. In contrast to MHCKs, however, it is broadly expressed among invertebrates (Fig. 1), it phosphorylates both threonine and serine residues [14], and has a distinct domain organization (Table 1). It lacks the C-terminal WD repeat domain and, consequently, the kinase domain is located at the extreme C-terminus (Table 1). Furthermore, the protein contains an N-terminal vWFa motif (aa121–233 in Dictyostelium) [40]. This domain is named after its homology to von Willebrand factor A, a plasma glycoprotein essential for proper blood clotting. The vWFa domain mediates receptor-ligand binding, protein–protein interactions, and multiprotein complex formation. Proteins containing a vWFa domain are involved in cell adhesion such as integrin–collagen interactions, but also in DNA repair and transcription (reviewed in [43]). Like for the heavy chain kinases in Dictyostelium, overexpression of vWKa leads to multinucleation, indicative of defects in cytokinesis due to disregulation of myosin II function [40]. Significant myosin II overassembly was observed after vWKA gene disruption, resembling the effect of MHCK gene disruption and introducing non-phosphorylatable threonine–alanine mutations into the myosin heavy chain (3*Ala) [19]. This is consistent with the notion that vWKa could be an additional myosin heavy chain kinase. However, several observations are in conflict with this hypothesis. First, overexpression of vWKa increased myosin II assembly [40]. Second, vWKa is unable to phosphorylate the myosin II heavy chain in vitro, although it autophosphorylates and has intrinsic catalytic activity [40]. Surprisingly, both overexpression and gene disruption of vWKa increased the myosin II protein levels by means of an unknown mechanism [40]. Altogether, these results do indicate a role for vWKa in the regulation of myosin II but not by direct phosphorylation of its heavy chain. vWKa localizes to contractile vacuoles, organelles involved in osmoregulation, and to the perinuclear golgi apparatus [40]. Hence, it is highly unlikely that, although varying vWKa expression levels affects growth kinetics, cell division, and development, its function is restricted to the dedicated functions of the previously identified MHCKs.
eEF2-kinase
Eukaryotic elongation factor-2 kinase (eEF2K) is a regulator of global protein synthesis. It phosphorylates eukaryotic elongation factor-2 (eEF2), a ribosome binding protein that catalyzes the movement of ribosomes along the mRNA during translation in most eukaryotic cells [44]. Phosphorylation of eEF2 on threonine residues Thr56 and Thr58 decreases its affinity for ribosomes and inhibits the global rate of protein synthesis [45–49]. As eEF2K activity is Ca2+ and calmodulin dependent, it was initially classified as a member of the Ca2+/calmodulin-dependent protein kinase family [48, 50]. However, sequence analysis revealed no homology to the other Ca2+/calmodulin kinases. Instead, its sequence showed remarkable homology to the previously described Dictyostelium MHCKs [8]. Additionally, eEF2K phosphorylates threonines whereas the other members of the CaM-kinase family predominantly target serine residues [48]. Of note is a publication by Jorgensen and colleagues showing that the targeted threonine residues are not located within an alpha-helix, as was determined by crystallography on the yeast eEF2 [12].
eEF2K comprises three functional domains (Table 1). The catalytic domain (aa100–350) is flanked at its N-terminus by a calmodulin binding domain (aa51–96), while its C-terminus contains the eEF2 binding domain (aa521–725) required for substrate targeting [23, 51]. Importantly, catalytic activity of eEF2K requires the binding of calmodulin to the calmodulin binding domain.
Since protein translation consumes a large amount of energy, the cell must carefully balance the activity of eEF2K with its need for protein translation. Therefore, eEF2K activity is modulated through multisite phosphorylation by kinases downstream of mTOR and AMPK, the central sensors of cellular nutrients and energy levels [52–56]. In the presence of sufficient nutrients, its kinase activity towards eEF2 is suppressed. Upon nutrient deprivation and hypoxia, the suppression of eEF2K is relieved which results in the phosphorylation of eEF2 and, consequently, inhibition of protein synthesis (reviewed in [55]).
eEF2K appears to be a major regulator of autophagy downstream from mTOR, which, besides the common degradation of organelles, serves as a cellular coping strategy to deal with long-term nutrient deprivation [57, 58]. When cells are deprived of nutrients, they will consume their own cytoplasmic content to obtain the energy needed to survive [59, 60]. During this state of metabolic stress, protein synthesis is inhibited by increased phosphorylation of eEF2 which terminates protein elongation [58, 61]. This may be further enhanced by a decrease in pH due to hypoxia which dramatically increases eEF2K activity [62]. As a consequence, a cell becomes more resistant to metabolic stress. An increased eEF2K activity has been observed in many cancers and may promote cancer cell survival, tumor growth and metastasis, processes that require the cell to cope with hypoxia and nutrient deprivation [57, 58, 63]. Moreover, silencing of eEF2K was shown to sensitize tumor cells to treatments that induce energy stress and cytotoxicity [64].
Another link between eEF2K and cancer is the close relationship between eEF2/eEF2K activity during cell-cycle progression (reviewed in [65]); during G1-phase, the stage in which cell growth takes place, eEF2K is inactivated and, consequently, eEF2 becomes active permitting protein synthesis. During DNA replication in the S-phase, eEF2K slowly dephosphorylates to fully inhibit eEF2 at the start of the G2 and M phase, where proofreading and mitosis take place. The common explanation for this decline in protein synthesis is that cellular energy sources are to be redirected from protein synthesis to proofreading and the actual cell division. Various reports describe delayed cell cycle progression or a complete arrest by deregulation of eEF2 or eEF2K [63, 66–68], which makes the eEF2/eEF2K pathway an attractive drug target for cancer treatment.
Alpha-kinase 1, 2 and 3
The vertebrate alpha-kinase 1, 2, and 3 were initially named after the tissues in which they were identified by Ryazanov and colleagues [4, 69]; lymphocyte alpha-kinase, heart alpha-kinase, and muscle alpha-kinase, respectively. All three carry an alpha-kinase domain at their extreme C-terminus that is particularly well conserved between ALPK2 and ALPK3 (Table 1 and Fig. 1).
ALPK1 has been implicated in epithelial cell polarity and exocytic vesicular transport towards the apical plasma membrane [70]. ALPK1 resides on golgi-derived vesicles where it phosphorylates myosin IA, an apical vesicle transport motor protein that regulates the delivery of vesicles to the plasma-membrane [70]. To our knowledge, no functional studies have been performed on ALPK2. Expression of ALPK3, also known as myocytic induction/differentiation originator (Midori), is restricted to fetal and adult heart and adult skeletal muscle. Overexpression of ALPK3 enhances differentiation of murine embryonic carcinoma cells into cardiomyocytes. As ALPK3 localizes to the nucleus, it is thought to act as a transcriptional regulator implicated in early cardiac development [71].
TRPM6 and TRPM7
Transient receptor potential (TRP) cation channels make up a large protein superfamily. Members of this family function as cellular sensors responding to stimuli such as temperature, taste, touch, pain, osmolarity, and mechanical stress [72–74]. TRPM6 and TRPM7 belong to the melastatin-related subfamily of TRP (TRPM) channels, which represents a group of eight proteins displaying a wide range of structural features, expression patterns, ion selectivity, gating properties, and functions [75]. Both TRPM6 and TRPM7 are unique in the sense that they encode divalent cation channels fused to an alpha-kinase domain. As these unusual channel-kinases have caught the attention of electrophysiologists worldwide, their channel properties have been studied in great detail. However, involvement of the alpha-kinase domain in regulating channel function remains subject to controversy. Before going into the proposed functions of the alpha-kinase domain within these channel-kinases, we will first discuss the regulation and function of their channel domains in cellular physiology.
Like other TRP channels, TRPM6 and TRPM7 possess six transmembrane domains flanked by large N- and C-terminal tails that reside in the cytosol. Within the N-terminal tail, four stretches of amino acids are present which are conserved between the different members of the TRPM subfamily but whose function is poorly understood [75]. Proximal to the last transmembrane domain in the C-terminal tail, there is the so-called TRP box, a conserved stretch of amino acids present in all TRP ion channels. In addition, there is a coiled-coil domain and, at the extreme C-terminus, the alpha-kinase domain [3] (Table 1). The transmembrane domains organize within the plasma membrane to form the channel with the pore formed by a short stretch of amino acids between the fifth and sixth transmembrane domains [76]. Functional channels are most likely organized as either homo- or heterotetramers which have their unique electrophysical properties and functions [77–79]. The exact mechanism of tetramerization is unknown. However, it has been demonstrated that several TRP channels including TRPM2 and TRPM8 require the presence of the coiled-coil domain for the assembly of active channels [80, 81].
TRPM7 is highly similar to TRPM6 displaying 50% identity at the amino acid level. Moreover, the kinase domains share 75% of their aminoacid sequence. Both channels are highly permeable to Mg2+ and Ca2+ [82, 83]. In spite of these similarities, these proteins appear to be functionally non-redundant. While TRPM7 is ubiquitously expressed, TRPM6 expression is mostly limited to brain, intestine, and kidneys [84]. Knockout studies have shown that both TRPM6 and TRPM7 are essential in development as the loss of these channels is embryonic lethal [85, 86]. Moreover, overexpression of TRPM6 cannot rescue cell growth arrest due to the ablation of TRPM7 [79]. Finally, mutations in TRPM6 cause familial hypomagnesemia with secondary hypocalcemia, a disease characterized by defects in renal and intestinal Ca2+ and Mg2+ absorption which progresses despite the expression of TRPM7 in both the kidney and intestine [87, 88].
The importance for TRPM6 in maintaining whole body Mg2+ levels, e.g. by renal Mg2+ reabsorbtion, is widely accepted [82, 84, 89]. The role of TRPM7 in Mg2+ homeostasis, however, remains controversial. Various publications have implicated TRPM7 in the regulation of cellular Mg2+ levels [83, 90, 91]. However, there is now increasing evidence that, in contrast to TRPM6, the TRPM7 channel might primarily be implicated in Ca2+ signaling [86, 92, 93]. Initially, TRPM7 was thought to mediate cellular Mg2+ homeostasis based on the growth arrest in TRPM7-deficient DT-40 B cells, which could be restored by culturing these cells in media supplemented with high Mg2+ [79, 83, 91]. However, in other cell types such as retinoblastoma cells, defects in cell growth due to loss of TRPM7 expression could not be reverted by the addition of Mg2+ [92]. Several investigations implicate TRPM7 in a number of other cellular functions including anoxic cell death, exocytosis, mechanotransduction, cell proliferation, cell adhesion, and cell migration [93–100], processes that depend on TRPM7-mediated Ca2+ influx rather than involving Mg2+. Importantly, it was recently shown that TRPM7 was not essential for maintaining Mg2+ levels in mice TRPM7 knockout thymocytes [86]. The current opinion is that TRPM7 regulates local intracellular Ca2+ concentrations and thereby affects the functioning of Ca2+ sensitive proteins in its close proximity [94, 98, 101].
Channel opening is regulated via multiple mechanisms. A unique feature of TRPM6 and TRPM7 channels is their inhibition by intracellular Mg2+ [82, 83]. Furthermore, cation conductance is regulated through signaling by G-protein coupled receptors such as the bradykinin receptor [102]. Additionally, TRP channels are well-known mechanosensors. Their conducting properties are modulated upon application of mechanical force which result in a variety of cellular responses [103–105]. Consistently, TRPM7 channel opening is regulated by mechanical force [94, 96, 106].
The role of the TRPM6 and TRPM7 alpha-kinase domain in mediating the influx of cations is still under debate. Both alpha-kinase domains have been suggested to function as intracellular Mg2+ sensors that mediate channel permeability [91, 107–110]. Additionally, the RACK1 scaffold protein inhibits Mg2+ conductance in a autophosphorylation-dependent manner through binding of the TRPM6 kinase domain [111]. Furthermore, the repressor of estrogen receptor activity (REA) inhibits TRPM6 channel opening in a similar manner. Of interest is the fact that the interaction between REA and the alpha-kinase domain can be abolished by estrogens, suggesting involvement of estrogens in Mg2+ homeostasis [112]. In contrast, phosphotransferase activity appears to be unrelated to channel opening in the case of TRPM7. For instance, TRPM7 channels where the entire kinase domain has been deleted, remain sensitive to inhibition by Mg2+- and Mg2+-nucleotide complexes [91, 110]. Furthermore, point mutations in the TRPM7 kinase domain that abolish phosphotransferase activity do not appear to affect TRPM7 channel gating [93] and have only a minor effect on the inhibition of TRPM7 channel activity by Mg2+- and Mg2+-nucleotides [113]. Hence, the consensus in the field is that the kinase activity is not essential for opening of TRPM7 channels [114]. Instead, our work and that of others points towards an alternative model where the opening of TRPM7 channels affects kinase function. Since Ca2+ does not directly influence kinase activity [115], we propose that a local increase in Ca2+ concentration regulates the recruitment/targeting of TRPM7 substrates [116].
Rather than modulating channel opening, the TRPM7 kinase domain transmits intracellular signals via the phosphorylation of downstream targets such as annexin 1 in a Ca2+-dependent manner. Annexin 1, a member of the annexin phospholipid binding family, binds Ca2+ which leads to a conformation change exposing the target residue to the TRPM7 kinase domain [117]. Although annexins are known to be involved in Ca2+-dependent actin dynamics and membrane subdomain organization [118], the physiological relevance of the TRPM7-annexin 1 interaction remains to be elucidated. TRPM7 function has been linked to vascular remodeling. Stimulation of vascular smooth muscle cells (VSMC) by the vasoactive, G-protein coupled receptor agonist bradykinin increases TRPM7 expression, autophosphorylation (indicative of kinase activity [116]), channel opening, annexin 1 activity, and cell migration [119, 120]. These findings appear to connect TRPM7 channel opening (Mg2+ and Ca2+ influx), kinase activity, and substrate targeting with cytoskeletal remodeling.
We recently reported the Ca2+- and kinase dependent interaction between TRPM7 and the actomyosin cytoskeleton [93]. Very similar to the Dictyostelium myosin II heavy chain kinases, TRPM6 and TRPM7 are capable of phosphorylating the three mammalian alpha-helical assembly domains of the myosin II heavy chain isoforms, MHC-A, B, and C, at least in vitro [13]. Consequently, kinase activity may induce myosin II filament disassembly, affecting stress fiber formation and reducing actomyosin contractility [121, 122]. Apparently, this property of alpha-kinases has been preserved during evolution. However, unlike the Dictyostelium MHCKs, we have shown that TRPM6 and TRPM7 can also phosphorylate the non-helical C-terminus of MHC-B and MHC-C, which suggests that residues phosphorylated by these mammalian alpha-kinases are not exclusively positioned within an alpha-helix [13].
Actomyosin contractility plays a central role in regulating the assembly and disassembly of adhesive contacts, such as focal adhesion, and invasive adhesions, such as podosomes [123–125]. The formation of podosomes requires a local inhibition of contractility [126]. Accordingly, activation of TRPM7 in neuroblastoma cells results in Ca2+- and kinase-dependent podosome formation suggesting a close link between TRPM7, cytoskeletal dynamics, and cell adhesion [93]. Wei and colleagues consistently showed that TRPM7 contributes to local Ca2+ flickers in response to mechanical stimulation, which mediate cell polarization and steer migration [94]. In conclusion, TRPM7 appears to act as a mechanosensor that conducts Ca2+ upon exposure to mechanical stress which elevates local Ca2+ levels allowing Ca2+- and kinase-dependent cytoskeletal rearrangements to take place [93, 101].
Regulation of (alpha-) kinase activity
Over the years, many different mechanisms have been proposed to regulate the activity of CPKs. In general, kinases adopt different conformations in their inactive and active states [127]. For several kinases, the inactive state is stabilized by intramolecular interactions with regulatory domains [128, 129]. Transition to the active state can occur by association with regulatory proteins, lipids, or second messengers such as cAMP [128, 130, 131], (de)phosphorylation by upstream phosphatases and kinases [129, 132], or autophosphorylation [133]. In addition to directly influencing the catalytic activity of the kinase, regulatory domains may also serve as a docking site to recruit substrates, which are subsequently phosphorylated by the kinase [134].
Initial studies in understanding the molecular mechanisms controlling the catalytic activity of alpha-kinases have revealed that members of this kinase family are regulated by mechanisms analogous to CPKs. The regulation of eEF2K has been the most extensively studied [52–56]. Depending on nutrient availability, eEF2K phosphorylates eEF2 leading to an inhibition of protein synthesis. This system is controlled by several protein kinase cascades that lead to the phosphorylation of eEF2K which can either inhibit or activate its catalytic activity. Two major kinases involved in the control of metabolic processes are mTOR and AMPK, both of which control eEF2K activity [53–56, 135]. Under conditions of nutrient deprivation, eEF2K activity remains high to suppress protein translation, and this occurs through the phosphorylation of Ser398 by AMPK, a kinase whose activity is induced by high AMP/ATP ratios in the cell [54]. Elevation of cAMP in cells also inhibits protein translation but this pathway involves the phosphorylation of eEF2K by PKA [136]. In contrast to AMPK, PKA phosphorylates Ser499 of eEF2K leading to increased enzymatic activity [137].
Other stimuli such as stress, growth factors, and mitogens must ensure the inhibition of eEF2K to promote protein translation. Growth factors activate a rapamycin-sensitive pathway involving the activation of p70S6K by mTOR and a rapamycin-resistant pathway where ERK1/2 activate p90RSK [138]. Both p70S6K and p90RSK phosphorylate Ser366 which correlates with a decrease in eEF2K activity and eEF2 phosphorylation in cell lysates. Cellular stress induces a set of protein kinase cascades including ones involving members of the p38 MAPK family. These signaling pathways target eEF2K leading to its phosphorylation on Ser359 and Ser377 [139, 140]. During mitosis, CDC2 phosphorylates Ser359 of eEF2K [52]. The phosphorylation of these residues correlates with dephosphorylation of eEF2 and protein translation in cells. Thus, eEF2K is a target of multiple protein kinase cascades to control protein synthesis under various cellular conditions. It remains unknown to what extent other alpha-kinases are also phosphorylated by protein kinases to regulate their activity.
In addition to phosphorylation, eEF2K is activated allosterically by Ca2+/calmodulin which binds a region directly N-terminal of the catalytic domain. The dependence of eEF2K on Ca2+/calmodulin for optimal catalytic activity led scientists to originally call this protein CaMKIII. Phosphorylation of activating residues by autophosphorylation or ‘upstream’ protein kinases generates an eEF2K activity that is less dependent on Ca2+/calmodulin [141]. However, other signaling cascades modulate the binding of Ca2+/calmodulin to eEF2K to regulate protein synthesis. Insulin activates a rapamycin-sensitive mTOR pathway leading to the phosphorylation of Ser78 nearby the calmodulin binding domain [53]. Phosphorylation of this residue inhibits the binding of Ca2+/calmodulin to eEF2K preventing its activation [53]. Finally, Ryazanov and colleagues postulated that eEF2K is activated at pH <7.4, also occurring during hypoxia and ischemia, and might thus represent another route in which metabolic stress inhibits protein synthesis [62].
Two regulatory mechanisms appear common to several members of the alpha-kinase family: (1) autophosphorylation, and (2) a requirement of additional domains beyond the catalytic core for efficient substrate phosphorylation. All alpha-kinases studied to date including MHCKs, eEF2K, TRPM6, and TRPM7 undergo autophosphorylation. Intriguingly, the level of autophosphorylation displays a wide dynamic range with stoichiometry of phosphorylation ranging from 6 to 30 mol Pi/mol kinase [17, 36, 37]. Complete autophosphorylation does not appear to be required for activation of MHC kinases since autophosphorylation of three sites (out of ten) is sufficient to activate MHCK-A [37], and myosin II phosphorylation proceeds in parallel to MHCK-C autophosphorylation [17]. In contrast, TRPM6 and TRPM7 only phosphorylate myosin II under conditions allowing complete autophosphorylation of the kinases [116]. TRPM6 and TRPM7 autophosphorylate a Ser/Thr-rich domain flanking the N-terminus of the catalytic domain. Autophosphorylation is not required for catalytic activity per se but provides a means for the kinase to recognize protein substrates such as myosin II. Notably, MHCKs and eEF2K also require additional domains for substrate recognition. The C-terminus of eEF2K [51] and MHCK [36] bind directly to their respective substrates. Deletion of these regions decreases the rate of protein phosphorylation without affecting the kinetics of phosphorylation of a synthetic peptide [23, 31, 36, 51].
As mentioned earlier, the crystal structure of the TRPM7 alpha-kinase domain has been solved [142]. Both in solution and in crystals the kinase forms a dimer which is essential for kinase activity [115, 142, 143]. Also, the TRPM6 and TRPM7 kinase domain can heteromerize to form a functional kinase [143]. The alpha-kinase domains of TRPM6 and TRPM7 dimerize where a short stretch of amino acids (aa1,553–1,562) at the N-terminus interacts with key catalytic residues in the opposite monomer [143]. Interestingly, a monomeric TRPM7 kinase domain can be fully activated by incubating it with a peptide corresponding to the activation sequence spanning amino acids 1,548–1,576. Thus, the activation sequence aa1,553–1,562 plays a key role in controlling TRPM6 and TRPM7 catalytic activity, and changes in conformation within this region of the kinase by posttranslational modifications or binding to a regulatory protein or lipid could provide an important regulatory mechanism to control enzymatic activity.
Alpha-kinases in health and disease
Protein kinases are important drug targets for the treatment of human diseases such as cancer as they are often the result of deregulated kinase-dependent signaling pathways (reviewed in [144–146]). The majority of protein kinase inhibitors bind to the ATP-binding pocket abolishing the access of ATP to the protein kinase. This strategy poses a major challenge in designing effective compounds with suitable specificity to prevent potential side-effects due to the blockade of other protein kinases. Since alpha-kinases bind to the sugar and base moieties of ATP using a different molecular interface than CPKs [21], it may be possible to take advantage of this feature to develop specific protein kinase inhibitors.
Among alpha-kinases, eEF2K, TRPM6, and TRPM7 have been linked to the pathogenesis of human disease. As we previously described, eEF2K dysfunction has been linked to cancer progression via its effect on eEF2 activity. Several clinically applied anti-cancer drugs, such as Doxorubicin and Ontak, inactivate eEF2 directly and thereby inhibit protein synthesis and cell cycling, leading to tumor cell death [65]. A few compounds have been reported to cause cell cycle arrest by inhibition of eEF2K of which NH125 is the most potent and specific in vitro [147]. However, its clinical value in cancer treatment has yet to be revealed.
TRPM6 has been identified as the gene mutated in patients suffering from familial hypomagnesemia with secondary hypocalcemia [87, 88]. Unfortunately, the majority of genetic lesions are frameshift mutations which lead to a premature arrest of the polypeptide chain and disrupt both the channel and kinase activities. Furthermore, Mg2+ conductance by the TRPM6 channel, rather than kinase activity, appears related to familial hypomagnesemia. Hence, modulation of TRPM6 protein kinase activity by pharmacological agents is unlikely to provide an effective treatment of this disease.
TRPM7 has been linked to ischemic brain damage [95, 148, 149]. Oxygen and glucose deprivation in neuronal cells is reported to mediate TRPM7 channel opening via a ROS-dependent mechanism, and the resulting Ca2+ influx induces cell death. Whether this is due to intracellular Ca2+ overload or a positive feedback loop in which Ca2+ influx triggers further ROS production is not known. A role for the alpha-kinase domain in anoxic cell death, if any, has not been established. As we described previously, kinase activity of TRPM7 is not required for channel activation. The opposite, however, is a possibility that cannot be excluded. The Ca2+ influx activates downstream signaling pathways that might implicate the TRPM7 alpha-kinase domain as well. Hence, specific TRPM7 kinase inhibitors are required to test this hypothesis. Although clinical application of TRPM7 kinase inhibitors is unlikely to be beneficial in cases of ischemic brain damage, TRPM7 may be a candidate drug target for the treatment of cancer. As was described before, and reported in recent publications [100, 150], changing TRPM7 expression levels affects cancer cell proliferation. But again, the role of its kinase domain therein remains to be established. Of current interest is the role of TRPM7 in cell migration and invasion. We have previously shown that the TRPM7 alpha-kinase associates with the cytoskeleton at sites of cell-ECM adhesion where it regulates the level of cytoskeletal tension, e.g. by myosin heavy chain phosphorylation, and the formation of adhesive structures called podosomes that permit cell invasion [93]. Knowing that TRPM7 responds to mechanical force, and steers cell polarity by initiating Ca2+ sparks [94], indicates that TRPM7 serves as mechanosensor that senses matrix properties and locally steers cytoskeletal remodeling to facilitate cell invasion. As the latter appears to be kinase dependent, it is tempting to speculate that inhibitors of the TRPM7 kinase may interfere with metastasis formation.
The development of relatively specific, small cell-permeable inhibitors of alpha-kinases could also provide important research tools to identify the physiological substrates and functions of these enzymes. These compounds can be used simply and rapidly and provide a complementary approach of genetic methods. An immediate question these inhibitors could help resolve is the role of the alpha-kinase domain in controlling the channel activity of TRPM6 and TRPM7, a debate that has been raging since the discovery of these channels at the turn of the century.
Conclusions and future perspectives
Alpha-kinases form a distinct family of protein kinases with a unique domain organization and specific substrate preference. A comprehensive search for alpha-kinase domains encoded by eukaryotic genomes has revealed a large number of novel alpha-kinases, many of which remain uncharacterized. Alpha-kinases show a remarkable functional diversity, which most likely arose by taxon specific gene duplications and recombination with other protein domains. To date, two members of this kinase family, eEF2K and TRPM7, have been linked to cancer growth and progression, while a loss of TRPM6 function contributes to familial hypomagnesia with secondary hypocalcemia. As the large majority of the alpha-kinase family remain to be characterized, additional roles for this intriguing set of proteins will undoubtedly be discovered.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Scheeff ED, Bourne PE. Structural evolution of the protein kinase-like superfamily. PLoS Comput Biol. 2005;1:e49. doi: 10.1371/journal.pcbi.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drennan D, Ryazanov AG. Alpha-kinases: analysis of the family and comparison with conventional protein kinases. Prog Biophys Mol Biol. 2004;85:1–32. doi: 10.1016/S0079-6107(03)00060-9. [DOI] [PubMed] [Google Scholar]
- 4.Ryazanov AG, Pavur KS, Dorovkov MV. Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr Biol. 1999;9:R43–R45. doi: 10.1016/s0960-9822(99)80006-2. [DOI] [PubMed] [Google Scholar]
- 5.Futey LM, Medley QG, Cote GP, Egelhoff TT. Structural analysis of myosin heavy chain kinase A from Dictyostelium. Evidence for a highly divergent protein kinase domain, an amino-terminal coiled-coil domain, and a domain homologous to the beta-subunit of heterotrimeric G proteins. J Biol Chem. 1995;270:523–529. doi: 10.1074/jbc.270.2.523. [DOI] [PubMed] [Google Scholar]
- 6.Cote GP, Luo X, Murphy MB, Egelhoff TT. Mapping of the novel protein kinase catalytic domain of Dictyostelium myosin II heavy chain kinase A. J Biol Chem. 1997;272:6846–6849. doi: 10.1074/jbc.272.11.6846. [DOI] [PubMed] [Google Scholar]
- 7.Clancy CE, Mendoza MG, Naismith TV, Kolman MF, Egelhoff TT. Identification of a protein kinase from Dictyostelium with homology to the novel catalytic domain of myosin heavy chain kinase A. J Biol Chem. 1997;272:11812–11815. doi: 10.1074/jbc.272.18.11812. [DOI] [PubMed] [Google Scholar]
- 8.Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, Erdjument-Bromage H, Tempst P, Parmer TG, Prostko CR, Germino FJ, Hait WN. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc Natl Acad Sci USA. 1997;94:4884–4889. doi: 10.1073/pnas.94.10.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinna LA, Ruzzene M. How do protein kinases recognize their substrates? Biochim Biophys Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 10.Vaillancourt JP, Lyons C, Cote GP. Identification of two phosphorylated threonines in the tail region of Dictyostelium myosin II. J Biol Chem. 1988;263:10082–10087. [PubMed] [Google Scholar]
- 11.Luck-Vielmetter D, Schleicher M, Grabatin B, Wippler J, Gerisch G. Replacement of threonine residues by serine and alanine in a phosphorylatable heavy chain fragment of Dictyostelium myosin II. FEBS Lett. 1990;269:239–243. doi: 10.1016/0014-5793(90)81163-i. [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen R, Ortiz PA, Carr-Schmid A, Nissen P, Kinzy TG, Andersen GR. Two crystal structures demonstrate large conformational changes in the eukaryotic ribosomal translocase. Nat Struct Biol. 2003;10:379–385. doi: 10.1038/nsb923. [DOI] [PubMed] [Google Scholar]
- 13.Clark K, Middelbeek J, Dorovkov MV, Figdor CG, Ryazanov AG, Lasonder E, van Leeuwen FN. The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. FEBS Lett. 2008;582:2993–2997. doi: 10.1016/j.febslet.2008.07.043. [DOI] [PubMed] [Google Scholar]
- 14.Crawley SW, Cote GP. Determinants for substrate phosphorylation by Dictyostelium myosin II heavy chain kinases A and B and eukaryotic elongation factor-2 kinase. Biochim Biophys Acta. 2008;1784:908–915. doi: 10.1016/j.bbapap.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Cote GP, Bukiejko U. Purification and characterization of a myosin heavy chain kinase from Dictyostelium discoideum . J Biol Chem. 1987;262:1065–1072. [PubMed] [Google Scholar]
- 16.Nagasaki A, Itoh G, Yumura S, Uyeda TQ. Novel myosin heavy chain kinase involved in disassembly of myosin II filaments and efficient cleavage in mitotic dictyostelium cells. Mol Biol Cell. 2002;13:4333–4342. doi: 10.1091/mbc.E02-04-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang W, Licate L, Warrick H, Spudich J, Egelhoff T. Differential localization in cells of myosin II heavy chain kinases during cytokinesis and polarized migration. BMC Cell Biol. 2002;3:19. doi: 10.1186/1471-2121-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo X, Crawley SW, Steimle PA, Egelhoff TT, Cote GP. Specific phosphorylation of threonine by the Dictyostelium myosin II heavy chain kinase family. J Biol Chem. 2001;276:17836–17843. doi: 10.1074/jbc.M009366200. [DOI] [PubMed] [Google Scholar]
- 19.Yumura S, Yoshida M, Betapudi V, Licate LS, Iwadate Y, Nagasaki A, Uyeda TQ, Egelhoff TT. Multiple myosin II heavy chain kinases: roles in filament assembly control and proper cytokinesis in Dictyostelium . Mol Biol Cell. 2005;16:4256–4266. doi: 10.1091/mbc.E05-03-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendoza L, Orozco E, Rodriguez MA, Garcia-Rivera G, Sanchez T, Garcia E, Gariglio P. Ehp53, an Entamoeba histolytica protein, ancestor of the mammalian tumour suppressor p53. Microbiology. 2003;149:885–893. doi: 10.1099/mic.0.25892-0. [DOI] [PubMed] [Google Scholar]
- 21.Yamaguchi H, Matsushita M, Nairn AC, Kuriyan J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol Cell. 2001;7:1047–1057. doi: 10.1016/s1097-2765(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 22.Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 23.Diggle TA, Seehra CK, Hase S, Redpath NT. Analysis of the domain structure of elongation factor-2 kinase by mutagenesis. FEBS Lett. 1999;457:189–192. doi: 10.1016/s0014-5793(99)01034-0. [DOI] [PubMed] [Google Scholar]
- 24.Somlyo AP, Somlyo AV. Ca2+sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 25.Collins JH, Korn ED. Actin activation of Ca2+ sensitive Mg2+ ATPase activity of Acanthamoeba myosin II is enhanced by dephosphorylation of its heavy chains. J Biol Chem. 1980;255:8011–8014. [PubMed] [Google Scholar]
- 26.Kuczmarski ER, Spudich JA. Regulation of myosin self-assembly: phosphorylation of Dictyostelium heavy chain inhibits formation of thick filaments. Proc Natl Acad Sci USA. 1980;77:7292–7296. doi: 10.1073/pnas.77.12.7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuznicki J, Albanesi JP, Cote GP, Korn ED. Supramolecular regulation of the actin-activated ATPase activity of filaments of Acanthamoeba Myosin II. J Biol Chem. 1983;258:6011–6014. [PubMed] [Google Scholar]
- 28.Egelhoff TT, Lee RJ, Spudich JA. Dictyostelium myosin heavy chain phosphorylation sites regulate myosin filament assembly and localization in vivo. Cell. 1993;75:363–371. doi: 10.1016/0092-8674(93)80077-r. [DOI] [PubMed] [Google Scholar]
- 29.Stites J, Wessels D, Uhl A, Egelhoff T, Shutt D, Soll DR. Phosphorylation of the Dictyostelium myosin II heavy chain is necessary for maintaining cellular polarity and suppressing turning during chemotaxis. Cell Motil Cytoskeleton. 1998;39:31–51. doi: 10.1002/(SICI)1097-0169(1998)39:1<31::AID-CM4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 30.Kolman MF, Futey LM, Egelhoff TT. Dictyostelium myosin heavy chain kinase A regulates myosin localization during growth and development. J Cell Biol. 1996;132:101–109. doi: 10.1083/jcb.132.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolman MF, Egelhoff TT. Dictyostelium myosin heavy chain kinase A subdomains. Coiled-coil and wd repeat roles in oligomerization and substrate targeting. J Biol Chem. 1997;272:16904–16910. doi: 10.1074/jbc.272.27.16904. [DOI] [PubMed] [Google Scholar]
- 32.Steimle PA, Licate L, Cote GP, Egelhoff TT. Lamellipodial localization of Dictyostelium myosin heavy chain kinase A is mediated via F-actin binding by the coiled-coil domain. FEBS Lett. 2002;516:58–62. doi: 10.1016/s0014-5793(02)02494-8. [DOI] [PubMed] [Google Scholar]
- 33.Egelhoff TT, Croft D, Steimle PA. Actin-activation of myosin heavy chain kinase A in Dictyostelium: a biochemical mechanism for the spatial regulation of myosin II filament disassembly. J Biol Chem. 2005;280:2879–2887. doi: 10.1074/jbc.M410803200. [DOI] [PubMed] [Google Scholar]
- 34.Russ M, Croft D, Ali O, Martinez R, Steimle PA. Myosin heavy-chain kinase A from Dictyostelium possesses a novel actin-binding domain that cross-links actin filaments. Biochem J. 2006;395:373–383. doi: 10.1042/BJ20051376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 36.Steimle PA, Naismith T, Licate L, Egelhoff TT. WD repeat domains target dictyostelium myosin heavy chain kinases by binding directly to myosin filaments. J Biol Chem. 2001;276:6853–6860. doi: 10.1074/jbc.M008992200. [DOI] [PubMed] [Google Scholar]
- 37.Medley QG, Gariepy J, Cote GP. Dictyostelium myosin II heavy-chain kinase A is activated by autophosphorylation: studies with Dictyostelium myosin II and synthetic peptides. Biochemistry. 1990;29:8992–8997. doi: 10.1021/bi00490a016. [DOI] [PubMed] [Google Scholar]
- 38.De la Roche MA, Smith JL, Betapudi V, Egelhoff TT, Cote GP. Signaling pathways regulating Dictyostelium myosin II. J Muscle Res Cell Motil. 2002;23:703–718. doi: 10.1023/a:1024467426244. [DOI] [PubMed] [Google Scholar]
- 39.Rico M, Egelhoff TT. Myosin heavy chain kinase B participates in the regulation of myosin assembly into the cytoskeleton. J Cell Biochem. 2003;88:521–532. doi: 10.1002/jcb.10361. [DOI] [PubMed] [Google Scholar]
- 40.Betapudi V, Mason C, Licate L, Egelhoff TT. Identification and characterization of a novel alpha-kinase with a von Willebrand factor A-like motif localized to the contractile vacuole and Golgi complex in Dictyostelium discoideum . Mol Biol Cell. 2005;16:2248–2262. doi: 10.1091/mbc.E04-07-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steimle PA, Yumura S, Cote GP, Medley QG, Polyakov MV, Leppert B, Egelhoff TT. Recruitment of a myosin heavy chain kinase to actin-rich protrusions in Dictyostelium . Curr Biol. 2001;11:708–713. doi: 10.1016/s0960-9822(01)00182-8. [DOI] [PubMed] [Google Scholar]
- 42.Egelhoff TT, Croft D, Steimle PA. Actin activation of myosin heavy chain kinase A in Dictyostelium: a biochemical mechanism for the spatial regulation of myosin II filament disassembly. J Biol Chem. 2005;280:2879–2887. doi: 10.1074/jbc.M410803200. [DOI] [PubMed] [Google Scholar]
- 43.Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryazanov AG, Rudkin BB, Spirin AS. Regulation of protein synthesis at the elongation stage. New insights into the control of gene expression in eukaryotes. FEBS Lett. 1991;285:170–175. doi: 10.1016/0014-5793(91)80798-8. [DOI] [PubMed] [Google Scholar]
- 45.Ryazanov AG, Natapov PG, Shestakova EA, Severin FF, Spirin AS. Phosphorylation of the elongation factor 2: the fifth Ca2+/calmodulin-dependent system of protein phosphorylation. Biochimie. 1988;70:619–626. doi: 10.1016/0300-9084(88)90245-3. [DOI] [PubMed] [Google Scholar]
- 46.Redpath NT, Price NT, Severinov KV, Proud CG. Regulation of elongation factor-2 by multisite phosphorylation. Eur J Biochem. 1993;213:689–699. doi: 10.1111/j.1432-1033.1993.tb17809.x. [DOI] [PubMed] [Google Scholar]
- 47.Carlberg U, Nilsson A, Nygard O. Functional properties of phosphorylated elongation factor 2. Eur J Biochem. 1990;191:639–645. doi: 10.1111/j.1432-1033.1990.tb19169.x. [DOI] [PubMed] [Google Scholar]
- 48.Ryazanov AG. Ca2+/calmodulin-dependent phosphorylation of elongation factor 2. FEBS Lett. 1987;214:331–334. doi: 10.1016/0014-5793(87)80081-9. [DOI] [PubMed] [Google Scholar]
- 49.Price NT, Redpath NT, Severinov KV, Campbell DG, Russell JM, Proud CG. Identification of the phosphorylation sites in elongation factor-2 from rabbit reticulocytes. FEBS Lett. 1991;282:253–258. doi: 10.1016/0014-5793(91)80489-p. [DOI] [PubMed] [Google Scholar]
- 50.Nairn AC, Bhagat B, Palfrey HC. Identification of calmodulin-dependent protein kinase III and its major Mr 1,00,000 substrate in mammalian tissues. Proc Natl Acad Sci USA. 1985;82:7939–7943. doi: 10.1073/pnas.82.23.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavur KS, Petrov AN, Ryazanov AG. Mapping the functional domains of elongation factor-2 kinase. Biochemistry. 2000;39:12216–12224. doi: 10.1021/bi0007270. [DOI] [PubMed] [Google Scholar]
- 52.Smith EM, Proud CG. cdc2-cyclin B regulates eEF2 kinase activity in a cell cycle- and amino acid-dependent manner. EMBO J. 2008;27:1005–1016. doi: 10.1038/emboj.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279:12220–12231. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 55.Proud CG. mTOR-mediated regulation of translation factors by amino acids. Biochem Biophys Res Commun. 2004;313:429–436. doi: 10.1016/j.bbrc.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 56.Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J. 2007;403:217–234. doi: 10.1042/BJ20070024. [DOI] [PubMed] [Google Scholar]
- 57.Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006;66:3015–3023. doi: 10.1158/0008-5472.CAN-05-1554. [DOI] [PubMed] [Google Scholar]
- 58.Hait WN, Wu H, Jin S, Yang JM. Elongation factor-2 kinase: its role in protein synthesis and autophagy. Autophagy. 2006;2:294–296. doi: 10.4161/auto.2857. [DOI] [PubMed] [Google Scholar]
- 59.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 60.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y, Matsushita M, Nairn AC, Damuni Z, Cai D, Frerichs KU, Hallenbeck JM. Mechanisms for increased levels of phosphorylation of elongation factor-2 during hibernation in ground squirrels. Biochemistry. 2001;40:11565–11570. doi: 10.1021/bi010649w. [DOI] [PubMed] [Google Scholar]
- 62.Dorovkov MV, Pavur KS, Petrov AN, Ryazanov AG. Regulation of elongation factor-2 kinase by pH. Biochemistry. 2002;41:13444–13450. doi: 10.1021/bi026494p. [DOI] [PubMed] [Google Scholar]
- 63.Parmer TG, Ward MD, Yurkow EJ, Vyas VH, Kearney TJ, Hait WN. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br J Cancer. 1999;79:59–64. doi: 10.1038/sj.bjc.6690012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu H, Zhu H, Liu DX, Niu TK, Ren X, Patel R, Hait WN, Yang JM. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009;69:2453–2460. doi: 10.1158/0008-5472.CAN-08-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White-Gilbertson S, Kurtz DT, Voelkel-Johnson C (2009) The role of protein synthesis in cell cycling and cancer. Mol Oncol (in press) [DOI] [PMC free article] [PubMed]
- 66.White SJ, Kasman LM, Kelly MM, Lu P, Spruill L, McDermott PJ, Voelkel-Johnson C. Doxorubicin generates a proapoptotic phenotype by phosphorylation of elongation factor 2. Free Radic Biol Med. 2007;43:1313–1321. doi: 10.1016/j.freeradbiomed.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakamura J, Aoyagi S, Nanchi I, Nakatsuka S, Hirata E, Shibata S, Fukuda M, Yamamoto Y, Fukuda I, Tatsumi N, Ueda T, Fujiki F, Nomura M, Nishida S, Shirakata T, Hosen N, Tsuboi A, Oka Y, Nezu R, Mori M, Doki Y, Aozasa K, Sugiyama H, Oji Y. Overexpression of eukaryotic elongation factor eEF2 in gastrointestinal cancers and its involvement in G2/M progression in the cell cycle. Int J Oncol. 2009;34:1181–1189. [PubMed] [Google Scholar]
- 68.Abdelmajid H, Leclerc-David C, Moreau M, Guerrier P, Ryazanov A. Release from the metaphase I block in invertebrate oocytes: possible involvement of Ca2+/calmodulin-dependent kinase III. Int J Dev Biol. 1993;37:279–290. [PubMed] [Google Scholar]
- 69.Ryazanov AG. Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett. 2002;514:26–29. doi: 10.1016/s0014-5793(02)02299-8. [DOI] [PubMed] [Google Scholar]
- 70.Heine M, Cramm-Behrens CI, Ansari A, Chu HP, Ryazanov AG, Naim HY, Jacob R. Alpha-kinase 1, a new component in apical protein transport. J Biol Chem. 2005;280:25637–25643. doi: 10.1074/jbc.M502265200. [DOI] [PubMed] [Google Scholar]
- 71.Hosoda T, Monzen K, Hiroi Y, Oka T, Takimoto E, Yazaki Y, Nagai R, Komuro I. A novel myocyte-specific gene Midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J Biol Chem. 2001;276:35978–35989. doi: 10.1074/jbc.M100485200. [DOI] [PubMed] [Google Scholar]
- 72.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 73.Corey DP. New TRP channels in hearing and mechanosensation. Neuron. 2003;39:585–588. doi: 10.1016/s0896-6273(03)00505-1. [DOI] [PubMed] [Google Scholar]
- 74.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 75.Perraud AL, Knowles HM, Schmitz C. Novel aspects of signaling and ion-homeostasis regulation in immunocytes. The TRPM ion channels and their potential role in modulating the immune response. Mol Immunol. 2004;41:657–673. doi: 10.1016/j.molimm.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 76.Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- 77.Chubanov V, Waldegger S, Mederos YSM, Vitzthum H, Sassen MC, Seyberth HW, Konrad M, Gudermann T. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci USA. 2004;101:2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol. 2006;127:525–537. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmitz C, Dorovkov MV, Zhao X, Davenport BJ, Ryazanov AG, Perraud AL. The channel kinases TRPM6 and TRPM7 are functionally nonredundant. J Biol Chem. 2005;280:37763–37771. doi: 10.1074/jbc.M509175200. [DOI] [PubMed] [Google Scholar]
- 80.Mei ZZ, Xia R, Beech DJ, Jiang LH. Intracellular coiled-coil domain engaged in subunit interaction and assembly of melastatin-related transient receptor potential channel 2. J Biol Chem. 2006;281:38748–38756. doi: 10.1074/jbc.M607591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Erler I, Al-Ansary DM, Wissenbach U, Wagner TF, Flockerzi V, Niemeyer BA. Trafficking and assembly of the cold-sensitive TRPM8 channel. J Biol Chem. 2006;281:38396–38404. doi: 10.1074/jbc.M607756200. [DOI] [PubMed] [Google Scholar]
- 82.Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 83.Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg-ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 84.Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, McNulty S. Tissue distribution profiles of the human TRPM cation channel family. J Recept Signal Transduct Res. 2006;26:159–178. doi: 10.1080/10799890600637506. [DOI] [PubMed] [Google Scholar]
- 85.Walder RY, Yang B, Stokes JB, Kirby PA, Cao X, Shi P, Searby CC, Husted RF, Sheffield VC (2009) Mice defective in Trpm6 show embryonic mortality and neural tube defects. Hum Mol Genet (in press) [DOI] [PMC free article] [PubMed]
- 86.Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31:166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 88.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet. 2002;31:171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 89.Chubanov V, Gudermann T, Schlingmann KP. Essential role for TRPM6 in epithelial magnesium transport and body magnesium homeostasis. Pflugers Arch. 2005;451:228–234. doi: 10.1007/s00424-005-1470-y. [DOI] [PubMed] [Google Scholar]
- 90.Paravicini TM, Yogi A, Mazur A, Touyz RM. Dysregulation of vascular TRPM7 and annexin-1 is associated with endothelial dysfunction in inherited hypomagnesemia. Hypertension. 2009;53:423–429. doi: 10.1161/HYPERTENSIONAHA.108.124651. [DOI] [PubMed] [Google Scholar]
- 91.Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- 92.Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, Sumimoto H, Ito Y, Mori Y, Inoue R. Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J Pharmacol Sci. 2004;95:403–419. doi: 10.1254/jphs.fp0040273. [DOI] [PubMed] [Google Scholar]
- 93.Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, Moolenaar WH, Jalink K, van Leeuwen FN. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 2006;25:290–301. doi: 10.1038/sj.emboj.7600931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 96.Numata T, Shimizu T, Okada Y. TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol. 2007;292:C460–C467. doi: 10.1152/ajpcell.00367.2006. [DOI] [PubMed] [Google Scholar]
- 97.Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98:245–253. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 98.Su LT, Agapito MA, Li M, Simonson WT, Huttenlocher A, Habas R, Yue L, Runnels LW. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J Biol Chem. 2006;281:11260–11270. doi: 10.1074/jbc.M512885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krapivinsky G, Mochida S, Krapivinsky L, Cibulsky SM, Clapham DE. The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron. 2006;52:485–496. doi: 10.1016/j.neuron.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 100.Jiang J, Li MH, Inoue K, Chu XP, Seeds J, Xiong ZG. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: role in cell proliferation. Cancer Res. 2007;67:10929–10938. doi: 10.1158/0008-5472.CAN-07-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clark K, Langeslag M, Figdor CG, van Leeuwen FN. Myosin II and mechanotransduction: a balancing act. Trends Cell Biol. 2007;17:178–186. doi: 10.1016/j.tcb.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 102.Langeslag M, Clark K, Moolenaar WH, van Leeuwen FN, Jalink K. Activation of TRPM7 channels by phospholipase C-coupled receptor agonists. J Biol Chem. 2007;282:232–239. doi: 10.1074/jbc.M605300200. [DOI] [PubMed] [Google Scholar]
- 103.Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci USA. 2006;103:15463–15468. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Clark K, Middelbeek J, van Leeuwen FN. Interplay between TRP channels and the cytoskeleton in health and disease. Eur J Cell Biol. 2008;87:631–640. doi: 10.1016/j.ejcb.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 105.Orr AW, Helmke BP, Blackman BR, Schwartz MA. Mechanisms of mechanotransduction. Dev Cell. 2006;10:11–20. doi: 10.1016/j.devcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 106.Numata T, Shimizu T, Okada Y. Direct mechano-stress sensitivity of TRPM7 channel. Cell Physiol Biochem. 2007;19:1–8. doi: 10.1159/000099187. [DOI] [PubMed] [Google Scholar]
- 107.Cao G, Hoenderop JG, Bindels RJ. Insight into the molecular regulation of the epithelial magnesium channel TRPM6. Curr Opin Nephrol Hypertens. 2008;17:373–378. doi: 10.1097/MNH.0b013e328303e184. [DOI] [PubMed] [Google Scholar]
- 108.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 109.Takezawa R, Schmitz C, Demeuse P, Scharenberg AM, Penner R, Fleig A. Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc Natl Acad Sci USA. 2004;101:6009–6014. doi: 10.1073/pnas.0307565101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Demeuse P, Penner R, Fleig A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127:421–434. doi: 10.1085/jgp.200509410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cao G, Thebault S, van der Wijst J, van der Kemp A, Lasonder E, Bindels RJ, Hoenderop JG. RACK1 inhibits TRPM6 activity via phosphorylation of the fused alpha-kinase domain. Curr Biol. 2008;18:168–176. doi: 10.1016/j.cub.2007.12.058. [DOI] [PubMed] [Google Scholar]
- 112.Cao G, van der Wijst J, van der Kemp A, van Zeeland F, Bindels RJ, Hoenderop JG. Regulation of the epithelial Mg2+ channel TRPM6 by estrogen and the associated repressor protein of estrogen receptor activity (REA) J Biol Chem. 2009;284:14788–14795. doi: 10.1074/jbc.M808752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matsushita M, Kozak JA, Shimizu Y, McLachlin DT, Yamaguchi H, Wei FY, Tomizawa K, Matsui H, Chait BT, Cahalan MD, Nairn AC. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/CHAK1. J Biol Chem. 2005;280:20793–20803. doi: 10.1074/jbc.M413671200. [DOI] [PubMed] [Google Scholar]
- 114.Penner R, Fleig A. The Mg2 + and Mg(2 +)-nucleotide-regulated channel-kinase TRPM7. Handb Exp Pharmacol. 2007;179:313–328. doi: 10.1007/978-3-540-34891-7_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG. Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to TRP ion channel. J Biol Chem. 2004;279:3708–3716. doi: 10.1074/jbc.M308820200. [DOI] [PubMed] [Google Scholar]
- 116.Clark K, Middelbeek J, Morrice NA, Figdor CG, Lasonder E, van Leeuwen FN. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS One. 2008;3:e1876. doi: 10.1371/journal.pone.0001876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dorovkov MV, Ryazanov AG. Phosphorylation of annexin I by TRPM7 channel-kinase. J Biol Chem. 2004;279:50643–50646. doi: 10.1074/jbc.C400441200. [DOI] [PubMed] [Google Scholar]
- 118.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 119.Callera GE, He Y, Yogi A, Montezano AC, Paravicini T, Yao G, Touyz RM. Regulation of the novel Mg2+ transporter transient receptor potential melastatin 7 (TRPM7) cation channel by bradykinin in vascular smooth muscle cells. J Hypertens. 2009;27:155–166. doi: 10.1097/hjh.0b013e3283190582. [DOI] [PubMed] [Google Scholar]
- 120.Touyz RM. Transient receptor potential melastatin 6 and 7 channels, magnesium transport, and vascular biology: implications in hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H1103–H1118. doi: 10.1152/ajpheart.00903.2007. [DOI] [PubMed] [Google Scholar]
- 121.Clark K, Middelbeek J, Lasonder E, Dulyaninova NG, Morrice NA, Ryazanov AG, Bresnick AR, Figdor CG, van Leeuwen FN. TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J Mol Biol. 2008;378:790–803. doi: 10.1016/j.jmb.2008.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ronen D, Ravid S. Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. J Biol Chem. 2009;284:24948–24957. doi: 10.1074/jbc.M109.023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bershadsky AD, Ballestrem C, Carramusa L, Zilberman Y, Gilquin B, Khochbin S, Alexandrova AY, Verkhovsky AB, Shemesh T, Kozlov MM. Assembly and mechanosensory function of focal adhesions: experiments and models. Eur J Cell Biol. 2006;85:165–173. doi: 10.1016/j.ejcb.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 124.Geiger B, Bershadsky A. Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell. 2002;110:139–142. doi: 10.1016/s0092-8674(02)00831-0. [DOI] [PubMed] [Google Scholar]
- 125.Gimona M, Buccione R. Adhesions that mediate invasion. Int J Biochem Cell Biol. 2006;38:1875–1892. doi: 10.1016/j.biocel.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 126.Burgstaller G, Gimona M. Actin cytoskeleton remodelling via local inhibition of contractility at discrete microdomains. J Cell Sci. 2004;117:223–231. doi: 10.1242/jcs.00839. [DOI] [PubMed] [Google Scholar]
- 127.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 128.Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 129.Roskoski R., Jr Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun. 2004;324:1155–1164. doi: 10.1016/j.bbrc.2004.09.171. [DOI] [PubMed] [Google Scholar]
- 130.Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24:7401–7409. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
- 131.Van Etten RA. c-Abl regulation: a tail of two lipids. Curr Biol. 2003;13:R608–R610. doi: 10.1016/s0960-9822(03)00528-1. [DOI] [PubMed] [Google Scholar]
- 132.Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005;118:3569–3572. doi: 10.1242/jcs.02470. [DOI] [PubMed] [Google Scholar]
- 133.Hubbard SR. Protein tyrosine kinases: autoregulation and small-molecule inhibition. Curr Opin Struct Biol. 2002;12:735–741. doi: 10.1016/s0959-440x(02)00383-4. [DOI] [PubMed] [Google Scholar]
- 134.Zhu G, Liu Y, Shaw S. Protein kinase specificity. A strategic collaboration between kinase peptide specificity and substrate recruitment. Cell Cycle. 2005;4:52–56. doi: 10.4161/cc.4.1.1353. [DOI] [PubMed] [Google Scholar]
- 135.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 136.Hovland R, Eikhom TS, Proud CG, Cressey LI, Lanotte M, Doskeland SO, Houge G. cAMP inhibits translation by inducing Ca2+/calmodulin-independent elongation factor 2 kinase activity in IPC-81 cells. FEBS Lett. 1999;444:97–101. doi: 10.1016/s0014-5793(99)00039-3. [DOI] [PubMed] [Google Scholar]
- 137.Diggle TA, Subkhankulova T, Lilley KS, Shikotra N, Willis AE, Redpath NT. Phosphorylation of elongation factor-2 kinase on serine 499 by cAMP-dependent protein kinase induces Ca2+/calmodulin-independent activity. Biochem J. 2001;353:621–626. doi: 10.1042/0264-6021:3530621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Knebel A, Morrice N, Cohen P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 2001;20:4360–4369. doi: 10.1093/emboj/20.16.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Knebel A, Haydon CE, Morrice N, Cohen P. Stress-induced regulation of eukaryotic elongation factor 2 kinase by SB 203580-sensitive and -insensitive pathways. Biochem J. 2002;367:525–532. doi: 10.1042/BJ20020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Redpath NT, Proud CG. Cyclic AMP-dependent protein kinase phosphorylates rabbit reticulocyte elongation factor-2 kinase and induces calcium-independent activity. Biochem J. 1993;293(Pt 1):31–34. doi: 10.1042/bj2930031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yamaguchi H, Matsushita M, Nairn AC, Kuriyan J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol Cell. 2001;7:1047–1057. doi: 10.1016/s1097-2765(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 143.Crawley SW, Cote GP. Identification of dimer interactions required for the catalytic activity of the TRPM7 alpha-kinase domain. Biochem J. 2009;420:115–122. doi: 10.1042/BJ20081405. [DOI] [PubMed] [Google Scholar]
- 144.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 145.Fabbro D, Garcia-Echeverria C. Targeting protein kinases in cancer therapy. Curr Opin Drug Discov Devel. 2002;5:701–712. [PubMed] [Google Scholar]
- 146.Johnson LN. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys. 2009;42:1–40. doi: 10.1017/S0033583508004745. [DOI] [PubMed] [Google Scholar]
- 147.Arora S, Yang JM, Kinzy TG, Utsumi R, Okamoto T, Kitayama T, Ortiz PA, Hait WN. Identification and characterization of an inhibitor of eukaryotic elongation factor 2 kinase against human cancer cell lines. Cancer Res. 2003;63:6894–6899. [PubMed] [Google Scholar]
- 148.Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, Kiyonaka S, Mori Y, Jones M, Forder JP, Golde TE, Orser BA, Macdonald JF, Tymianski M (2009) Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci (in press) [DOI] [PubMed]
- 149.Wei WL, Sun HS, Olah ME, Sun X, Czerwinska E, Czerwinski W, Mori Y, Orser BA, Xiong ZG, Jackson MF, Tymianski M, MacDonald JF. TRPM7 channels in hippocampal neurons detect levels of extracellular divalent cations. Proc Natl Acad Sci USA. 2007;104:16323–16328. doi: 10.1073/pnas.0701149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol. 2009;297:C493–C502. doi: 10.1152/ajpcell.00624.2008. [DOI] [PubMed] [Google Scholar]
- 151.Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 2004;32:D142–D144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 154.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 155.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]