

Abstract
Traumatic brain injury (TBI) survivors often suffer from a post-traumatic syndrome with deficits in learning and memory. Calcium (Ca2+) has been implicated in the pathophysiology of TBI-induced neuronal death. However, the role of long-term changes in neuronal Ca2+ function in surviving neurons and the potential impact on TBI-induced cognitive impairments are less understood. Here we evaluated neuronal death and basal free intracellular Ca2+ ([Ca2+]i) in acutely isolated rat CA3 hippocampal neurons using the Ca2+ indicator, Fura-2, at seven and thirty days after moderate central fluid percussion injury. In moderate TBI, cognitive deficits as evaluated by the Morris Water Maze (MWM), occur after injury but resolve after several weeks. Using MWM paradigm we compared alterations in [Ca2+]i and cognitive deficits. Moderate TBI did not cause significant hippocampal neuronal death. However, basal [Ca2+]i was significantly elevated when measured seven days post-TBI. At the same time, these animals exhibited significant cognitive impairment (F2,25 = 3.43, p < 0.05). When measured 30 days post-TBI, both basal [Ca2+ ]i and cognitive functions had returned to normal. Pretreatment with MK-801 blocked this elevation in [Ca2+]i and also prevented MWM deficits. These studies provide evidence for a link between elevated [Ca2+]i and altered cognition. Since no significant neuronal death was observed, the alterations in Ca2+ homeostasis in the traumatized, but surviving neurons may play a role in the pathophysiology of cognitive deficits that manifest in the acute setting after TBI and represent a novel target for therapeutic intervention following TBI.
Keywords: Calcium dynamics, Fura-2, Acute isolation of hippocampal neurons, Morris water maze, Neuronal death, Sprague, Dawley rats
Traumatic brain injury (TBI) survivors often suffer from a post-traumatic syndrome in the sub-acute period following injury [1,28] that include headaches, fatigue, altered mood and cognitive impairments [1,18,25,28]. Symptoms can linger long after injury before resolving. Animal models of head injury have been utilized to characterize the temporal nature of TBI-induced cognitive deficits [15,28,36]. In a more severe TBI paradigm, hippocampal cell loss has been associated with cognitive deficits [2,8,12]; however, little is understood of the underlying molecular mechanisms that cause cognitive impairments following mild to moderate TBI that do not cause significant neuronal death.
Calcium (Ca2+) is a major signaling molecule in neurons. While, brief elevations in free intracellular calcium concentration ([Ca2+]i) are required for normal brain function, as in synaptic transmission and learning-memory, prolonged elevations in [Ca2+]i have been implicated in triggering cell death [27] and are also thought to underlie memory disturbances associated with aging [24,30]. Calcium has long been implicated in the pathophysiology of TBI [7,8,14,19,33]. We recently demonstrated that TBI causes a long-lasting plateau of elevated [Ca2+]i and altered Ca2+ homeostatic mechanisms in hippocampal neurons that survive moderate TBI [29]. These alterations in Ca2+ homeostasis following TBI may play an important role in mediating both transient and long-term changes in neuronal function.
The majority of TBI studies have examined alterations in Ca2+ regulation in the acute time period of the injury. However, few studies have investigated potential longer lasting changes in [Ca2+]i levels in traumatized, surviving neurons that may underlie some of the sequalae associated with TBI in the sub-acute setting [29]. To address this issue, we investigated alterations in neuronal Ca2+ levels during the time period when cognitive deficits are manifested secondary to TBI. Using the moderate rather than the severe TBI injury model, it is possible to induce cognitive deficits in rats that resolve several weeks after the injury and do not cause significant hippocampal cell death. Thus, moderate TBI in rats provides a useful model to compare alterations in [Ca2+]i and cognitive deficits. Basal [Ca2+]i levels were evaluated in acutely isolated CA3 neurons 7 and 30-days after moderate central fluid percussion injury (c-FPI) using the ratiometric, fluorescent Ca2+ indicator, Fura-2 [23,29]. In addition, we utilized the Morris Water Maze (MWM) [20] to assess cognitive deficits at these same time points after TBI. Exploration of Ca2+ dynamics in the sub-acute setting following TBI may help elucidate the basis of some of the longer-lasting symptoms of the post-traumatic syndrome.
All animal use procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by VCU’s Institutional Animal Care and Use Committee. The FPI device used to produce experimental TBI was identical to that previously used on rodents and described in details elsewhere [5,15]. Animals were surgically prepared 24-h before sham injury or TBI as described previously [5,15]. Animals in the TBI group received a moderate c-FPI (2.0 ± 0.02 atm). Animals in the sham group were coupled to the injury device, but no fluid pulse was delivered. Control animals received neither anesthesia nor surgery.
Hippocampal CA3 neurons were acutely isolated from control, sham-operated and TBI-injured animals at different times after TBI using methods previously described [23,29]. The CA3 region tissue chucks from the hippocampal slices (450 μM) obtained from the rapidly dissected brains of TBI-injured and sham-operated rats was triturated with a series of Pasteur pipettes of decreasing diameter at 4°C, resulting in an even suspension of CA3 hippocampal cells that were then plated on poly-L-lysine coated glass coverslip chambers (Nunc, Naperville, IL) and used for experimentation.
Changes in neuronal [Ca2+]i were measured using the ratiometric, high affinity (Kd ≈ 224 nM) fluorescent Ca2+ indicator, Fura-2 [23,29]. The Temporal Module of the Perkin-Elmer Life Sciences Imaging Suite was used to control image acquisition and processing. Ratio measurements were taken at 10-s intervals for 60 s, averaged, and calibrated according to the Grynkiewicz equation [9]. An in situ Ca2+ calibration curve was employed to convert fluorescent ratios to Ca2+ concentrations using standard procedures [23,29].
For cell death analysis, animals were anesthetized and brains were fixed using 4% paraformaldehyde on post-injury day 7 and 30 [23,29]. Coronal brain slices (40 μm) were taken at least 80-μm apart and cresyl violet-stained neurons were counted per unit area (150 μm × 230 μm) from at least six serial sections and averaged as neurons per region for each animal.
The MWM was used to assess cognitive performance on post-injury days 7 and 30 as described previously [15]. Rats completed four trials per day. On each trial, the starting location was randomized to one of four starting positions (North, East, South, and West). The rat was placed in the maze facing the wall, and the latency to reach the hidden goal platform and swim speeds were recorded on each trial. The mean of these four daily trials was calculated and is presented in Fig. 2.
Fig. 2.

Performance in the MWM seven and thirty day post-moderate central fluid percussion injury. (A) Seven days post-TBI (open circles), animals demonstrated a significant (p < 0.015) deficit in the MWM as indicated by an increased goal latency compared to sham animals (filled circles). In contrast, 30 days post-TBI (filled triangles), animals did not demonstrate a significant deficit in the MWM compared to sham animals. (B) No significant differences (p = 0.132) in the mean swim speed were observed between sham-operated and TBI animals 7 and 30-days post-injury. (n = 9 animals each for 7 and 30-days post-TBI and n =10 for sham-operated animals, *p <0.05). Data represented as mean ± S.E.M.
Data analyses of [Ca2+]i imaging was performed as described previously [29]. The means of each group of neurons from each animal were used to evaluate results and conduct statistical analysis. Student’s t-test or One-Way ANOVA were applied when appropriate to compare [Ca2+]i among groups. The Tukey test was used for all post hoc multiple comparisons of [Ca2+]i. A split-plot ANOVA [3 (group) × 5 (day)] was used to analyze maze latencies. The post hoc Fishers test was utilized to make group comparisons when appropriate. Statistical analyses were performed using SigmaStat 2.0 and SPLUS (Insightful Corp., Seattle, WA) and graphs generated with SigmaPlot 8.0 (SPSS Inc., Chicago, IL).
To investigate the potential role of Ca2+ in the sub-acute period of TBI, we measured neuronal [Ca2+]i 7-days after moderate c-FPI. Acutely isolated CA3 neurons harvested 7-days after TBI manifested a mean basal [Ca2+]i of 250.72 ± 12.50 nM (n = 47 neurons, 5 animals), significantly higher than mean basal [Ca2+]i in CA3 hippocampal neurons harvested from age-matched control (143.92 ± 11.36 nM, n = 60, 6 animals, p<0.05) and sham animals (145.10 ± 10.25 nM, n = 62, 6 animals, p <0.05, Fig. 1). No statistical differences were observed between the control and sham groups. Analysis of the population distributions of basal [Ca2+]i revealed values ≤200 nM in approximately 75% of control and sham neurons, respectively. In contrast, only 40% neurons 7-days post-TBI had basal values ≤200 nM. These histograms demonstrated a shift in the population of neurons from the TBI animals to higher basal [Ca2+]i levels (data not shown, [29]). We have also measured [Ca2+]i in CA3 hippocampal neurons harvested at 1-day after TBI and observed that [Ca2+]i was also elevated (292 ± 12.45 nM, n = 50, 5 animals) at this early time point compared to age-matched or sham-operated animals [29].
Fig. 1.
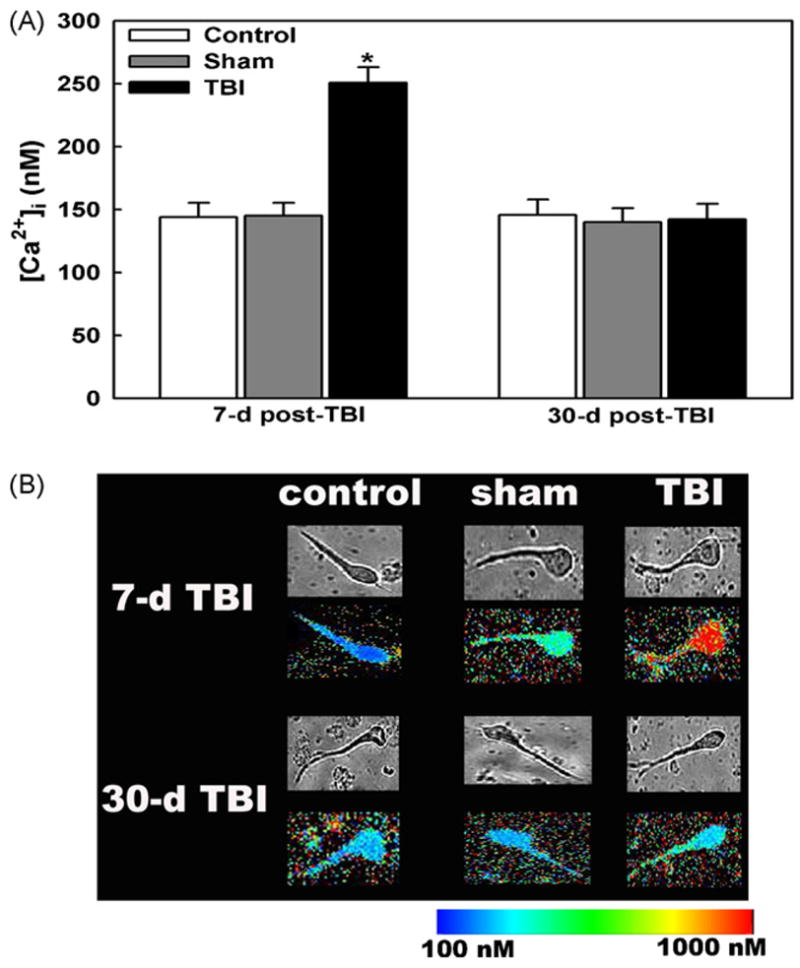
Basal neuronal [Ca2+]i in CA3 hippocampal neurons seven and thirty days after moderate central fluid percussion injury. (A) Acutely isolated CA3 hippocampal neurons 7-days post-TBI manifested significant elevations in [Ca2+]i compared to sham-operated or age-matched control neurons. Basal neuronal [Ca2+]i 30-days after TBI did not manifest significant differences in [Ca2+]i compared to sham or control neurons (p = 0.71). There were no significant differences between age-matched control and sham-operated neurons at both the time points. “p <0.05, ANOVA and Tukey post hoc test, n = 5–6 animals for each condition with a minimum 5–7 neurons per animal for the time points studied. Data are represented as mean ± S.E.M. (B) Pseudocolor images of representative control, sham and TBI neurons 7 and 30-days post-TBI. Basal [Ca2+ ]i were low in the control, sham-operated and 30-day TBI neurons but were higher in 7-day TBI neurons. The top panel at each time point shows the phase-bright photomicrographs of the representative control, sham and TBI neurons. The data shown were the representative of 5 animals with over 20 neurons sampled per condition.
We then measured neuronal [Ca2+]i 30-days after moderate c-FPI to determine if the alterations in Ca2+ levels observed 7-days post-injury persisted into a chronic setting. In contrast to the sub-acute period, the basal [Ca2+]i of acutely isolated CA3 neurons 30-days post-TBI (142.31 ± 12.26 nM, n = 63, 6 animals) were not significantly different (p = 0.71, t-test, Fig. 1) from 30-days post-shams (139.82 ± 11.42 nM, n = 65, 6 animals) or age-matched control neurons (145.56 ± 12.26 nM, n = 60, 6 animals). The distribution of [Ca2+]i in neurons from sham and 30-day post-TBI animals were similar with greater than 80% of basal neuronal [Ca2+]i ≤ 200nM, respectively (data not shown, [29]).
To investigate the potential significance of alterations in Ca2+ levels measured in traumatized neurons, we assessed cognitive function in rats 7 and 30-days after moderate c-FPI or sham operation using the MWM paradigm [20]. With the MWM, a shorter latency to find the goal platform indicates better cognitive performance. A split-plot ANOVA [3 (group) × 5 (day)] revealed significant differences between the three groups in (F2,25 = 3.43, p<0.05). To examine specific group differences in the MWM, the Fisher LSD test was used. The results of this test indicated that the 7-day post-TBI group exhibited longer goal latencies than the sham-injured group (p< 0.015). However, the 30-day post-TBI group’s goal latency was not significantly different from the sham group’s goal latency (Fig. 2A). In addition, a single-factor ANOVA (group) revealed none of the groups’ swim speeds significantly differed (F225 = 2.20, p<0.05) suggesting that the observed differences in goal latency were not due to injury-induced motor impairment (Fig. 2B). Thus, the TBI-induced cognitive deficits indicated by MWM were similar in temporal profile to the alterations in Ca2+ levels after trauma.
To establish a link between elevated [Ca2+]i and poor performance on MWM, we measured [Ca2+]i in neurons dissociated from animals treated with NMDA-Ca2+ channel antagonist MK-801 (0.3mg/kg, i.p.) 15-min prior to or 15-min after TBI. Calcium imaging studies revealed that MK-801 pretreatment blocked the elevated [Ca2+]i due to TBI and gave [Ca2+]i values (148 ± 11.38 nM, n = 50, 6 animals) that were not significantly different (p = 0.65) from sham controls 7-days post-injury. In contrast, administering MK-801 15-min after the TBI did not block the rise of [Ca2+]i and these values (261 ± 14.11 nM, n = 55, 5 animals) were not significantly different (p = 0.55) from untreated TBI animals, but were significantly different (p < 0.01) from sham controls. We have previously demonstrated that administration of MK-801 only before but not after the TBI significantly reduced the goal latency in MWM paradigm [10]. Together, these results demonstrate a link between elevated [Ca2+]i and poor performance on MWM following TBI.
Moderate c-FPI has been well characterized and shown not to induce neuronal death in the rat hippocampus [5,15,29]. To con firm this finding under the conditions of our study, we estimated neuronal survival in various regions of hippocampus 7 and 30-days after injury in sham and TBI animals. As shown in Fig. 3, there were no significant differences (p > 0.25, t-test) in the number of cresyl violet-stained neurons per unit area in sham-operated or TBI animals in CA1, CA3 or dentate gyrus. Further, we have demonstrated that the dissociation process or apoptosis does not significantly contribute to neuronal death at 7-days following TBI [29]. These results demonstrated that neuronal death was not the cause of the elevated [Ca2+]i levels or cognitive deficits observed 7-days post-TBI.
Fig. 3.
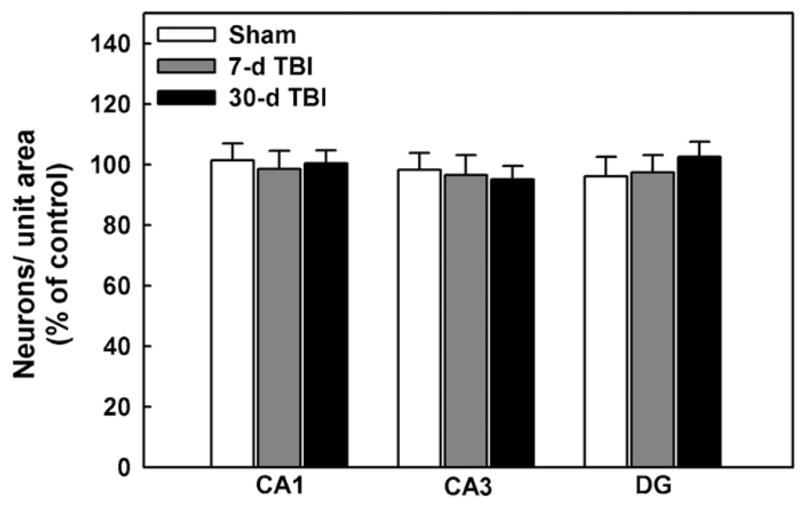
Moderate central fluid percussion injury did not cause significant neuronal death in various regions of hippocampus 7 and 30-days post-TBI. TBI did not cause a significant difference in the number of cresyl-violet stained CA1, CA3 and dentate gyrus (DG) hippocampal neurons per field observed 7 or 30-days after injury in sham-operated and TBI animals compared to naïve controls (p > 0.25, t-test, n = 5 animals). Data are expressed as percent of neuronal density of naïve control animals that did not undergo sham or TBI treatments.
TBI is a common cause of morbidity and mortality. Survivors of TBI often suffer with difficulties in attention, memory and learning [1,28] and these symptoms also manifest as a post-concussive syndrome [3,26,35]. Cognitive deficits such as sensorimotor and working memory deficits, delay-dependent and spatial memory impairments have been characterized in humans and animal models of TBI [31]. These cognitive deficits have been observed in the absence of significant neuronal death and are reported to reverse in the chronic period after TBI [5,15,28,36], which parallels with alterations in [Ca2+]i dynamics in our study. These findings suggest that some alteration in the physiology of traumatized, surviving neurons must exist to underlie these reversible impairments in cognition. Indeed, we have recently demonstrated that lasting alterations in Ca2+ homeostasis occur in the sub-acute time period in neurons following TBI [29]. These long-term, but reversible changes in Ca2+ dynamics mirrored the TBI-induced cognitive impairments in learning and memory 7-days post-TBI, but not observed in shams or 30-days post-TBI.
Delayed neuronal death is associated with massive elevations in [Ca2+]i and typically occurs 1–3 days after injury. In this study, neuronal cell loss was evaluated in the CA1, CA3 dentate gyrus regions of the hippocampus in sham-operated and TBI animals at 7 and 30-days post-injury. The results are shown in Fig. 3 and are presented as percent cell loss in comparison to naïve control animals that did not receive sham or TBI treatments. The data demonstrate no significant differences between sham and TBI animals at 7 or 30-days post-TBI in any hippocampal region under conditions that did cause changes in cognitive and [Ca2+]i levels at 7-days post-injury. This data is in agreement with prior studies [5,15,29], showing that moderate c-FPI in the rat did not cause significant hippocampal cell death. These findings demonstrate that the elevations in neuronal [Ca2+]i after TBI were not occurring due to delayed neuronal death, but rather were occurring in surviving neurons in a state of altered Ca2+ homeostasis.
Calcium is a major second messenger in neurons and plays a pivotal role in various aspects of cognition such as learning, memory consolidation and long-term potentiation (LTP)–a cellular correlate of learning and memory in brain. It has been reported that hippocampal slices derived from lateral fluid percussion injured mice demonstrate an inability to induce LTP 7-days post-injury [27] contributing to the cognitive impairments associated with TBI. Age-related declines in working memory have been attributed to greater Ca2+ currents via increased expression of L-type Ca2+ channel protein [32]. Further, these alterations in memory reverse with chronic administration of Ca2+ channel antagonist, nimodipine [13,32]. In addition, Ca2+ chelation has been reported to lower elevated Ca2+ in aged animals that improved spatial learning in MWM paradigm and also enhanced LTP in hippocampal slices [30]. These studies, in agreement with our findings that pretreatment with MK-801 blocked elevations in [Ca2+]i following TBI and that this pharmacological manipulation also prevents MWM deficits [10] indicate that elevated [Ca2+]i are associated with memory impairments and restoration of altered Ca2+ dynamics ameliorates the cognitive deficits.
Calcium regulates numerous enzyme systems that affect neuronal physiology and gene transcription [34]. Prolonged, but reversible and sub-excitotoxic, elevations in [Ca2+]i that do not cause neuronal death have been demonstrated to cause alterations in neuronal excitability [27] and long-term changes in gene expression [6]. Similarly, changes in the expression of transcription factors have also been reported in the acute setting of TBI [11,21,22]. It is possible that the prolonged alterations in Ca2+ homeostasis following TBI may induce persistent changes in gene expression and neuronal function that may ultimately underlie some of the pathophysiological sequelae of TBI in the sub-acute setting.
We recently demonstrated elevated [Ca2+]i and altered Ca2+ homeostatic mechanisms in hippocampal neurons that survive moderate TBI [29]. This investigation extends these previous observations and demonstrate a reversible functional pathology and how it mirrors alterations in [Ca2+]i dynamics. Further this study provides pharmacological data that demonstrates a direct correlation between [Ca2+]i alterations and cognitive impairments in the absence of significant neuronal death.
In the clinical setting of repeated, mild TBI, cognitive deficits can become more long-term in nature [4,16,17]. Similarly, alterations in neuronal Ca2+ homeostasis in this setting may become more persistent and underlie more significant cognitive impairments. The results of this study suggest that prolonged, but reversible alterations in the Ca2+ levels of traumatized, surviving neurons may underlie reversible cognitive deficits of the post-traumatic syndrome. An enhanced understanding of the mechanisms of TBI-induced alterations in Ca2+ homeostasis may provide attractive targets for therapeutic interventions and contribute to the treatment and relief of some of the deficits associated with TBI.
Acknowledgments
NINDS Grants RO1NS051505 and RO1NS052529, and award UO1NS058213 from the NIH CounterACT Program through NINDS to R.J.D. supported this study. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
Footnotes
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit:
References
- 1.Capruso DX, Levin HS. Cognitive impairment following closed head injury. Neurol Clin. 1992;10:879–893. [PubMed] [Google Scholar]
- 2.Clausen F, Lewen A, Marklund N, Olsson Y, McArthur DL, Hillered L. Correlation of hippocampal morphological changes and morris water maze performance after cortical contusion injury in rats. Neurosurgery. 2005;57:154–163. doi: 10.1227/01.neu.0000163412.07546.57. [DOI] [PubMed] [Google Scholar]
- 3.Corrigan JD, Whiteneck G, Mellick D. Perceived needs following traumatic brain injury. J Head Trauma Rehabil. 2004;19:205–216. doi: 10.1097/00001199-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 4.DeFord SM, Wilson MS, Rice AC, Clausen T, Rice LK, Barabnova A, Bullock R, Hamm RJ. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma. 2002;19:427–438. doi: 10.1089/08977150252932389. [DOI] [PubMed] [Google Scholar]
- 5.Delahunty TM, Jiang JY, Gong QZ, Black RT, Lyeth BG. Differential consequences of lateral and central fluid percussion brain injury on receptor coupling in rat hippocampus. J Neurotrauma. 1995;12:1045–1057. doi: 10.1089/neu.1995.12.1045. [DOI] [PubMed] [Google Scholar]
- 6.DeLorenzo RJ, Morris TA. Long-term modulation of gene expression in epilepsy. Neuroscientist. 1999;5:86–99. [Google Scholar]
- 7.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 8.Fineman I, Hovda DA, Smith M, Yoshino A, Becker DP. Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res. 1993;624:94–102. doi: 10.1016/0006-8993(93)90064-t. [DOI] [PubMed] [Google Scholar]
- 9.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 10.Hamm RJ, O’Dell DM, Pike BR, Lyeth BG. Cognitive impairment following traumatic brain injury: the effect of pre- and post-injury administration of scopolamine and MK-801. Brain Res Cogn Brain Res. 1993;1:223–226. doi: 10.1016/0926-6410(93)90006-q. [DOI] [PubMed] [Google Scholar]
- 11.Hayes RL, Yang K, Raghupathi R, McIntosh TK. Changes in gene expression following traumatic brain injury in the rat. J Neurotrauma. 1995;12:779–790. doi: 10.1089/neu.1995.12.779. [DOI] [PubMed] [Google Scholar]
- 12.Hong J, Cui J, Zhou Y, Gao J. Study on cognition disorder and morphologic change of neurons in hippocampus area following traumatic brain injury in rats. Chin J Traumatol. 2002;5:36–39. [PubMed] [Google Scholar]
- 13.Levere TE, Walker A. Old age and cognition: enhancement of recent memory in aged rats by the calcium channel blocker nimodipine. Neurobiol Aging. 1992;13:63–66. doi: 10.1016/0197-4580(92)90010-u. [DOI] [PubMed] [Google Scholar]
- 14.Lusardi TA, Smith DH, Wolf JA, Meaney DF. The separate roles of calcium and mechanical forces in mediating cell death in mechanically injured neurons. Biorheology. 2003;40:401–409. [PubMed] [Google Scholar]
- 15.Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, Young HF, Hayes RL. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. [DOI] [PubMed] [Google Scholar]
- 16.Macciocchi SN, Barth JT, Littlefield LM. Outcome after mild head injury. Clin Sports Med. 1998;17:27–36. doi: 10.1016/s0278-5919(05)70058-2. [DOI] [PubMed] [Google Scholar]
- 17.Matser JT, Kessels AG, Jordan BD, Lezak MD, Troost J. Chronic traumatic brain injury in professional soccer players. Neurology. 1998;51:791–796. doi: 10.1212/wnl.51.3.791. [DOI] [PubMed] [Google Scholar]
- 18.McAllister TW, Arciniegas D. Evaluation and treatment of postconcussive symptoms. NeuroRehabilitation. 2002;17:265–283. [PubMed] [Google Scholar]
- 19.McIntosh TK, Saatman KE, Raghupathi R. Calcium and the pathogenesis of traumatic CNS injury: cellular and molecular mechanisms. Neuroscientist. 1997;3:169–175. [Google Scholar]
- 20.Morris RG, Garrud P, Rawlins JN, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 21.Morrison B, III, Eberwine JH, Meaney DF, McIntosh TK. Traumatic injury induces differential expression of cell death genes in organotypic brain slice cultures determined by complementary DNA array hybridization. Neuroscience. 2000;96:131–139. doi: 10.1016/s0306-4522(99)00537-0. [DOI] [PubMed] [Google Scholar]
- 22.Raghupathi R, McIntosh TK. Regionally and temporally distinct patterns of induction of c-fos, c-jun and junB mRNAs following experimental brain injury in the rat. Brain Res Mol Brain Res. 1996;37:134–144. doi: 10.1016/0169-328x(95)00289-5. [DOI] [PubMed] [Google Scholar]
- 23.Raza M, Blair RE, Sombati S, Carter DS, Deshpande LS, DeLorenzo RJ. Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc Natl Acad Sci USA. 2004;101:17522–17527. doi: 10.1073/pnas.0408155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raza M, Deshpande LS, Blair RE, Carter DS, Sombati S, DeLorenzo RJ. Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci Lett. 2007;418:77–81. doi: 10.1016/j.neulet.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenthal M, Christensen BK, Ross TP. Depression following traumatic brain injury. Arch Phys Med Rehabil. 1998;79:90–103. doi: 10.1016/s0003-9993(98)90215-5. [DOI] [PubMed] [Google Scholar]
- 26.Ryan LM, Warden DL. Post concussion syndrome. Int Rev Psychiatry. 2003;15:310–316. doi: 10.1080/09540260310001606692. [DOI] [PubMed] [Google Scholar]
- 27.Schwarzbach E, Bonislawski DP, Xiong G, Cohen AS. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006;16:541–550. doi: 10.1002/hipo.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw NA. The neurophysiology of concussion. Prog Neurobiol. 2002;67:281–344. doi: 10.1016/s0301-0082(02)00018-7. [DOI] [PubMed] [Google Scholar]
- 29.Sun DA, Deshpande LS, Baranova A, Wilson MS, Hamm RJ, DeLorenzo RJ. Traumatic brain injury causes a long-lasting calcium-plateau of elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons surviving the brain injury. Eur J Neurosci. 2008;27:1659–1672. doi: 10.1111/j.1460-9568.2008.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tonkikh A, Janus C, El-Beheiry H, Pennefather PS, Samoilova M, McDonald P, Ouanounou A, Carlen PL. Calcium chelation improves spatial learning and synaptic plasticity in aged rats. Exp Neurol. 2006;197:291–300. doi: 10.1016/j.expneurol.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Vakil E. The effect of moderate to severe traumatic brain injury (TBI) on different aspects of memory: a selective review. J Clin Exp Neuropsychol. 2005;27:977–1021. doi: 10.1080/13803390490919245. [DOI] [PubMed] [Google Scholar]
- 32.Veng LM, Mesches MH, Browning MD. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein [alpha] 1D (Cav1.3) in area CA1 of the hippocampus and both are amelio-rated by chronic nimodipine treatment. Mol Brain Res. 2003;110:193–202. doi: 10.1016/s0169-328x(02)00643-5. [DOI] [PubMed] [Google Scholar]
- 33.Weber JT, Rzigalinski BA, Ellis EF. Traumatic injury of cortical neurons causes changes in intracellular calcium stores and capacitative calcium influx. J Biol Chem. 2001;276:1800–1807. doi: 10.1074/jbc.M009209200. [DOI] [PubMed] [Google Scholar]
- 34.West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang CC, Tu YK, Hua MS, Huang SJ. The association between the postconcussion symptoms and clinical outcomes for patients with mild traumatic brain injury. J Trauma. 2007;62:657–663. doi: 10.1097/01.ta.0000203577.68764.b8. [DOI] [PubMed] [Google Scholar]
- 36.Zohar O, Schreiber S, Getslev V, Schwartz JP, Mullins PG, Pick CG. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience. 2003;118:949–955. doi: 10.1016/s0306-4522(03)00048-4. [DOI] [PubMed] [Google Scholar]