

1. Introduction
Inhibition of cellular replication is one characteristic of cancer cells that has been effectively exploited in the past for the development of anticancer agents. Most of the drugs currently used to kill cancer cells inhibit the synthesis of DNA or interfere with its function in one manner or another. For a cell to divide into two cells, it must replicate all components including its genome, and unlike the synthesis of other major macromolecules (protein, RNA, lipid, etc.), the synthesis of DNA does not occur to a great degree in quiescent cells. Since most cells in an adult organism are quiescent and are not in the process of duplicating their genome, targeting DNA replication affords some level of selectivity. Of course, certain tissues (bone marrow, gastrointestinal, hair follicles, etc.) are in a replicative state, and all cells must continually repair their DNA. Therefore, inhibition of DNA replication in normal tissues results in considerable toxicity that limits the amount of drug that can be tolerated by the patient. In spite of this problem, very effective anticancer drugs have been developed that increase survival and, in some cases, cure the patient of his or her disease.
Human cells have the capacity to salvage purines and pyrimidines for the synthesis of deoxyribonucleotides that are used for DNA synthesis, and analogues of these nucleotide precursors have proven to be an important class of anticancer agents. There are a total of 14 purine and pyrimidine antimetabolites that are approved by the FDA for the treatment of cancer (Table 1), which account for nearly 20% of all drugs that are used to treat cancer. Some of the first compounds approved by the FDA for the treatment of cancer were in this class of compounds. 6-Mercaptopurine was approved in 1953 for the treatment of childhood leukemia, where it is curative and is still the standard of treatment for this disease. Since 1991, nine nucleoside analogues have been approved by the FDA for the treatment of various malignancies. Four of these new agents were approved since 2004, and there are numerous agents that are currently being evaluated in clinical trials. These recent FDA approvals indicate that the design and synthesis of new nucleoside analogues is still a productive area for discovering new drugs for the treatment of cancer. In general, these compounds have been most useful in the treatment of hematologic malignancies, and even though there is still room for significant improvements in the treatment of these diseases, some of the newer agents are finding use in the treatment of solid tumors.
Table 1.
FDA Approved Purine and Pyrimidine Antimetabolites
| drug | date approved |
|---|---|
| 5-aza-2′-deoxycytidine (decitabine) | 2006 |
| O6-methylarabinofuranosyl guanine (nelarabine) | 2005 |
| 2′-fluoro-2′-deoxyarabinofuranosyl-2-chloroadenine (clofarabine) | 2004 |
| 5-aza-cytidine (vidaza) | 2004 |
| N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine (capecitabine) | 1998 |
| 2,2-difluoro-2′-deoxycytidine (gemcitabine) | 1996 |
| 2-chloro-2′-deoxyadenosine (cladribine) | 1992 |
| arabinofuranosyl-2-fluoroadenine (fludarabine) | 1991 |
| 2′-deoxycoformycin (pentostatin) | 1991 |
| 5-fluoro-2′-deoxyuridine (floxuridine) | 1970 |
| arabinofuranosylcytosine (cytarabine) | 1969 |
| 6-thioguanine | 1966 |
| 5-fluorouracil | 1962 |
| 6-mercaptopurine | 1953 |
The basic mechanism of action of purine and pyrimidine antimetabolites is similar. These compounds diffuse into cells (usually with the aid of a membrane transporter1) and are converted to analogues of cellular nucleotides by enzymes of the purine or pyrimidine metabolic pathway. These metabolites then inhibit one or more enzymes that are critical for DNA synthesis, causing DNA damage and induction of apoptosis.2 Even though the compounds in this class are structurally similar and share many mechanistic details, it is clear that subtle quantitative and qualitative differences in the metabolism of these agents and their interactions with target enzymes can have a profound impact on their antitumor activity. As noted by Plunkett and Gandhi,3 “one of the remarkable features of purine and pyrimidine nucleoside analogues that remains unexplained is how drugs with such similar structural features, that share metabolic pathways, and elements of their mechanism of action show such diversity in their clinical activities”. Possibly the best example of this fact is the newly approved drug, clofarabine, which differs from cladribine by only one fluorine atom, because it has demonstrated excellent efficacy in the treatment of relapsed and refractory pediatric acute lymphoblastic leukemia, whereas cladribine is not effective against this disease. These clinical results indicate that the biochemical actions of clofarabine are sufficiently different from that of cladribine to impart unique clinical activities. This and other examples indicate that small structural modifications of nucleoside analogues can have profound effects on the chemical stability and biological activity of nucleoside analogues.
1.1. Primary Enzymes Involved in the Metabolism and Activity of Purine and Pyrimidine Analogues
To adequately understand the mechanism of action of this class of compounds it is necessary to be familiar with the enzymes that are involved in the metabolism of natural purines and pyrimidines. Human cells have all the enzymes needed for de novo synthesis of purine and pyrimidine nucleotides; however, other than orotate phosphoribosyl transferase with fluorouracil, these enzymes are not involved in the activation of the purine and pyrimidine antimetabolites and are only secondary targets responsible for antitumor activity of these compounds. Although salvage of purines and pyrimidines is not required for growth, human cells express many enzymes that can utilize purines and pyrimidines as substrates, and it is these enzymes (shown in Figures 1 and 2) that are most important to the anabolism and catabolism of the purine and pyrimidine antimetabolites that are used in the treatment of cancer. The catabolic enzymes are important because they are often responsible for detoxifying the nucleoside analogues, and these enzymes are expressed thoughout the body. Dihydropyrimidine dehydrogenase and xanthine oxidase are the initial enzymes in the degradation pathways of pyrimidines and purines. Adenosine deaminase and purine nucleoside phosphorylase are two important enzymes in the inactivation of purine nucleoside analogues but have also been successful targets of two agents, pentostatin and forodesine.
Figure 1.

Primary enzymes involved in the metabolism of pyrimidine analogues.
Figure 2.

Primary enzymes involved in the metabolism of purine analogues.
Phosphoribosyl transferases are responsible for activating the 3 base analogues (mercaptopurine, thioguanine, and fluorouracil), and there are five enzymes in human cells that can phosphorylate deoxynucleoside analogues4–6 (deoxycytidine kinase, thymidine kinase 1, thymidine kinase 2, deoxyguanosine kinase, and 5′-nucleotidase). The primary rate-limiting enzyme for activation of most of the approved nucleoside analogues is deoxycytidine kinase. Although deoxycytidine is the preferred natural substrate for this enzyme, it also recognizes deoxyadenosine and deoxyguanosine as substrates. The purine analogues are also substrates for deoxyguanosine kinase expressed in mitochondria, and this enzyme can contribute to the activation of these agents. Once formed, the monophosphate metabolites are phosphorylated by the appropriate monophosphate kinases7 to the diphosphate metabolite, which is phosphorylated by nucleoside diphosphate kinase. The first step in the formation of the 5′-triphosphates is typically the rate-limiting step and is, therefore, the most important step in activation of deoxynucleoside analogues. The X-ray crystal structure of deoxycytidine kinase has recently been solved,8 and given its importance in the activation of deoxynucleoside analogues, its structure is used for design of new agents.
The primary target of the deoxynucleoside analogues are the DNA polymerases involved in DNA replication. There are at least 14 eukaryotic DNA polymerases expressed in human cells,9 three of which are primarily involved in chromosomal replication (DNA polymerases α, δ, and ε) and are the primary targets for the anticancer nucleoside analogues. The other major cellular polymerases are DNA polymerase β, which is involved in DNA repair; DNA polymerase γ, which is the polymerase responsible for mitochondrial DNA replication; and telomerase, which is responsible for the replication of DNA telomeres, but these enzymes are not primary targets for the anticancer antimetabolites. Inhibition of DNA polymerase γ or telomerase activity does not result in the immediate inhibition of cell growth.
A deoxynucleotide triphosphate analogue could theoretically interact with a DNA polymerase in one of three ways: (i) it could compete with the natural substrate, but not be used as a substrate; (ii) it could substitute for the natural substrate with little effect on subsequent DNA synthesis; or (iii) it could substitute for the natural substrate and interfere with subsequent DNA synthesis, causing chain termination. The second two possibilities are the primary manners in which the anticancer nucleotide analogues interact with DNA polymerases, and all of these analogues have been shown to be good substrates for the replicative DNA polymerases. The primary differences in these compounds are (i) how easily the DNA chain is elongated after the incorporation of the analogue and (ii) how easily they can be removed from the DNA by the proof-reading exonucleases. The incorporation of these agents into DNA is one of the most important aspects of their mechanism of action resulting in antitumor activity, because the incorporation is difficult to repair and causes a lasting inhibition of DNA synthesis or disruption of DNA function. The inhibition of DNA synthesis by agents, such as aphidicolin, that only inhibit DNA polymerase activity without being incorporated into the DNA chain have not made good anticancer agents, because the DNA is not damaged by these agents and DNA synthesis resumes after the removal of the agent. Indeed, aphidicolin is used to synchronize cell populations,10 because of its ability to temporarily inhibit DNA synthesis without inducing cell death.
2. FDA Approved Purine And Pyrimidine Antimetabolites Used In The Treatment of Cancer
The FDA approved purine and pyrimidine antimetabolites can be grouped into three primary classes (thiopurines, fluoropyrimidines, and the deoxynucleoside analogues) based on structural and mechanistic considerations. The deoxynucleoside analogues are the largest class and are where most of the design of new compounds has occurred recently. A massive amount of literature on the mechanism of action of these established agents is available, and there will be no attempt in this review to include all that has been done with these compounds. Instead, a description of the important metabolic features of each compound, the primary enzymatic targets responsible for their antitumor activity, and the unique features of the various compounds will be presented.
2.1. Thiopurines (Mercaptopurine and Thioguanine)
6-mercaptopurine (MP) was one of the first agents approved by the FDA for the treatment of cancer,11 where it proved to be effective in the treatment of childhood acute lymphocytic leukemia. MP is an analogue of hypoxanthine (Figure 3), and like hypoxanthine, it is a good substrate for hypoxanthine/guanine phosphoribosyl transferase (Figure 4). The product of the reaction, 6-thio-inosine monophosphate (T-IMP), is a substrate for IMP dehydrogenase and is subsequently converted to guanine nucleotides. The primary intracellular metabolite of MP is 6-thioguanosine-5′-triphosphate, and it is readily incorporated into RNA. However, since specific inhibition of RNA synthesis does not affect the activity of MP,12 the incorporation of thioguanine (TG) into RNA does not appear to play an important role in the antitumor activity of MP.
Figure 3.

Structures of thiopurines.
Figure 4.

Metabolism of mercaptopurine and thioguanine.
MP is also converted via ribonucleotide reductase to 6-thio-2′-deoxyguanosine-5′-triphosphate, which is incorporated into DNA. Unlike most of the other cytotoxic purine and pyrimidine antimetabolites used in the treatment of cancer, treatment of cells with MP does not result in the immediate inhibition of DNA synthesis in that cells continue to divide before dying. This result is consistent with studies that indicate that T-dGTP is a good substrate for the DNA polymerases involved in DNA replication.14,15 It is utilized as effectively as dGTP as a substrate for DNA polymerase α, and once incorporated, it is readily extended by the polymerase and is incorporated into internal positions in the DNA chain. Although treatment with MP does not inhibit DNA polymerase activity, its incorporation into DNA resulting in DNA damage is believed to be primarily responsible for the antitumor activity of MP. It is thought that TG in DNA, as well as its methylated counterpart, is recognized by mismatch repair enzymes, which causes a futile cycle of repair that results in lethal DNA damage.13
The sulfur atom of T-IMP is methylated by thiopurine S-methyltransferase (TPMT) present in mammalian tissues, and methyl mercaptopurine riboside monophosphate (methyl-T-IMP) is also an important metabolite in cells. This metabolite is a potent inhibitor of PRPP amidotransferase, the first enzyme in de novo purine biosynthesis, and its inhibition results in a decrease in purine nucleotide pools. Therefore, there are two primary biochemical actions that contribute to the anticancer activity of MP; its inhibition of de novo purine synthesis and its incorporation into DNA as 6-thio-2′-deoxyguanosine.
No adenine nucleotide analogues of MP are formed in cells, because T-IMP is not a substrate for adenylosuccinate synthetase, the first enzyme in the formation of adenine nucleotides from IMP. Even if it were a substrate for this enzyme, the mechanism of action of this enzyme would remove the 6 sulfur atom and replace it with an aspartic acid to form adenylosuccinic acid, which is the natural product of this reaction. A small amount of T-ITP is formed in cells, but this metabolite is not believed to be important in the mechanism of activity of MP.
The metabolism of thioguanine (TG) is much simpler than that of MP. TG is also a substrate for hypoxanthine/guanine phosphoribosyl transferase and large concentrations of TG nucleotides accumulate in cells treated with TG. T-GMP is also methylated by S-methyl transferase, but the product of the reaction, methyl-T-GMP, is not a potent inhibitor of PRPP amidotransferase. Therefore, inhibition of de novo purine biosynthesis is less important to the action of TG, and the mechanism of cytotoxicity of TG is believed to be primarily due to its incorporation into DNA and subsequent DNA damage.13 Thioguanine (TG) is approved for use in acute myelogenous leukemia.
In patients, the methylation of the purine bases, MP and TG, by thiopurine S-methyltransferase (TPMT) is a major mechanism of detoxification of these agents.16,17 The products of the reaction, S6-methyl-mercaptopurine and S6-methyl-thioguanine, are not substrates for hypoxanthine/guanine phosphoribosyl transferase (HGPRT) and are, therefore, not toxic to human cells. Approximately 0.3% of the population does not express functional TPMT activity, and treatment of these people with either thiopurine can result in severe toxicity.
2.2. Fluoropyrimidines
2.2.1. Fluorouracil
5-Fluorouracil (FUra, Figure 5) is one of the first examples of an anticancer drug that was designed based on the available biochemical information. It was known that (i) a fluorine atom was of similar size to a hydrogen atom; (ii) a carbon–fluorine bond was much stronger than a carbon–hydrogen bond; (iii) the reaction mechanism of thymidylate synthase replaces the 5-hydrogen of deoxyuridine mono-phosphate with a methyl group obtained from methylene tetrahydrofolate to make thymidylate (TMP); and (iv) rat hepatoma cells, but not normal liver cells, could utilize uracil (although it was subsequently found that this observation did not extend to other cell types). Utilizing this information, Heidelberger18 and colleagues hypothesized that FUra would selectively kill tumor cells because of its selective metabolism in tumor cells to F-dUMP, which would inhibit thymidylate synthetase due to the inability of the enzyme to remove the 5-fluorine atom. Much of the original hypothesis has been shown to be true,19 and FUra is used for palliative treatment of colorectal, breast, stomach, and pancreatic cancer. It also has utility as a topical treatment of superficial basal cell carcinoma that cannot be treated with surgery and actinic keratosis, a precancerous skin condition. Much work has been done since the approval of this agent that has enhanced our understanding of its mechanism of action, and this work has been extensively reviewed.20,21
Figure 5.
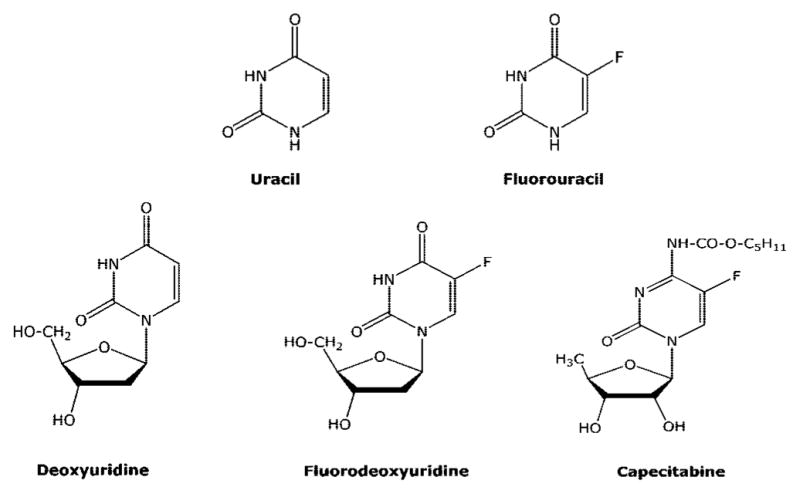
Structures of fluoropyrimidines.
As shown in Figure 6 the metabolism of FUra is very complex. FUra is converted into F-UMP by orotate phosphoribosyl transferase, which is the first step in its activation. Nucleotide kinases then convert F-UMP to F-UTP, which is the primary intracellular metabolite of FUra. F-UTP is used as a substrate for RNA synthesis in place of uridine triphosphate (UTP), and a considerable amount of FUra is incorporated into all species of RNA. The incorporation of FUra into various species of RNA has been shown to disrupt the function of these species of RNA, but these effects have only been observed at high concentrations. There are various types of RNA molecules, and the effect of FUra on many of the newer functions of RNA has not yet been evaluated. It is believed that the incorporation of FUra into RNA does contribute to its cytotoxic activity, but because of the complexity of RNA, the precise RNA-directed action(s) has not been defined. It is likely that the incorporation into RNA causes more than one defect and that inhibition of numerous RNA activities contribute to its RNA-directed activity. Although incorporation into RNA is an important component of the mechanism of action of FUra, the RNA-directed actions are believed to be secondary to its DNA-directed actions described below, which is similar to the case with the thiopurines.
Figure 6.

Metabolism of fluoropyrimidines.
F-UDP is a substrate for ribonucleotide reductase, which removes the 2′-OH group. F-dUDP is a good substrate for nucleoside diphosphate (NDP) kinase that forms F-dUTP, which is an excellent substrate for DNA polymerases. F-dUTP (as well as dUTP) is used by DNA polymerases for the synthesis of DNA as effectively as thymidine triphosphate (TTP). Therefore, if F-dUTP accumulates in cells, it will be incorporated into the DNA by the DNA polymerases. Human cells have developed a mechanism to recognize uracil in DNA and remove it, because a considerable amount of uracil is formed in the DNA of any cell due to the spontaneous deamination of cytosine and since uracil base-pairs as thymine, this deamination of cytosine in DNA would result in mutation. The enzyme responsible for the removal of uracil from DNA is uracil glycosylase, and it recognizes FUra in DNA as a substrate and readily removes it from the DNA, resulting in an apyrimidinic site, which is recognized by apurinic/apyrimidinic endonuclease 1, causing a single strand break. The single strand break is recognized by DNA repair enzymes, and in a manner similar to TG, the repair and resynthesis of DNA sets up a futile cycle that results in inhibition of DNA synthesis and cell death.
Another mechanism the cell uses to keep uracil out of DNA is to prevent its use as a substrate by DNA polymerases. Since human cells contain the enzymes necessary to make dUTP, human cells also express dUTPase, which converts dUTP to dUMP and keeps levels of dUTP very low in the cell. dUMP is a substrate for thymidylate synthase and is utilized for the synthesis of thymidine nucleotides. dUTPase also recognizes F-dUTP and is responsible for the formation of F-dUMP, which is a potent inhibitor of thymidylate synthase, as hypothesized by Heidelberger. The inhibition of thymidylate synthase results in decreases in TTP levels and large increases in deoxyuridine nucleotides, including both dUTP and F-dUTP. As indicated above, an increase in dUTP levels can result in the incorporation of uracil in DNA and its subsequent removal by uracil glycosylase. Therefore, the inhibition of DNA synthesis in cells treated with FUra is a result of two actions: depression of intracellular TTP levels due to inhibition of thymidylate synthetase and incorporation and removal of uracil (and FUra) in DNA. Therefore, inhibition of thymidylate synthesis by F-dUMP results in a nonproductive incorporation and removal of uracil and FUra from DNA, which results in inhibition of DNA synthesis and DNA damage.
An important enzyme in the catabolism of FUra is dihydropyrimidine dehydrogenase. This enzyme is the rate-limiting enzyme in the conversion of FUra to fluoro-β-alanine and is, therefore, very important in the detoxification of FUra. Three to five percent of Caucasians express low levels of dihydropyrimidine dehydrogenase, and if these people are treated with FUra, severe toxicity, including death, can occur.17
2.2.2. Capecitabine
Capecitabine (Figure 5) is a prodrug of FUra that is administered orally.22 It has almost 100% oral bioavailability and is converted in three enzymatic reactions to FUra (Figure 6). The N4-pentyloxycarbonyl moiety is first removed by carboxylesterases in the liver to generate 5′-deoxy-5-fluorocytidine, which is a good substrate for cytidine deaminase, and is converted to 5′-deoxy-5-fluorouridine. Because of the absence of a 5′-OH group, 5′-deoxy-5-fluorouridine cannot be activated to FUra nucleotides by nucleoside kinases; however, it is a good substrate for thymidine phosphorylase and is converted to FUra. Because thymidine phosphorylase is overexpressed in tumor tissues, capecitabine should have a better selective index than FUra. In addition, thymidine phosphorylase activity is stimulated by radiation therapy, and combination treatment with capecitabine plus radiation can further enhance selectivity of this compound for tumor cells. As a prodrug of FUra, capecitabine has two advantages over intravenous FUra: ease of administration (oral vs IV) and a potential increased therapeutic effect. It is currently approved for use in the treatment of stage III colon cancer and metastatic breast cancer.
2.2.3. Floxuridine
Floxuridine (F-dUrd) is an excellent substrate for thymidine kinase, and it is converted by this enzyme directly to F-dUMP. In vitro, this compound is a much more potent inhibitor of cell growth than FUra and is not converted to ribonucleotide metabolites to a significant degree at cytotoxic concentrations. However, F-dUrd is also a good substrate for thymidine phosphorylase, which converts it to FUra, and a significant amount of F-dUrd is converted to FUra in vivo. Therefore, when used in the treatment of patients, F-dUrd is not a specific inhibitor of thymidylate synthesis. F-dUrd has demonstrated some efficacy when given by hepatic arterial infusion to treat liver metastases.23 Although approved by the FDA for this purpose, it is not widely used.
2.3. Deoxynucleoside Analogues
There are numerous deoxynucleoside analogues that are useful in the treatment of cancer. Other than cytarabine, which was approved in 1969 for the treatment of acute leukemias, these agents are relatively new, having been approved for use since 1991, and except for deoxycoformycin, which is a potent inhibitor of adenosine deaminase, the mechanisms of action of these agents are quite similar. They are converted to their respective nucleotide analogues, which inhibit DNA synthesis by inhibition of DNA polymerases and/or ribonucleotide reductase. However, in spite of these similarities, there are differences in the interaction of these agents and their metabolites with the various metabolic enzymes and intracellular targets that imparts unique properties to each of these agents and results in unique clinical activity.
2.3.1. Deoxycytidine Analogues (Cytarabine, Gemcitabine, Decitabine, Vidaza)
2.3.1.1. Cytarabine (araC)
The metabolism of the deoxycytidine analogues (Figure 7) is much simpler than that of the thiopurines and fluoropyrimidines. They are good substrates for deoxycytidine kinase and the primary intracellular metabolite is their respective triphosphates, which accumulate to high intracellular concentrations (Figure 8). AraCTP is a good substrate for DNA polymerases,24 but once incorporated into the 3′-end of the DNA chain, further extension of the DNA chain by the DNA polymerase is significantly inhibited.25 Because araCTP has a 3′-OH group, it is not an absolute inhibitor of DNA chain elongation, as is seen with the anti-HIV nucleoside analogues, and it is incorporated into internal positions in DNA chain. However, treatment of cells with araC causes an immediate and significant inhibition of DNA replication, and it is this action that is primarily responsible for the cytotoxicity of araC to tumor cells.
Figure 7.

Structures of deoxycytidine analogues.
Figure 8.

Deoxycytidine metabolism.
2.3.1.2. Gemcitabine (dFdC)
As indicated above, dFdC-TP is also a good substrate for the DNA polymerases responsible for DNA replication; however, the DNA chain is more easily extended after its incorporation than is seen with araC.26 Interestingly, DNA chain elongation after incorporation of dFdC-TP was inhibited after incorporation of the next nucleoside after the incorporation of dFdC-MP. Moreover, a significant number of DNA chains were extended beyond dFdC incorporation, and in cells treated with dFdC, more than 90% of the dFdC incorporated into DNA was incorporated in internal positions, which is much greater than that seen with araC. These results indicate that dFdC-TP is less of a chain terminator than is araCTP, and that the incorporation of dFdC into DNA and the subsequent disruption of its function are more important to the activity of dFdC than araC. Recent studies have indicated that the incorporation of dFdC into DNA is the most important determinant of its ability to kill cells.27 The 3′ to 5′ proofreading exonuclease associated with DNA polymerase ε was not able to remove dFdC from the 3′ end of DNA chains but was able to remove a substantial amount of araC,28 which indicated that dFdC in DNA was poorly repaired once incorporated. In addition, dFdC-DP is an important metabolite, because it is a potent inhibitor of ribonucleotide reductase.29 Unlike araC, inhibition of this enzyme is a significant action of dFdC that contributes to its anticancer activity. Inhibition of ribonucleotide reductase results in depletion of the natural deoxynucleotides used as substrates for DNA synthesis and thereby enhances the use of dFdC-TP as a substrate for DNA polymerases due to decreases in dCTP pools.
An important theme with the new deoxynucleoside analogues starting with dFdC29 is the long intracellular half-life of the nucleotide metabolites of these agents. This attribute of dFdC nucleotides and the production of DNA damage that is less easily repaired are believed to be the major actions of dFdC that are responsible for its activity against solid tumors. Currently dFdC is the only deoxynucleoside analogue that is approved for use against solid tumors, where it is approved for treatment of both pancreatic cancer and nonsmall cell lung cancer.
2.3.1.3. Decitabine (aza-dCyd) and Vidaza (aza-Cyd)
Like araC and dFdC, aza-dCyd, which was recently approved for use in the treatment of myelodysplastic syndromes, is converted via deoxycytidine kinase to aza-dCTP.30–33 However, unlike araC and dFdC, its interaction with the DNA polymerase is more similar to TG in that it is readily incorporated into DNA and extended into internal positions in the DNA by the DNA polymerase α.34 Therefore, treatment with aza-dCyd does not immediately result in the inhibition in DNA synthesis. Instead, the therapeutic activity of aza-dCyd is due to the inhibition of DNA methylation once it has been incorporated into the DNA chain. Methylation of the 5 position of cytosine residues in DNA is a major mechanism that is used by human cells to control gene expression. Methylation of cytosine residues causes a repression of gene expression, so replacement of deoxycytidine residues by aza-dCyd results in inhibition of DNA methylation and enhanced gene expression in daughter cells, resulting in the activation of epigenetically repressed genes. Although the primary mechanism of action is due to inhibition of DNA methylation, at high doses aza-dCyd can cause other effects that may contribute to its antitumor activity.35 Aza-dCyd is not chemically stable, and this chemical instability may contribute to its cytotoxicity once it is incorporated into the DNA. Aza-Cyd is a ribonucleoside analogue (Figure 7) and is activated by uridine/cytidine kinase, but it is included with the deoxycytidine analogues because its primary activity is due to its conversion to deoxynucleotides via ribonucleotide reductase and its incorporation into DNA. Even though a considerable amount of aza-Cyd is incorporated into RNA, the antitumor activity of aza-Cyd is believed to be primarily due to its incorporation into DNA and inhibition of DNA methyltranferase as is seen with the thiopurines and fluoropyrimidines.
All of the deoxycytidine analogues are good substrates for cytidine deaminase, and this enzyme plays an important role in the mechanism of action of these agents. Although deamination of a deoxycytidine analogue results in a deoxyuridine analogue, which could also be cytotoxic, deamination of the deoxycytidine analogues used in the treatment of cancer is not an activating step but is instead an important route in the detoxification of these compounds, because the respective deoxyuridine analogues are poorly activated to cytotoxic nucleotides by thymidine kinase. The monophosphates of the deoxycytidine analogues, particularly dFdC-MP, are substrates for dCMP deaminase and can also be detoxified by this enzyme. dFdU-MP is formed in cells, but there is little evidence concerning its interaction with thymidylate synthetase. There is no evidence of dFdT-TP in cells, indicating that dFdU-MP is not a substrate for thymidylate synthetase. There is some evidence that aza-dUMP may be an inhibitor of thymidylate synthetase,36 and this could contribute to the cytotoxicity of aza-dCyd at high concentrations. Because of the role of deaminases in the detoxification of these cytosine analogues, the design of new analogues usually seeks compounds that are poor substrates for these enzymes. 5-F-deoxycytidine is an example of a deoxycytidine analogue that is activated by deamination, and it has been suggested to be used as a prodrug of F-dUrd,37 but it has not been approved for human use.
2.3.2. Purine Deoxynucleoside Analogues (Fludarabine, Nelarabine, Cladribine, Clofarabine, Pentostatin)
2.3.2.1. Fludarabine (F-araA) and Nelarabine
There are five purine deoxynucleoside analogues that have been approved for the treatment of cancer since 1991. Two of these agents are arabinoside analogues (fludarabine phosphate (F-araAMP) and nelarabine) and, therefore, incorporate the same structural feature responsible for the anticancer activity of araC. F-araAMP is a deoxy-AMP analogue that is approved for the treatment of chronic lymphocytic leukemia.38,39 F-araAMP is a prodrug of F-araA (Figure 9) and is used clinically because of the poor solubility of F-araA. F-araAMP is rapidly converted by plasma phosphatases to F-araA, which is the primary circulating form of the drug. Adenosine deaminase is ubiquitously expressed and is an important detoxifying enzyme of deoxyadenosine analogues. Many years ago it was discovered that replacement of the 2-hydrogen atom of adenosine with a halogen atom (e.g., fluorine or chlorine) results in a molecule that is resistant to deamination,40,41 and this feature has been incorporated into all of the approved anticancer deoxyadenosine analogues (F-araA, cladribine, and clofarabine) to prevent their inactivation by adenosine deaminase.
Figure 9.

Structures of deoxyadenosine analogues.
Nelarabine (Figure 10) is a prodrug of 9-β-D-arabinofuranosyl guanine (araG) and has recently been approved for the treatment of T-cell malignancies.42 Adenosine deaminase is also an important enzyme in the metabolism of nelarabine. However, in the case of nelarabine, adenosine deaminase is critical in its activation to cytotoxic metabolites, which replaces the 6-methoxy substituent with oxygen forming araG. Like F-araA, nelarabine is used instead of araG, because of the poor solubility of araG, which was first synthesized in 196443 and shown to have good activity against T cells in vitro.44 T-cells have been shown to accumulate more araGTP than B-cells,45,46 and it is this accumulation that is believed to be responsible for the selective activity of araG in T-cell malignancies. The reason that T-cells accumulate higher levels of purine nucleotides than most cells is not well-understood but is related to the enhanced sensitivity of T-cells to inhibitors of purine nucleoside phosphorylase (see forodesine below).
Figure 10.

Structure of nelarabine.
Both F-araA and araG are substrates for deoxycytidine kinase (Figure 11), which is the primary enzyme responsible for conversion of these compounds to active metabolites, although araG is also a substrate for mitochondrial deoxyguanosine kinase,47 and this enzyme could also contribute to its activation in some cell types. Like araCTP, F-araATP and araGTP are good substrates for the replicative DNA polymerases. The incorporation of either F-araAMP or araGMP into the 3′-end of DNA inhibits the further elongation of the DNA by these enzymes,48–50 resulting in the inhibition of DNA replication. Therefore, the mechanism of cell kill of these three arabinofuranosyl analogues is similar.
Figure 11.

Deoxyadenosine and deoxyguanosine metabolism.
F-araATP is also a weak inhibitor of ribonucleotide reductase.51 The activity of ribonucleotide reductase in cells is tightly controlled by the natural deoxynucleoside triphosphates to ensure that the cell has all of the deoxynucleotides needed for DNA synthesis in the correct concentrations. dATP is a potent regulator of ribonucleotide reductase activity and inhibits the reduction of ADP, UDP, and CDP.52 F-araATP binds to ribonucleotide reductase in the allosteric binding site as an analogue of dATP. As in the case with dFdC, inhibition of ribonucleotide reductase activity by F-araATP could potentiate the DNA polymerase directed activity of this compound by reducing the intracellular levels of dATP, the natural substrate that competes with F-araATP for the DNA polymerase active site. Inhibition of ribonucleotide reductase activity does not appear to play an important role in the anticancer activity of araG.45
2.3.2.2. Cladribine (Cl-dAdo)
Cl-dAdo (Figure 9) is a deoxyadenosine analogue that was approved in 1992 for the treatment of hairy-cell leukemia.53 The sugar component of this compound is the normal deoxyribose as opposed to an arabinose, and this compound is readily phosphorylated by deoxycytidine kinase to Cl-dAdo nucleotides. Cl-dATP is a good substrate for DNA polymerases, where it is incorporated into the growing DNA chain and is extended better than arabinoside analogues such as F-araA.48,53 DNA polymerase α easily extended the DNA chain past the incorporation of a single Cl-dAdo residue but was stopped by three successive incorporated Cl-dAdo residues.53 Cl-dATP is a much more potent inhibitor of ribonucleotide reductase than is F-araATP,48,53 and therefore, inhibition of this enzyme is more important to its mechanism of action. As with dFdC and F-araA, inhibition of ribonucleotide reductase can potentiate the inhibition of DNA polymerases by nucleotide analogues. Since the incorporation of three successive dAdo residues is a likely event in the replication of the genome, Cl-dAdo could still cause considerable chain termination. Like F-araA, Cl-dAdo is not a substrate for adenosine deaminase, because of the presence of chlorine at the 2 position.
2.3.2.3. Clofarabine (Cl-F-araA)
Cl-F-araA was approved for the treatment of relapsed and refractory pediatric acute lymphoblastic leukemia in 2004.54,55 The structure of Cl-F-araA differs from that of Cl-dAdo in that it contains a fluorine atom at the 2′ position in the deoxyribose portion of the molecule (Figure 9). Comparison of these two FDA approved drugs is the best example of how small structural differences can result in dramatic clinical differences. This modest structural difference significantly increases the stability of the glycosidic bond, resulting in enhanced acid stability of the compound as well as good oral bioavailability. The mechanism of action of Cl-F-araA is similar to that of Cl-dAdo and F-araA in that it is activated by deoxycytidine kinase to Cl-F-araA 5′-triphosphate, which inhibits DNA replication due to its potent inhibition of both ribonucleotide reductase and DNA polymerase.48,56,57 The potency of Cl-F-araA with respect to inhibition of ribonucleotide reductase is similar to that of Cl-dAdo. Furthermore, it is readily incorporated into the DNA chain but has a chain-terminating effect more similar to F-araA than Cl-dAdo. Therefore, Cl-F-araA combines into one molecule the features of Cl-dAdo (potent inhibition of ribonucleotide reductase) and F-araA (potent inhibition of DNA polymerase) that are responsible for their antitumor activity. Like dFdC, Cl-F-araA-TP has been shown to have a long intracellular retention time,56 and Cl-F-araA has demonstrated good activity against numerous human solid tumor xenografts in mice.58–60 Similar to Cl-dAdo, Cl-F-araA is not a substrate for adenosine deaminase.
2.3.2.4. Pentostatin
Pentostatin (deoxycoformycin, Figure 9), like Cl-dAdo, is used in the treatment of hairy-cell leukemia.61,62 It is a potent inhibitor of adenosine deaminase and is the only purine or pyrimidine antimetabolite approved for use by the FDA that is active without metabolism. Adenosine deaminase deficiency in humans results in a severe combined immunodeficiency syndrome characterized by a profound deficiency in B and T lymphocytes, which indicates that these cells are particularly sensitive to the inhibition of this enzyme. Inhibition of adenosine deaminase activity by pentostatin causes an increase in circulating deoxyadenosine and is responsible for the accumulation of deoxyadenosine nucleotides particularly dATP, which inhibits ribonucleotide reductase activity and inhibits DNA synthesis due to the decline in dCTP and other deoxynucleotides substrates needed for DNA synthesis.
3. New Compounds
3.1. Troxacitabine
Troxacitabine (OddC) is a deoxycytidine analogue with two unique structural features (Figure 12): It is an L nucleoside analogue and it lacks both the 2′ and 3′ hydroxyl groups. This compound originated out of the drug discovery efforts to identify nucleoside analogues that are active against human immunodeficiency virus. Because deoxycytidine kinase can phosphorylate the unnatural L conformation of nucleosides,18,19 OddC is phosphorylated very well in human cells. However, unlike most other analogues, the major intracellular metabolite is OddC-DP,63 which is then converted to the triphosphate by 3-phosphoglycerate kinase, not nucleoside diphosphate kinase.64,65 Unlike most other dideoxynucleotides, OddC-TP is a good substrate for DNA polymerase α and is incorporated into the DNA chain where it is an absolute DNA chain terminator because of its lack of a 3-OH group.66 Because of the chiral preference for 3′–5′ proof-reading exonucleases associated with DNA polymerase, once incorporated into DNA, OddC is not easily removed from the DNA chain,67 although OddC is recognized by apurinic/apyrimidinic endonuclease.68 OddC is a very poor substrate for cytidine deaminase. OddC has demonstrated efficacy in both solid and hematological malignancies in clinical trials.69
Figure 12.

Structures of troxacitabine, thio-araC, CNDAC, and forodesine.
3.2. Thiarabine
Although thiarabine (T-araC) is structurally similar to araC (Figure 12), the antitumor activity of T-araC against a variety of human tumor xenografts in mice is dramatically better than that of araC,70 a compound that does not demonstrate solid tumor activity in these animal models or in patients. T-araC has also demonstrated better activity than gemcitabine against various human tumor xenografts in mice. Although the basic mechanism of action of T-araC71–75 is similar to that of araC (both compounds are phosphorylated to their respective triphosphates (T-araCTP or araCTP) and inhibit DNA synthesis), there are several quantitative differences in the metabolism and biochemical activity of these two compounds that can explain their differences in antitumor activity. Most importantly, the half-life of T-araCTP in solid tumor cells is approximately 10 times longer than that of araCTP,76 and T-araCTP is a much more potent inhibitor of DNA synthesis than is araCTP.71 As with gemcitabine, these two activities are believed to be very important to the activity demonstrated in mice against solid tumor xenografts. In addition, the interaction of T-araC with numerous other enzymes involved with the activation of deoxycytidine analogues differs from araC, and these differences may also contribute to the in vivo activity of T-araC. With respect to araC and its metabolites, T-araC is a poor substrate for deoxycytidine kinase and deoxycytidine deaminase activities. T-araCMP is a poor substrate for dCMP deaminase activity, but it is a better substrate for CMP/UMP kinase than is araCMP, a difference that may help explain the long half-life of T-araCTP.76 Like araC, T-araC has only a modest effect on ribonucleotide reductase activity. T-araC has been evaluated in two clinical trials to treat solid tumors77,78 and is currently being prepared for further clinical evaluation. T-araC demonstrated partial responses in some of the heavily pretreated patients with relapsed solid tumors in these trials.
3.3. Sapacitabine
1-[2-C-cyano-2-deoxy-β-D-arabinofuranosyl]-cytosine (CN-DAC) is a deoxycytidine analogue with a structure that is similar to araC (Figure 12). However, instead of a 2′-hydroxy group, CNDAC has a 2′-cyano group. Similar to araC, CNDAC is phosphorylated via deoxycytidine kinase to CNDAC-TP, which is a good substrate for DNA polymerases involved in DNA replication. Once incorporated into the DNA chain, CNDAC is a powerful chain terminator.79 Chain elongation by DNA polymerase α was severely inhibited by the incorporation of CNDAC into the 3′-terminus, which was greater than that observed with either araC and gemcitabine. If CNDAC is incorporated into the internal DNA linkages, it has a secondary affect on DNA integrity. Once the DNA chain is extended after the incorporation of CNDAC, the 3′-phosphodiester link between CNDAC and the next nucleotide is not stable and the DNA chain is spontaneously cleaved through a β elimination reaction that generates a DNA chain that is terminated with 2′-C-cyano-2′,3′-didehydro-2′,3′-dideoxycytidine. Therefore, incorporation of CNDAC into DNA chains can result in single strand breaks in the DNA. This mechanistic consideration contributed to the design of this molecule, and the dideoxy analogue has been detected in the DNA of cells treated with CNDAC.81,82 Like araC, treatment with CNDAC does not inhibit ribonucleotide reductase activity. An N4 palmitoyl derivative of CNDAC (Sapacitabine) is being evaluated in the clinic for antitumor activity.83
3.4. Forodesine
People born with a deficiency of purine nucleoside phosphorylase (PNP) are healthy except that they do not produce T-cells, which results in a severe immunodeficiency disease that usually causes death early in life.84,85 This condition suggests that inhibitors of PNP would have selective activity against T-cell malignancies. PNP is an important enzyme in the salvage of purine nucleosides, and in its absence, intracellular deoxyguanosine is not cleaved to guanine but is instead converted to deoxyguanosine 5′-triphosphate (dGTP), which is a feedback inhibitor of ribonucleotide reductase activity. Therefore, the expanded dGTP pool in T-cells results in the inhibition of ribonucleotide reductase activity and depletion of intracellular deoxynucleotides that are required for DNA synthesis. The sensitivity of T cells to PNP inhibition is believed to be due to relatively high levels of nucleoside kinase activity and low levels of nucleotidase activity in these cells. Forodesine (Figure 12) is a potent inhibitor of PNP activity with a Ki of 72 pM.86,87 The affinity of this compound for the enzyme is approximately 1 million times that for inosine, the natural substrate. Forodesine was potent enough to result in a profound inhibition of PNP activity in intact animals and has demonstrated excellent activity against human peripheral blood lymphocytes engrafted into SCID mice. Forodesine is similar to pentostatin in that it is active without metabolism. The FDA granted orphan drug status to forodesine in February of 2004, and it is being evaluated in human clinical trials for the treatment of cutaneous T-cell lymphoma and chronic lymphocytic leukemia.88–90
3.5. Suicide Gene Therapy of Cancer Using Purine and Pyrimidine Analogues
There are a few gene therapy approaches to the treatment of cancer (Figure 13) that involve the selective activation of purine or pyrimidine analogues by foreign genes that are delivered to and expressed in tumor cells.91–94 In this strategy, the selective transfection and expression of nonhuman genes in tumor cells creates a difference in the tumor cells that can be exploited to selectively kill the tumor cells. Theoretically, this approach to the treatment of cancer should kill cancer cells with much less toxicity than is seen with conventional therapy. The genes for these enzymes are first delivered to tumor cells by various viral or bacterial vectors that have been engineered for this purpose, and then the patient is treated systemically with prodrugs that are activated to cytotoxic compounds by the enzymes expressed from the genes.
Figure 13.

Gene therapy approaches to the treatment of cancer.
The gene that has received the most attention is the herpes simplex virus (HSV) thymidine kinase (TK), and numerous clinical trials have been conducted to evaluate this approach without much success. The expression of this gene in cells infected with HSV is the basis of the selective antiviral therapy for the treatment of HSV infections, and its expression in tumor cells has been used to activate ganciclovir, a purine nucleoside analogue used in the treatment of CMV, to cytotoxic nucleotide metabolites. The HSV-TK strategy, however, has a limited ability to kill neighboring tumor cells that do not express the gene (bystander cells), because the product of the reaction of HSV-TK with ganciclovir is ganciclovir-5′-monophosphate, which does not easily diffuse out of the cell in which it was formed. The bystander activity seen with the HSV-TK approach is dependent upon gap junctions and requires cell-to-cell contact. Since current technology is not able to deliver foreign genes to the majority of the tumor cells, the limited bystander activity of ganciclovir monophosphate is a major limiting factor of the HSV-TK approach in the treatment of cancer.
In addition, ganciclovir-TP kills tumor cells by inhibiting DNA polymerases involved in DNA replication, much like conventional nucleoside analogues. Therefore, ganciclovir primarily targets proliferating cells. Since solid tumors often have a low growth fraction, the lack of activity of this approach against nonproliferating tumor cells is another deficiency of this approach to the treatment of solid tumors.
5-Fluorocytosine (F-Cyt) is approved for the treatment of fungal diseases because of its selective deamination in fungal cells to FUra and has also been evaluated in gene therapy strategies in which E. coli or yeast cytosine deaminase is expressed in tumor cells. Human cells do not express cytosine deaminase, and F-Cyt is well tolerated in people. Delivery of cytosine deaminase to tumor cells has been shown to sensitize them to F-Cyt, and a few clinical trials are underway to evaluate this gene therapy strategy.
E. coli purine nucleoside phosphorylase (PNP), unlike human PNP, accepts adenosine as a substrate and cleaves the glycosidic bond to produce adenine and ribose-1-phosphate. This difference in substrate specificity between these two enzymes has been exploited to create a gene therapy strategy to activate deoxyadenosine analogues to very active adenine analogues in tumor cells. The adenine analogues produced from E. coli PNP can readily diffuse to and kill surrounding tumor cells that do not express E. coli PNP, which is an important attribute for gene therapy approaches to the treatment of cancer due to the difficulty of delivering genes to tumor cells. Because human cells contain nucleoside and nucleobase transporters in their membranes that facilitate the diffusion of purines across membranes in either direction, the bystander activity for purine and pyrimidine bases is not dependent upon gap junctions and does not require cell-to-cell contact, as is the case with ganciclovir nucleotides. Excellent antitumor activity has been observed with F-araAMP against human tumor xenografts in mice, even when only 2.5% of the tumor cells express E. coli PNP.95 Two clinical trials are scheduled to begin in 2009 to evaluate the safety and efficacy of the use of E. coli PNP with F-araAMP in treatment of solid tumors.
In addition to the high bystander activity of the E. coli PNP approach, the toxic adenine analogue formed from F-araAMP and E. coli PNP (2-fluoroadenine) has a unique mechanism of action that results in the killing of both proliferating and nonproliferating tumor cells.96 2-Fluoro-adenine is converted to an ATP analogue, that inhibits RNA and/or protein synthesis. This mechanism of cell kill is different from that of all currently used anticancer agents and would not be tolerated if the agent was administered systemically. Fluoroadenine has been evaluated in mouse models of cancers and has not demonstrated selective antitumor activity. The activity of this antitumor strategy against nonproliferating cells is of particular importance to the treatment of solid tumors, which often have a very low growth fraction. The ability to kill nonproliferating tumor cells is a major characteristic of the E. coli PNP approach that distinguishes it from both the cytosine deaminase and the thymidine kinase approach.
The use of gene therapy to deliver genes to tumor cells solves the problem associated with the lack of selectivity of the current chemotherapy, but it introduces another difficult problem to solve, i.e., selective delivery of genes to tumor cells with sufficient enzyme expression. The vectors available in 2009 do not express enough enzyme activity in enough tumor cells after systemic administration to activate enough prodrug. Therefore, gene therapy approaches in the clinic have been limited to tumors that can be injected with the vector. It is hoped that, with continued study, new vectors will be developed that will be able to selectively deliver sufficient amount of genes to tumors after systemic administration, allowing for activity against metastatic disease. However, because of the difficulty of delivering vectors to tumor cells throughout the body, gene therapy may only prove to be useful for the treatment of localized tumors.
4. Drug Design Considerations
There is clearly an important role for nucleosides in the treatment of cancer, and the design of new agents within this class of compounds is still warranted. However, design, synthesis, and evaluation of new analogues as potential anticancer agents is not currently a major emphasis in the drug development community. The reasons for this lack of activity include (a) concerns about the toxicity of nucleoside analogues and the important goal of designing new drugs with less toxicity than the classical agents and (b) concerns that perhaps new nucleoside analogues would not be sufficiently different from those already known and approved for human use, and therefore no further advances were likely. Although toxicity is still a problem and is an issue that is hard to circumvent with antimetabolites (or other classical cytotoxic agents), the information provided in the preceding pages clearly indicates that small structural changes can have profound effects on the biological activity of nucleoside analogues and suggests that new agents with useful activities can still be identified.
An important aspect of the design of purine and pyrimidine antimetabolites is that the drug design process is largely empirical in nature. Compounds are designed that are structurally similar to existing agents based on a thorough understanding of the past work in this field, and they are tested in various biological assays. As indicated in this review, a considerable amount of structure activity relationship (SAR) data is available from the many years of work with this class of compounds that helps guide the design of new compounds. Although this review has focused on the success stories, there are many more examples of antimetabolites that have been designed and synthesized that have not been successful, and a thorough understanding of both the successes and failures is critical to the rational development of new agents of this class. The analysis of the existing FDA-approved anticancer nucleosides indicates a clear and simple guideline that should be considered in the design of new agents in this class. The new compounds should include structural changes that are as small as possible, and as few changes as possible should be made to the natural molecule, with 1–3 changes being the most desirable number.
Because all of the purine and pyrimidine analogues used in the treatment of cancer are prodrugs (except pentostatin), their mechanism of action is very complex and involves interaction with many different anabolic and catabolic enzymes. Therefore, it will not be easy to replace this empirical process with a more “rational” drug design process. Although the empirical approach used in the design of new nucleosides is also a “rational” way to design new drugs, the newer “rational” drug design concepts optimally involve the use of the three-dimensional structure of the protein target coupled with biochemical results and in silico modeling methodology. This approach is most useful when a drug is envisioned to act largely by affecting a single enzymatic target. Although structural information with the various enzymes (particularly deoxycytidine kinase) is increasingly being utilized to aid in the design of new antimetabolites, the design of new antimetabolites is not driven by the desire to interact with just one enzyme. For example, it would not be helpful to design a potent nucleotide inhibitor of DNA polymerase based on structural information, if the nucleoside component of the designed nucleotide inhibitor cannot readily penetrate the cell and be converted to the triphosphate. Nucleotide analogues do not make good drugs, because they do not easily penetrate cell membranes and the phosphates are rapidly removed by plasma phosphatases, although in recent years phosphonate nucleotides have proven to be useful in the treatment of certain viral diseases97 and some research groups are developing strategies to deliver nucleotides to tumor cells.98
Another complicating factor in the “rational” design of new analogues is that the known active agents often inhibit more than one intracellular target. This attribute can be viewed as a strength of this class of compounds and is one of the reasons that antimetabolites have been so successful in the clinic. The multiple points of mechanistic action, however, bring with them serious challenges in terms of “rational” drug design. As difficult as it is to develop a new drug that inhibits only one intracellular target, it is much more than twice as difficult to design one compound that can inhibit two or more enzymes.
The process of evaluating a new analogue for antitumor activity is fairly simple. Once a new antimetabolite has been designed and synthesized, the first and most important experiment is to determine whether or not the compound can kill cancer cells in in vitro assays. A positive result indicates that the compound is able to interact with the metabolic enzymes of either the purine or pyrimidine pathway to create a metabolite that inhibits an enzyme important to DNA replication. Because of the considerable knowledge around this class of compounds, the biochemical details of the mechanism of action can be sketched out with a fair degree of accuracy based on knowledge of the structure of the new agent. However, biochemical studies must still be done to determine how the new agent differs from structurally similar compounds and whether the new agent has characteristics that may be beneficial.
Because of the similarity of structure and mechanism of action of nucleoside analogues, there is no in vitro assay that can predict whether or not any new agent will have sufficient selectivity to be useful in the clinic. An analogue must be able to kill cells in vitro, but to determine whether it will have the appropriate antitumor selectivity, the compound must be evaluated in in vivo studies against various mouse models of cancer. Since selectivity for tumor cells is the most important aspect of a new antitumor agent and in vitro studies cannot predict for selectivity, new analogues must be evaluated in in vivo tumor models as soon as possible. Of course there are serious issues with the ability of the currently used in vivo mouse models to predict for clinical activity, but they are currently the best method to determine whether an agent has antitumor activity at doses that are tolerated in an intact animal. Because large amounts of compound are needed for in vivo studies, compound availability is often a significant hurdle to conducting these studies. Regardless, the importance of in vivo studies cannot be overstated and they should be initiated as soon as sufficient compound is available to proceed. Through many years of experience in the drug discovery process at Southern Research Institute, we have learned that activity against one type of human tumor xenograft (for instance breast tumors) does not predict for activity against that particular tumor type in human disease. The current best predictor for clinical activity for any compound against any tumor type is the demonstration of robust antitumor activity (ability to cause regressions and cures) in numerous human tumor xenografts in mice. If an agent demonstrates robust antitumor activity in mouse models, then clinical trials are necessary to determine which tumor types, if any, are sensitive to the agent in people.
5. Summary
Purine and pyrimidine analogues remain an important class of drugs in the treatment of cancer. Although these agents share many structural and biochemical characteristics, each compound has unique activities that make it a useful drug. The basis of selectivity of these agents is not clearly defined (because the molecular targets exist in both tumor cells and normal host tissues) but is believed to be primarily due to differences in metabolism and proliferative states between tumor cells and normal cells. For instance, the greater expression of deoxycytidine kinase in leukemias and lymphomas is believed to contribute to the sensitivity of these malignancies to nucleoside analogues that are activated by this enzyme. Furthermore, most normal cells in a patient are quiescent and, therefore, are not sensitive to these agents. However, the selectivity of antimetabolites is still poor and better agents are needed with fewer toxicities.
Analysis of the existing agents identifies three primary characteristics of antimetabolites that are important to their ability to kill tumor cells: sufficient metabolism to active metabolite; long retention of active metabolite; and potent and sustained inhibition of DNA replication or function. The analogue should be a reasonable substrate for the activating enzymes, although clearly this aspect of the activity of an analogue can be affected by the potency of the active metabolite against the enzymatic target. For instance, an analogue that produces a very potent active metabolite would not be as dependent on activation. In addition to all the anabolic enzymes involved in the activation of nucleoside analogues, there are numerous catabolic enzymes that interact with these compounds, and these enzymes can also have profound impact on their biological activity and are important in the activity of all of the purine and pyrimidine antimetabolites.
The compound should be a good selective inhibitor of DNA replication and have minimal effects on RNA and protein synthesis, as inhibition of these activities leads to toxicity. The primary intracellular targets of the existing purine and pyrimidine antimetabolites are DNA polymerases, thymidylate synthetase, and ribonucleotide reductase. Although some of the currently approved agents (FUra, mercaptopurine, thioguanine, and aza-Cyd) are converted to ribonucleotide metabolites and are extensively incorporated into RNA, the primary activity of these compounds that results in their antitumor activity is their inhibition of DNA synthesis or disruption of DNA function. Unless there is selective activation in tumor cells, nucleoside analogues that target RNA synthesis or function should be extremely cytotoxic, since all cells require RNA for vitality.
As with most other classical antitumor agents, the inhibition of DNA replication is the most important action of purine and pyrimidine metabolites responsible for their antitumor activity. Disruption of de novo purine biosynthesis or RNA effects are secondary to activities that disrupt DNA replication or cause DNA damage. However, inhibition of DNA synthesis is not sufficient to kill a tumor cell. For example, an agent such as aphidicolin, which is a potent inhibitor of DNA replication, is a good cell synchronizer, because it only inhibits DNA synthesis and, unlike nucleoside analogues, it does not cause any lasting inhibition. Once it is removed from the cell, DNA synthesis readily resumes without lasting toxicity. Nucleoside analogues have two attributes that result in a lasting inhibition of DNA replication after removal of the drug by natural processes within the body. First, the active metabolites of these agents are nucleotide analogues, which do not readily penetrate cell membranes and, therefore, are retained in the cell after the drug has been removed, which is an attribute that is unique to this class of antitumor agents. The half-life for the removal of the triphosphates from cells can be quite long, which leads to continued use by the polymerases and, thus, continued inhibition of DNA replication. The intracellular retention time of the active metabolites (nucleoside triphosphate) can vary considerably between the various analogues, and this can have an important effect on the activity of an agent against solid tumor cells. The much longer half-life of dFdC-TP than araCTP is believed to be a primary contributing factor to the solid tumor activity of gemcitabine and the lack of solid tumor activity of araC. Second, nucleosides are incorporated into DNA, resulting in a DNA molecule that is not easily extended and must be repaired before synthesis can resume. Therefore, an agent that causes DNA damage that is poorly or slowly repaired will result in prolonged damage to the DNA, which will lead to the induction of apoptosis.
In conclusion, purine and pyrimidine antimetabolites are an important class of drugs used in the treatment of cancer and viral diseases. Although the toxicity of these compounds can limit their usefulness, the antimetabolites will continue to play an important role in the treatment of cancer for the foreseeable future. It is likely that some of the new nucleoside analogues that are currently in the pipeline will be approved for use in the coming years. Although drug discovery is being pursued of new anticancer agents that target enzyme activities more closely associated with the cancer phenotype, the unpredicted toxicity of these new agents could still be a major issue of these agents as well. The design, synthesis, and evaluation of new purine and pyrimidine analogues is still a productive area for discovering new drugs for the treatment of cancer, since many years of knowledge with respect to their potential actions and toxicity has accumulated. Novel nucleoside analogues with unique actions are continuously being identified, and the information provided in this review indicates that small structural modifications of nucleoside analogues can have profound effects on their chemical stability and spectrum of biological activity.
Biography

Dr. Parker obtained a Ph.D. in Pharmacology in 1984 from George Washington University and received further postdoctoral research training at The University of North Carolina at Chapel Hill with Dr. Yung-Chi Cheng. In 1988, he joined Southern Research Institute, where he is currently a Senior Research Scientist and Director of the Biochemistry and Molecular Biology Department. For the past 20 years, Dr. Parker has been part of a research team at Southern Research Institute that designs, synthesizes, and evaluates nucleoside analogues in an effort to identify new anticancer drugs. A recent success of this team is the FDA approval in 2004 of clofarabine for the treatment of childhood leukemia. He is an expert in the study of purine and pyrimidine antimetabolites used in the treatment of cancer and infectious diseases, and his work has led to more than 85 publications related to the understanding of the mechanism of action of various analogues in this important class of drugs.
References
- 1.Zhang J, Visser F, King KM, Baldwin SA, Young JD, Cass CE. Cancer Metastasis Rev. 2007;26:85. doi: 10.1007/s10555-007-9044-4. [DOI] [PubMed] [Google Scholar]
- 2.Sampath D, Rao VA, Plunkett W. Oncogene. 2003;22:9063. doi: 10.1038/sj.onc.1207229. [DOI] [PubMed] [Google Scholar]
- 3.Plunkett W, Gandhi V. Cancer Chemother Biol Response Modif. 2001;19:21. [PubMed] [Google Scholar]
- 4.Arner ESJ, Eriksson S. Pharmacol Ther. 1995;67:155. doi: 10.1016/0163-7258(95)00015-9. [DOI] [PubMed] [Google Scholar]
- 5.Johansson NG, Eriksson S. Acta Biochim Pol. 1996;43:143. [PubMed] [Google Scholar]
- 6.Eriksson S, Munch-Peterson B, Johansson K, Eklund H. Cell Mol Life Sci. 2002;59:1327. doi: 10.1007/s00018-002-8511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rompay ARV, Johansson M, Karlsson A. Pharmacol Ther. 2000;87:189. doi: 10.1016/s0163-7258(00)00048-6. [DOI] [PubMed] [Google Scholar]
- 8.Sabini E, Ort S, Monnerjahn C, Konrad M, Lavie A. Nat Struct Biol. 2003;10:513. doi: 10.1038/nsb942. [DOI] [PubMed] [Google Scholar]
- 9.Shcherbakova PV, Bebenek K, Kunkel TA. Sci Aging Knowledge Environ. 2003;2003:1. doi: 10.1126/sageke.2003.8.re3. [DOI] [PubMed] [Google Scholar]
- 10.Matherly LH, Schuetz JD, Westin E, Goldman ID. Anal Biochem. 1989;182:338. doi: 10.1016/0003-2697(89)90605-2. [DOI] [PubMed] [Google Scholar]
- 11.Elion GB. Science. 1989;244:41. doi: 10.1126/science.2649979. [DOI] [PubMed] [Google Scholar]
- 12.Nelson JA, Carpenter JW, Rose LM, Adamson DJ. Cancer Res. 1975;35:2872. [PubMed] [Google Scholar]
- 13.Karran P. Br Med Bull. 2007;79:153. doi: 10.1093/bmb/ldl020. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida S, Yamada M, Masaki S, Saneyoshi M. Cancer Res. 1979;39:3955. [PubMed] [Google Scholar]
- 15.Ling YH, Nelson JA, Cheng YC, Anderson RS, Beattie KL. Mol Pharmacol. 1991;40:508. [PubMed] [Google Scholar]
- 16.Aarbakke J, Janka-Schaub G, Elion GB. Trends Pharmacol Sci. 1997;18:3. doi: 10.1016/s0165-6147(96)01007-3. [DOI] [PubMed] [Google Scholar]
- 17.Tomalik-Scharte D, Lazar A, Fuhr U, Kirchheiner J. Pharmacogenomics J. 2008;8:4. doi: 10.1038/sj.tpj.6500462. [DOI] [PubMed] [Google Scholar]
- 18.Heidelberger C, Chaudhuri NK, Danenberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E, Scheiner J. Nature. 1957;179:663. doi: 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- 19.Danenberg PV. Biochim Biophys Acta. 1977;473:73. doi: 10.1016/0304-419x(77)90001-4. [DOI] [PubMed] [Google Scholar]
- 20.Myers CE. Pharmacol Rev. 1981;33:1. [PubMed] [Google Scholar]
- 21.Parker WB, Cheng YC. Pharmacol Ther. 1990;48:381. doi: 10.1016/0163-7258(90)90056-8. [DOI] [PubMed] [Google Scholar]
- 22.Walko CM, Lindley C. Clin Ther. 2005;27:23. doi: 10.1016/j.clinthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Homsi J, Garrett CR. Cancer Control. 2006;13:42. doi: 10.1177/107327480601300106. [DOI] [PubMed] [Google Scholar]
- 24.Grant S. Adv Cancer Res. 1998;72:197. doi: 10.1016/s0065-230x(08)60703-4. [DOI] [PubMed] [Google Scholar]
- 25.Townsend AJ, Cheng YC. Mol Pharmacol. 1987;32:330. [PubMed] [Google Scholar]
- 26.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Cancer Res. 1991;51:6110. [PubMed] [Google Scholar]
- 27.Ostruszka LJ, Shewach DS. Cancer Chemother Pharmacol. 2003;52:325. doi: 10.1007/s00280-003-0661-5. [DOI] [PubMed] [Google Scholar]
- 28.Gandhi V, Legha J, Chen F, Hertel LW, Plunkett W. Cancer Res. 1996;56:4453. [PubMed] [Google Scholar]
- 29.Plunkett W, Huang P, Searcy CE, Gandhi V. Semin Oncol. 1996;23(Suppl 10):3. [PubMed] [Google Scholar]
- 30.Stresemann C, Lyko F. Int J Cancer. 2008;123:8. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 31.Cihak A, Vesely J, Skoda J. Adv Enzyme Regul. 1985;24:335. doi: 10.1016/0065-2571(85)90085-8. [DOI] [PubMed] [Google Scholar]
- 32.Momparler RL. Pharmacol Ther. 1985;30:287. doi: 10.1016/0163-7258(85)90053-1. [DOI] [PubMed] [Google Scholar]
- 33.Oki Y, Aoki E, Issa JJ. Crit Rev Oncol/Hematol. 2007;61:140. doi: 10.1016/j.critrevonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Bouchard J, Momparler RL. Mol Pharmacol. 1983;24:109. [PubMed] [Google Scholar]
- 35.Link PA, Baer MR, James SR, Jones DA, Karpf AR. Cancer Res. 2008;68:9358. doi: 10.1158/0008-5472.CAN-08-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vesely J, Cihak A, Sorm F. Collect Czech Chem Commun. 1969;34:901. [Google Scholar]
- 37.Newman EM, Santi DV. Proc Natl Acad Sci USA. 1982;79:6419. doi: 10.1073/pnas.79.21.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plunkett W, Huang P, Gandhi V. Semin Oncol. 1990;17:3. [PubMed] [Google Scholar]
- 39.Gandhi V, Plunkett W. Drug Dispos. 2002;41:93. doi: 10.2165/00003088-200241020-00002. [DOI] [PubMed] [Google Scholar]
- 40.Chilson OP, Fisher JR. Arch Biochem Biophys. 1963;102:77. doi: 10.1016/0003-9861(63)90322-9. [DOI] [PubMed] [Google Scholar]
- 41.Frederickson S. Arch Biochem Biophys. 1966;113:383. doi: 10.1016/0003-9861(66)90202-5. [DOI] [PubMed] [Google Scholar]
- 42.Buie LW, Epstein SS, Lindley CM. Clin Ther. 2007;29:1887. doi: 10.1016/j.clinthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Reist EJ, Goodman L. Biochemistry. 1964;3:15. doi: 10.1021/bi00889a004. [DOI] [PubMed] [Google Scholar]
- 44.Cohen A, Lee JW, Gelfand EW. Blood. 1983;61:660. [PubMed] [Google Scholar]
- 45.Shewach DS, Daddona PE, Ashcraft E, Mitchell BS. Cancer Res. 1985;45:1008. [PubMed] [Google Scholar]
- 46.Rodriguez CO, Jr, Stellrecht CM, Gandhi V. Blood. 2003;102:1842. doi: 10.1182/blood-2003-01-0317. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez CO, Jr, Mitchell BS, Ayres M, Eriksson S, Gandhi V. Cancer Res. 2002;62:3100. [PubMed] [Google Scholar]
- 48.Parker WB, Bapat AR, Shen JX, Townsend AJ, Cheng YC. Mol Pharmacol. 1988;34:485. [PubMed] [Google Scholar]
- 49.Huang P, Chubb S, Plunkett W. J Biol Chem. 1990;265:16617. [PubMed] [Google Scholar]
- 50.Gandhi V, Mineishi S, Huang P, Chapman AJ, Yang Y, Chen F, Nowak B, Chubb S, Hertel LW, Plunkett W. Cancer Res. 1995;55:1517. [PubMed] [Google Scholar]
- 51.Nutter LM, Cheng YC. Pharmacol Ther. 1984;26:191. doi: 10.1016/0163-7258(84)90016-0. [DOI] [PubMed] [Google Scholar]
- 52.Goodman GR, Beutler E, Saven A. Best Pract Res, Clin Haematol. 2003;16:101. doi: 10.1016/s1521-6926(02)00089-0. [DOI] [PubMed] [Google Scholar]
- 53.Parker WB, Shaddix SC, Chang CH, White EL, Rose LM, Brockman RW, Shortnancy AT, Montgomery JA, Secrist JA, III, Bennett LL., Jr Cancer Res. 1991;51:2386. [PubMed] [Google Scholar]
- 54.Bonate PL, Arthaud L, Jr, Stephenson K, Secrist JA, III, Weitman S. Nat Rev Drug Discovery. 2006;5:855. doi: 10.1038/nrd2055. [DOI] [PubMed] [Google Scholar]
- 55.Faderl S, Gandhi V, Keating MJ, Jeha S, Plunkett W, Kantarjian HM. Cancer. 2005;103:1985. doi: 10.1002/cncr.21005. [DOI] [PubMed] [Google Scholar]
- 56.Xie C, Plunkett W. Cancer Res. 1995;55:2847. [PubMed] [Google Scholar]
- 57.Xie KC, Plunkett W. Cancer Res. 1996;56:3030. [PubMed] [Google Scholar]
- 58.Waud WR, Schmid SM, Montgomery JA, Secrist JA., III Nucleosides Nucleotides Nucleic Acids. 2000;19:447. doi: 10.1080/15257770008033020. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi T, Shimizu M, Akinaga S. Cancer Chemother Pharmacol. 2002;50:193. doi: 10.1007/s00280-002-0472-0. [DOI] [PubMed] [Google Scholar]
- 60.Carson DA, Wasson DB, Esparza LM, Carrera CJ, Kipps TJ, Cottam HB. Proc Natl Acad Sci USA. 1992;89:2970. doi: 10.1073/pnas.89.7.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheson B. Semin Oncol. 1992;19:695. [PubMed] [Google Scholar]
- 62.Tallman MS, Hakimian D. Blood. 1995;86:2463. [PubMed] [Google Scholar]
- 63.Grove KL, Guo X, Liu SH, Gao Z, Chu CK, Cheng YC. Cancer Res. 1995;55:3008. [PubMed] [Google Scholar]
- 64.Krishnan P, Fu Q, Lam W, Liou JY, Dutschman G, Cheng YC. J Biol Chem. 2002;277:5453. doi: 10.1074/jbc.M109025200. [DOI] [PubMed] [Google Scholar]
- 65.Krishnan P, Gullen EA, Lam W, Dutschman GE, Grill SP, Cheng YC. J Biol Chem. 2003;278:36726. doi: 10.1074/jbc.M307052200. [DOI] [PubMed] [Google Scholar]
- 66.Kukhanova M, Liu SH, Mozzherin D, Lin TS, Chu CK, Cheng YC. J Biol Chem. 1995;270:23055. doi: 10.1074/jbc.270.39.23055. [DOI] [PubMed] [Google Scholar]
- 67.Grove KL, Cheng YC. Cancer Res. 1996;56:4187. [PubMed] [Google Scholar]
- 68.Chou KM, Kukhanova M, Cheng YC. J Biol Chem. 2000;275:31009. doi: 10.1074/jbc.M004082200. [DOI] [PubMed] [Google Scholar]
- 69.Swords R, Giles F. Hematology. 2007;12:219. doi: 10.1080/10245330701406881. [DOI] [PubMed] [Google Scholar]
- 70.Waud WR, Gilbert KS, Shepherd RV, Montgomery JA, Secrist JA., III Cancer Chemother Pharmacol. 2003;51:422. doi: 10.1007/s00280-003-0589-9. [DOI] [PubMed] [Google Scholar]
- 71.Parker WB, Shaddix SC, Rose LM, Waud WR, Shewach DS, Tiwari KN, Secrist JA., III Biochem Pharmacol. 2000;60:1925. doi: 10.1016/s0006-2952(00)00520-7. [DOI] [PubMed] [Google Scholar]
- 72.Richardson F, Black C, Richardson K, Franks A, Wells E, Karimi S, Sennello G, Hart K, Meyer D, Emerson D, Brown E, LeRay J, Nilsson C, Tomkinson B, Bendele R. Cancer Chemother Pharmacol. 2005;55:213. doi: 10.1007/s00280-004-0844-8. [DOI] [PubMed] [Google Scholar]
- 73.Clarke ML, Damaraju VL, Zhang J, Mowles D, Tackaberry T, Lang T, Smith KM, Young JD, Tomkinson B, Cass CE. Mol Pharmacol. 2006;70:303. doi: 10.1124/mol.105.021543. [DOI] [PubMed] [Google Scholar]
- 74.Richardson KA, Vega TP, Richardson FC, Moore CL, Rohloff JC, Tomkinson B, Bendele RA, Kuchta RD. Biochem Pharmacol. 2004;68:2337. doi: 10.1016/j.bcp.2004.07.042. [DOI] [PubMed] [Google Scholar]
- 75.Someya H, Shaddix SC, Tiwari KN, Secrist JA, III, Parker WB. J Pharmacol Exp Ther. 2003;304:1314. doi: 10.1124/jpet.102.045435. [DOI] [PubMed] [Google Scholar]
- 76.Someya H, Waud WR, Parker WB. Cancer Chemother Pharmacol. 2006;57:772. doi: 10.1007/s00280-005-0126-0. [DOI] [PubMed] [Google Scholar]
- 77.Goss G, Siu LL, Gauthier I, Chen EX, Oza AM, Goel R, Maroun J, Powers J, Walsh W, Maclean M, Drolet DW, Rusk J, Seymour LK. Cancer Chemother Pharmacol. 2006;58:703. doi: 10.1007/s00280-006-0201-1. [DOI] [PubMed] [Google Scholar]
- 78.Lee CP, de Jonge MJ, O’Donnell AE, Schothorst KL, Hanwell J, Chick JB, Brooimans RA, Adams LM, Drolet DW, de Bono JS, Kaye SB, Judson IR, Verweij J. Clin Cancer Res. 2006;12:2841. doi: 10.1158/1078-0432.CCR-05-1932. [DOI] [PubMed] [Google Scholar]
- 79.Azuma A, Huang P, Matsuda A, Plunkett W. Biochem Pharmacol. 2001;61:1497. doi: 10.1016/s0006-2952(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 80.Matsuda A, Nakajima Y, Azuma A, Tanaka M, Sasaki T. J Med Chem. 1991;34:2917. doi: 10.1021/jm00113a034. [DOI] [PubMed] [Google Scholar]
- 81.Hanaoka K, Suzuki M, Kobayashi T, Tanzawa F, Tanaka K, Shibayama T, Miura S, Ikeda T, Iwabuchi H, Nakagawa A, Mitsuhashi Y, Hisaoka M, Kaneko M, Tomida A, Wataya Y, Nomura T, Sasaki T, Matsuda A, Tsuruo T, Kurakata S. Int J Cancer. 1999;82:226. doi: 10.1002/(sici)1097-0215(19990719)82:2<226::aid-ijc13>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 82.Wang Y, Liu X, Matsuda A, Plunkett W. Cancer Res. 2008;68:3881. doi: 10.1158/0008-5472.CAN-07-6885. [DOI] [PubMed] [Google Scholar]
- 83.Gilbert J, Carducci MA, Baker SD, Dees EC, Donehower R. Invest New Drugs. 2006;24:499. doi: 10.1007/s10637-006-8219-0. [DOI] [PubMed] [Google Scholar]
- 84.Markert ML. Immunodefic Rev. 1991;3:45. [PubMed] [Google Scholar]
- 85.Montgomery JA. Med Res Rev. 1993;13:209. doi: 10.1002/med.2610130302. [DOI] [PubMed] [Google Scholar]
- 86.Schramm VL. Biochem Biophys Acta. 2002;1587:107. doi: 10.1016/s0925-4439(02)00073-x. [DOI] [PubMed] [Google Scholar]
- 87.Gandhi V, Balakrishnan K. Semin Oncol. 2007;34(suppl 5):S8. doi: 10.1053/j.seminoncol.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 88.Larson RA. Semin Oncol. 2007;34(suppl 5):S13. doi: 10.1053/j.seminoncol.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 89.Furman RR, Hoelzer D. Semin Oncol. 2007;34(suppl 5):S29. doi: 10.1053/j.seminoncol.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 90.Gore L, Stelljes M, Quinones R. Semin Oncol. 2007;34(suppl 5):S35. doi: 10.1053/j.seminoncol.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 91.Springer CJ, Niculescu I. J Clin Invest. 2000;105:1161. doi: 10.1172/JCI10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Denny WA. J Biomed Biotechnol. 2003;2003:48. doi: 10.1155/S1110724303209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dachs GU, Tupper J, Tozer GM. Anti-Cancer Drugs. 2005;16:349. doi: 10.1097/00001813-200504000-00001. [DOI] [PubMed] [Google Scholar]
- 94.Altaner C. Cancer Let. 2008;270:191. doi: 10.1016/j.canlet.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 95.Hong JS, Waud WR, Levasseur DN, Townes TM, Wen H, McPherson SA, Moore BA, Bebok Z, Allan PW, Secrist JA, III, Parker WB, Sorscher EJ. Cancer Res. 2004;64:6610. doi: 10.1158/0008-5472.CAN-04-0012. [DOI] [PubMed] [Google Scholar]
- 96.Parker WB, Allan PW, Shaddix SC, Rose LM, Speegle HF, Gillespie GY, Bennett LL., Jr Biochem Pharmacol. 1998;55:1673. doi: 10.1016/s0006-2952(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 97.De Clercq E. Antiviral Res. 2007;75:1. doi: 10.1016/j.antiviral.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 98.Boyer SH, Sun Z, Jiang H, Esterbrook J, Gómez-Galeno JE, Craigo W, Reddy KR, Ugarkar BG, MacKenna DA, Erion MD. J Med Chem. 2006;49:7711. doi: 10.1021/jm0607449. [DOI] [PubMed] [Google Scholar]