

Abstract
The tracheal system of Drosophila embryos achieves its archetypal branching pattern through a series of cell migration events requiring the FGF, Dpp, and Wg/WNT signaling pathways. To gain insight into tracheal cell migration, we performed an F4 EMS mutagenesis screen to generate and characterize new mutations resulting in tracheal defects. From 2591 mutagenized third chromosome lines, we identified 33 mutations with defects in tracheal development, corresponding to 12 distinct complementation groups. The new mutations included novel hypomorphic alleles of the FGF receptor gene, breathless, and the ETS-domain transcription factor gene, pointed. We show that reduced function of either breathless or pointed specifically affects migration of the dorsal and ventral tracheal branches, more specific functions than previously described for these genes. Our analysis reveals that breathless and pointed control dorsal branch migration through transcriptional regulation of the Dpp pathway effectors, Knirps and Knirps-related, which are necessary for migration of this branch. We further show that expression of knirps or knirps-related rescues dorsal but not ventral branch migration in the breathless hypomorph. These studies support a model in which both the Dpp- and the FGF-signaling pathways control expression of knirps and knirps-related, thereby regulating cell migration during dorsal branch formation.
Keywords: Breathless, Cell migration, Decapentaplegic, Drosophila melanogaster, knirps, knirps-related, Pointed, Trachea
Introduction
Directed cell migration occurs repeatedly throughout the development of multicellular organisms to give rise to many tissues and organs. Over the past decade, the developing tracheal (respiratory) system of Drosophila embryos has become one of the models of choice for studying the molecular mechanisms of directed movement. The simple structure of the Drosophila trachea combined with its accessible genetics has led to the identification of multiple signaling molecules, their receptors, and the intracellular signaling pathways involved in the migration of tracheal cells. The entire tracheal system originates from 10 placodes, or clusters, of approximately 90 cells each on either side of the embryo (reviewed in Manning and Krasnow, 1993). After invaginating into the underlying mesoderm, tracheal cells form a stereotypical pattern of tubular branched structures. Some tracheal branches, such as the dorsal trunk, grow along the anterior–posterior axis and others, such as the dorsal, lateral, and ganglionic branches, grow along the dorsal–ventral axis of the embryo. Subsequent fusion of the dorsal trunk branches and the lateral branches of each individual tracheal metamere connects the primary branches on each side of the embryo. Following primary branch formation, secondary and terminal branches form, some of which fuse, ultimately giving rise to a highly branched network of interconnected tubes responsible for transporting oxygen and other gases throughout the animal.
Fibroblast growth factor (FGF) signaling through the FGF receptor, Breathless (Btl), is one of the key signaling pathways involved in tracheal cell migration. Branchless (Bnl), an FGF-like ligand, is expressed in non-tracheal cells surrounding the invaginated tracheal cells in a pattern that prefigures the formation of the primary tracheal branches (Sutherland et al., 1996). Tracheal cells expressing the Btl receptor migrate towards the source of Bnl, initiating the formation of the six primary branches. In either bnl or btl null mutants, all primary branches fail to form, whereas expression of bnl in novel locations is sufficient to induce migration of new branches to ectopic sites (Klambt et al., 1992; Sutherland et al., 1996). Bnl acts as a chemoattractant for tracheal cells and induces the formation of filopodia at the tips of budding primary branches (Ribeiro et al., 2002; Wolf et al., 2002). One downstream component essential for FGF-signaling is heartbroken (also known as downstream of FGF and stumps), which encodes a novel intracellular protein that functions as a signaling adaptor (Petit et al., 2004). In hbr mutants, filopodial extension and primary branch formation are defective, as in bnl and btl mutants (Imam et al., 1999; Michelson et al., 1998; Vincent et al., 1998).
Although the branchless signaling pathway sets the general pattern of tracheal branching, additional signaling events are required for individual branches to acquire their distinctive characteristics, such as their size, shape, and branching patterns. These characteristics are controlled by molecules expressed or activated in a region- or branch-specific pattern, such as components of the Decapentaplegic (Dpp) and Wingless/WNT (WG) signaling pathways. Dpp signaling is required for tracheal cell migration during formation of the dorsal, ganglionic, and lateral trunk branches (Llimargas and Casanova, 1997; Vincent et al., 1997; Wappner et al., 1997). Dpp, which is expressed in cells dorsal and ventral to the trachea, signals through its receptors Thick veins and Punt (types I and II TGFβ receptor family members) to induce expression of the zinc finger transcription factor genes, knirps (kni) and knirps-related (knrl), in the responding tracheal cells (Affolter et al., 1994; Chen et al., 1998; Ruberte et al., 1995; Vincent et al., 1997). When Dpp signaling is specifically inhibited in tracheal cells, filopodia still extend from the tips of primary buds; however, the dorsal branches fail to form (Ribeiro et al., 2002). Thus, for dorsal branch migration to occur, Dpp signaling in combination with Bnl induction of filopodial activity is required.
Although previous studies have revealed how the general pattern of tracheal migration is achieved, we are only beginning to identify downstream target genes of the signaling pathways involved in either general or branch-specific tracheal cell migration. For example, genes required for Bnl/Btl induction of filopodial activity or that function downstream of Kni and Knrl for proper dorsal and ventral branch formation have yet to be identified. Furthermore, it is unclear how the signaling pathways involved in branch-specific migration are integrated with the Bnl/Btl pathway, which regulates migration of all tracheal branches. To better understand tracheal cell migration, we generated a collection of EMS mutants and screened for defects in tracheal development. From 2591 mutant lines, we identified mutations in 12 complementation groups that fall into three categories: mutations in genes previously known to affect tracheal migration, mutations in genes for which tracheal defects have not been described previously and novel mutations corresponding to as yet unidentified genes. Among genes known to affect tracheal migration, we identified novel hypomorphic alleles of btl and pointed (pnt) and show that Btl and Pnt play roles in branch-specific migration through transcriptional regulation of the Dpp-signaling effectors, Kni and Knrl.
Materials and methods
Fly stocks
The wild-type fly strain Oregon-R was used for controls. The following strains were obtained from the Bloomington Stock Center and are described in FlyBase (http://flybase.bio.indiana.edu): rhoΔ38, hh21, btlLG19, btlH82Δ3, btldev1, bnlP1, hbr, pntΔ88, punt10460, kni6, and the third chromosome deficiency kit (DK3). btlH82Δ11 was kindly provided by B. Shilo. For the UAS-GAL4 expression system (Brand and Perrimon, 1993), we used btl-GAL4 (kindly provided by S. Hayashi) to drive expression throughout the tracheal system and 69B-GAL4 (provided by the Bloomington Stock Center) to drive expression throughout the ectoderm. The following are GAL-4-dependent UAS expression lines: UAS-btl, UAS-tkv1a1BX, UAS-tkvQD(147.1) (kindly provided by M. Affolter), UAS-kni and UAS-knrl (kindly provided by R. Schuh), and UAS-bnl (kindly provided by M. Krasnow). rp395 is a rosy+ P-element enhancer-trap line provided by C. Goodman.
Whole-mount antibody staining and in situ hybridizations
Embryo fixation and antibody staining were performed as previously described (Reuter et al., 1990). Antisera were used at the following dilutions: mouse monoclonal anti-Crumbs (Developmental Studies Hybridoma Bank) at 1:100; mouse anti-β-galactosidase (β-gal) (Promega: Madison, WI) at 1:10,000; and rat anti-Knirps (D. Kosman) at 1:200. Appropriate biotinylated secondary antibodies (Vector Labs, Burlingame, CA) were used at a dilution of 1:500. Stained embryos were mounted in methylsalicylate (Sigma, St. Louis, MO) and visualized and photographed on a Zeiss Axiophot microscope (Carl Zeiss, Thornwood, NY) with Nomarski optics using either Kodak print film (Eastman Kodak, Rochester, NY) or a Nikon CoolPix 4500 digital camera (Image Systems Inc., Columbia, MD). Whole-mount in situ hybridizations to detect mRNA were performed as previously described (Lehmann and Tautz, 1994). The following cDNAs were used as templates for generating antisense digoxygenin-labeled RNA probes: trh (our lab), bnl (S. Hayashi), btl (D. Montell), dpp (W. Gelbart), tkv and kni (Research Genetics), and knrl (P. Beachy). Embryos were mounted in 70% glycerol and visualized and photographed as described above for antibody-stained embryos. Homozygous mutant embryos were unambiguously identified by the absence of β-gal staining, which detects lacZ expression from the Ubx-lacZ insert on either the TM6B or TM3 balancer chromosome or by lack of hybridization with anti-sense lacZ probes.
Mutagenesis and complementation analysis
One hundred males homozygous for a recently isogenized third chromosome (iso-3 red e) were collected 3–5 days after eclosion, starved for 6–12 h, and subsequently treated with 25 mM ethyl methane sulfonate (EMS; Sigma) in 1% sucrose for 18–24 h using standard procedures (Grigliatti, 1986). The mutagenized males were mated en masse to an equivalent number of virgin females homozygous for the rp395 P-element insertion on the second chromosome and carrying the third chromosome balancers TM6B and TM3-UbxLacZ (TM3-UL; Fig. 1A). Single F1 males heterozygous for the rp395 insertion and carrying a mutagenized third chromosome over a balancer chromosome (either TM6B or TM3-UL) were mated to between three and five virgin females of the parental female strain. The F2 cross, identical to the F1 cross, was repeated to eliminate mosaicism. Male and female progeny of the F3 generation homozygous for the rp395 insertion and also carrying the mutagenized third chromosome over the TM3-UL balancer chromosome were used to establish stocks. Once the stocks were sufficiently expanded, embryos from each stock were collected and stained for β-gal expression to visualize defects in tracheal development. Homozygous embryos were easily distinguished from their heterozygous siblings by the lack of β-gal expression conferred by the Ubx-lacZ insert on the TM3-UL balancer chromosome. Mutant lines that did not show tracheal defects were not further characterized.
Fig. 1.

An F4 EMS screen of the third chromosome to isolate mutations causing defects in tracheal development. A schematic outline of the mutagenesis screen is shown in A. Parent (P) males with isogenized third chromosomes (iso red e) were fed EMS sucrose and subsequently crossed to females homozygous for the rp395 insertion on their second chromosome and carrying the third chromosome balancers TM6B and TM3-UbxlacZ (TUL). The resultant mutagenized third chromosome is marked with an “*” in all subsequent generations. A balanced stock was established and F4 embryos were immunostained with antiserum to β-galactosidase (β-gal) and analyzed for defects in tracheal migration. See Materials and methods for details. β-gal staining of the original rp395 line (B and C) shows expression in the developing tracheal network, including cells of the dorsal branch (DB), dorsal trunk (DT), visceral branch (VB), lateral trunk anterior (Lta), and ganglionic branch (GB). Embryos shown are at stage 14 (Campos-Ortega and Hartenstein, 1997).
To identify potential complementation groups, mutant lines from the EMS screen that showed defects in tracheal development were crossed to each other. Allelism was determined as failure to produce viable adults when transheterozygous. A mutant line from each complementation group was then tested for complementation with known tracheal mutations that map to the third chromosome. The third chromosome deficiency collection (DK3) is comprised of 74 lines that delete approximately 77% of the chromosome. Sixty-five of the 74 deficiency lines could be maintained over the TM6B-UL balancer chromosome where the β-gal expression from the Ubx-lacZ insert allowed homozygous embryos to be easily distinguished from their heterozygous siblings. Embryos from the 65 deficiency lines with the marked balancer chromosomes were stained for the apical membrane protein Crumbs (Crb) and β-gal and screened for defects in tracheal development. Deficiency lines with tracheal defects were crossed to the EMS mutant lines that were not allelic to known tracheal genes and scored for complementation. Transheterozygous embryos of an EMS mutant and a deficiency line that showed non-complementation were stained with αCrb and analyzed for tracheal defects to confirm that the tracheal mutant phenotype mapped to the interval deleted by the deficiency. The deficiency lines that could not be maintained over a marked balancer were also stained. Six of these lines had tracheal defects and were also tested for complementation with the new tracheal mutations.
Sequence analysis of new btl mutations
To determine the molecular lesion in btl95 and btl469 mutants, genomic DNA was isolated and overlapping fragments spanning the btl ORF were amplified by PCR. The resultant products were sequenced and analyzed for allelic differences. The btl gene from an unrelated mutation generated in the same EMS screen and maintained over the same TM3-UL balancer as btl95 and btl469 (h674; Myat and Andrew, 2002) was also sequenced as a control. Synthesis of PCR primers and DNA sequencing were performed at the Johns Hopkins University Biosynthesis and Sequencing Facility.
Results
A screen for mutations affecting tracheal development
To systematically identify mutations affecting tracheal development, we performed an F4 EMS mutagenesis screen to isolate third chromosome mutations. The overall scheme for the mutagenesis is shown in Fig. 1A and is described in detail in the Materials and methods. To visualize the developing tracheal network, we utilized a second chromosome P-element insertion line, rp395, which drives expression of β-galactosidase (β-gal) in all tracheal cells, as well as in a number of other tissues, such as the salivary gland (Figs. 1B and C; data not shown). From the 2591 lines screened, we identified 33 mutations that disrupted tracheal development when homozygous. These mutations corresponded to 12 complementation groups based on lethality and tracheal defects in the trans-heterozygotes. Complementation tests between third chromosome mutations known to affect trachea formation and each of the 12 complementation groups revealed that the EMS screen created one novel allele of rhomboid, two of hedgehog, three of breathless, one of branchless, seven of heartbroken, and one of pointed (Tables 1 and 2). The mutagenesis screen also generated new alleles of hairy and klarsicht, both of which affect the size and morphology of the tracheal lumen (M.M. Myat and D.J. Andrew, in preparation).
Table 1.
Tracheal mutations generated by the EMS mutagenesis screen of chromosome 3
| Gene/mutant | Tracheal function | Allele name(s) |
|---|---|---|
| rhomboid | invagination | rho207 |
| hedgehog | patterning | hh520, hh688 |
| breathless | migration | btl95, btl469, btl1928 |
| branchless | migration | bnl708 |
| heartbroken | migration | hbr65, hbr878, hbr1468, hbr1653, hbr1887, hbr2407, hbr2756 |
| pointed | migration | pnt737 |
| hairy | patterning/lumen size | h674 |
| klarsicht | migration/lumen size | klar2679 |
| anduril | migration | aul589, aul3999 |
| gimli | migration | gim2388 |
| gloin | migration | glo99, glo319, glo427, glo1041, glo1043, glo1273, glo1721, glo1786, glo1986, glo2001, glo2009, glo2056 |
| fangorn | migration | fang586 |
Table 2.
Complementation analysis of EMS mutations with known tracheal mutations on chromosome 3
| Mutant | rhoΔ38 | rho207 | hh21 | btlLG19 | bnlP1 | bnl708 | hbr | hbr1653 | hbr65 | pntΔ88 | pnt737 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rho207 | 167:0 | ||||||||||
| hh520 | 176:0 | ||||||||||
| hh688 | 131:0 | ||||||||||
| btl95 | 153:2 | ||||||||||
| btl469 | 203:0 | ||||||||||
| btl1928 | 95:0 | ||||||||||
| bnl708 | 124:0 | ||||||||||
| hbr65 | 104:0 | ||||||||||
| hbr878 | 140:0 | ||||||||||
| hbr1468 | 107:0 | ||||||||||
| hbr1653 | 104:0 | ||||||||||
| hbr1887 | 78:0 | ||||||||||
| hbr2407 | 137:0 | ||||||||||
| hbr2756 | 107:0 | ||||||||||
| pnt737 | 135:0 | ||||||||||
| aul589 | 19:1 | 36:24 | 23:12 | 16:22 | 64:10 | 16:10 | |||||
| aul3999 | 54:12 | ||||||||||
| gim2388 | 25:10 | 4:10 | 17:10 | 22:11 | 31:10 | 8:10 | |||||
| glo319 | 26:16 | 56:12 | 29:16 | 56:16 | 44:24 | 70:15 | |||||
| fang586 | 34:14 | 20:11 | 59:22 | 34:14 | 112:21 | 33:10 |
The mutagenesis screen also identified four novel mutations corresponding to as yet unidentified genes affecting tracheal migration (Fig. 2). Names for the novel mutations were chosen from characters in J. R. R. Tolkein’s Lord of the Rings and were based on their respective tracheal phenotypes. In embryos homozygous for anduril (aul), certain branches appeared broken (Figs. 2B and B′, arrow) or failed to form (arrowheads). In embryos homozygous for gimli (gim) and gloin (glo; Figs. 2C, C′, D, and D′), most of the primary branches failed to migrate normally; however, the dorsal trunk (DT; arrows) and the visceral branches (VB; arrowheads) were the most affected. In contrast, in fangorn (fang) embryos, the dorsal branches (DB; Figs. 2E and E′, arrow) and ventral branches (Lta, Ltp; arrowhead) were most severely affected. As a first step towards identifying the wild-type genes corresponding to the aul, gim, glo, and fang mutations, we attempted to map each mutation to a discrete genomic interval by complementation analysis using the third chromosome deficiency collection available from the Bloomington Stock Center. This collection consists of 74 lines that delete DNA corresponding to approximately 77% of the chromosome (FlyBase). To expedite the complementation analysis between the novel mutations causing tracheal defects and the deficiency collection, we first screened lines from the third chromosome deficiency collection for defects in tracheal development. Deficiency embryos were stained for Crumbs (Crb), a transmembrane protein that localizes to the apical domain of tracheal cells as well as other epithelia (Tepass et al., 1990). Crb staining was used to assess the extent of invagination as well as migration of individual tracheal branches. From the deficiency collection, we identified 34 with clear defects in either invagination or migration of tracheal cells, or defects in tracheal lumen morphology (Table 3). We then tested for complementation between each of the deficiency lines with tracheal defects and a mutant line from each new complementation group (anduril, gimli, gloin, and fangorn). aul3999 and aul589 failed to complement Df(3R)by62 (Table 3 and data not shown). Furthermore, transheterozygous embryos showed tracheal phenotypes similar to those of aul homozygous embryos (data not shown), indicating that aul maps to a region from 85D11–14 to 85F6. Mutations in the remaining complementation groups, gim, glo, and fang, complemented the 34 deficiencies with tracheal defects (data not shown), indicating that the corresponding mutations map outside the intervals deleted by the 34 deficiency lines we tested.
Fig. 2.

EMS-induced mutations in new genes affect tracheal migration. In a wild-type embryo (A and A′), all primary branches migrate normally to form a connected tracheal network. In an aul3999 embryo (B and B′), formation of the DT (arrow) and the ventral branches (arrowheads) is affected. In gim2388 (C and C′) and glo319 (D and D′) embryos, migration of all tracheal branches is severely affected, particularly the DT (C′ and D′, arrows) and the VB (C′, arrowhead). In a fang586 homozygous embryo (E and E′), the DB (arrow) and Lta (arrowhead) are most affected. All embryos shown are at stage 14 except for gim2388, which is at stage 13. All embryos were stained for the apical membrane protein, Crumbs, to mark the tracheal lumen and β-gal to distinguish homozygous from heterozygous embryos.
Table 3.
Third chromosome deficiencies with defects in tracheal morphogenesis
| Deficiency | Breakpoints | Tracheal defect | Tracheal genes in interval |
|---|---|---|---|
| Df(3L)emc-E12 | 61A;61D3 | Invagination | trachealess, klarsicht |
| Df(3L)Ar14-8 | 61C5-8;62A8 | Invagination | rhomboid |
| Df(3L)R-G7 | 62B8-9;62F2-5 | Migration | |
| Df(3L)M21 | 62F;63D,62A;64C | Disrupted branches | |
| Df(3L)GN24 | 63F6-7;64C13-15 | Migration, specific metameres | |
| Df(3L)ZN47 | 64C;65C | Invagination | drifter |
| Df(3L)pbl-X1 | 65F3;66B10 | Lumen size | |
| Df(3L)66C-G28 | 66B8-9;66C9-10 | Migration | |
| Df(3L)hi22 | 66D10-11;66E1-2 | Patterning/lumen size | hairy |
| Df(3L)Ac1 | 67A2;67D7-13 | Migration | |
| Df(3L)vin5 | 68A2-3;69A1-3 | Migration | |
| Df(3L)vin7 | 68C8-11;69B4-5 | Migration | |
| Df(3L)fz-GF3b | 70C1-2;70D4-5 | Migration | breathless |
| Df(3L)fz-M21 | 70D2-3;71E4-5 | Migration | |
| Df(3L)brm1 | 71F1-4;72D1-10 | Migration | |
| Df(3L)81K19 | 73A3;74F | Delayed migration | |
| Df(3L)VW3 | 76A3;76B2 | Migration | |
| Df(3L)kto2 | 76B1-2;76D5 | Migration | |
| Df(3L)XS533 | 76B04;77B | Migration | |
| Df(3L)ri-79C | 77B-C;77F-78A | Disrupted migration | knirps/knirps-related |
| Df(3L)ME107 | 77F3;78C8-9 | Migration | |
| Df(3L)Pc-2q | 78C5-6;78E3-79A1 | Migration | |
| Df(3R)e1025 | 82F8-10;83A1-3 | Migration | |
| Df(3R)Scr | 84A1-2;84B1-2 | Migration | |
| Df(3R)Antp17 | 84B1-2;84D11-12 | Migration | |
| Df(3R)pXT103 | 85A2;85C1-2 | Disrupted branches | |
| Df(3R)by62 | 85D11-14;85F06 | Migration | anduril |
| Df(3R)M-Kx1 | 86E2-4;87C6-7 | Lumen size | |
| Df(3R)Dl-BX12 | 90F1-2;92D3-6 | Migration | branchless |
| Df(3R)mbc-30 | 95A5-7;95C10-11 | Migration | dally, dally-like |
| Df(3R)mbc-R1 | 95A5-7;95D6-11 | Migration | |
| Df(3R)Espl3 | 96F1;97B1 | Migration | |
| Df(3R)3450 | 98E3;99A6-8 | Lumen size | |
| Df(3R)Dr-rv1 | 99A1-2;99B6-11 | Migration |
breathless and pointed regulate dorsal–ventral migration through knirps and knirps-related
The mutagenesis screen generated three new alleles of breathless (btl), which normally directs the migration of all tracheal branches in response to its ligand, Bnl (Klambt et al., 1992; Sutherland et al., 1996; Fig. 3). With two of the new btl alleles identified in this screen, btl469 and btl1928, the expected null phenotypes, in which all branches completely fail to migrate, were observed (Fig. 3C; data not shown). The third new allele, btl95, affected only the migration of specific tracheal branches, the dorsal branch (DB), and the lateral trunk anterior (Lta; Figs. 3E and F). A previously identified btl hypomorph, btlH82Δ11, affects migration of all primary branches, as observed with btl null alleles; some branches, however, such as the dorsal trunk (DT), ganglionic (GB), and visceral branches (VB), form rudimentary structures (Fig. 3G). btl95 in trans to the null btl allele, btlLG19 (Fig. 3H), the hypomorphic allele, btlH82Δ11 (Fig. 3I), or Df(3L)fz-Gf3b, which removes btl (Fig. 3J), showed intermediate phenotypes, again with the DB and Lta completely failing to migrate. This analysis suggests that btl95 is a hypomorphic allele of btl and causes milder tracheal defects than the pre-existing hypomorphic alleles, btlH82Δ11, btlH82Δ3, and btldev1 (Fig. 3G and data not shown). To confirm that the tracheal migration defects observed in btl95 mutants are due to reduced btl function, we expressed wild-type btl specifically in the tracheal cells of btl95 homozygous embryos using the UAS-GAL4 responder system (Brand and Perrimon, 1993). Both the DB and Lta defects were rescued by the ubiquitous tracheal expression of wild-type btl, confirming that the tracheal defects of btl95 mutants are indeed due to loss of btl function (Fig. 5B and Table 4). This analysis suggests that although all tracheal branches require btl function for their migration, the DB and LTa are the most sensitive to reduction of btl activity.
Fig. 3.

breathless and pointed mutations cause defects in dorsal–ventral migration. In wild-type embryos (A), all primary branches migrate normally. In embryos homozygous for Df(3L)fz-Gf3b (B), which deletes btl, or null alleles of btl, btl469 (C) and btlLG19 (D), all primary branches fail to migrate and only elongated sacs of tracheal cells form (arrows). In btl95 mutants (E and F), migration of the DBs (arrows) and the Ltas (arrowheads) is specifically affected. In btlH82Δ11 embryos (G), a few primary branches, such as the GB (arrow), DT (arrowhead), and VB (slightly out of focal plane) initiate but do not complete their migration. btl95 in trans to btlLG19 (H), btlH82Δ11 (I), or Df(3L)fz-Gf3b (J) result in an intermediate tracheal phenotype, where migration of all primary branches is affected to varying degrees. In pnt737 embryos (K and L), migration of the DBs (arrows) and the ventral branches (arrowheads) is affected, whereas in pntΔ88 embryos (M and N), migration of all primary branches is incomplete. pnt737 in trans to pntΔ88 (O) results in an intermediate phenotype. All embryos shown are stage 14 and were stained for Crumbs (A–C, E, J, K, M, and O) or Trachealess (F, H, I, L, and N) or both (D and G) and β-gal.
Fig. 5.

knirps and knirps-related are sufficient for dorsal-branch migration. btl95 mutant embryos (A) show defects in DB (arrows) and Lta migration (arrowheads). Expression of wild-type btl in btl95 embryos (B) rescues the DB (arrows) and Lta (arrowheads) migration defects. Both the DB (arrows) and Lta (arrowheads) fail to migrate in btl95 embryos expressing wild-type tkv (C). In btl95 embryos expressing either activated tkv (D), wild-type knrl (E), or wild-type kni (F), the DBs (arrows) migrate normally, although the Lta (arrowheads) still fail to migrate. All embryos shown are at stage 14 and were stained for Crumbs and β-gal.
Table 4.
Rescue of dorsal migration defects in btl95 mutants
| Genotype | DB2 | DB3 | DB4 | DB5 | DB6 | DB7 | DB8 | DB9 | Lta2 | Lta3 | Lta4 | Lta5 | Lta6 | Lta7 | Lta8 | Lta9 | n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| btl95 | 40 | 60 | 15 | 2 | 0 | 0 | 0 | 2 | 42 | 30 | 8 | 24 | 13 | 4 | 0 | 8 | 45 |
| btl95 btl-GAL4 | 77 | 87 | 42 | 20 | 0 | 22 | 32 | 39 | 81 | 71 | 39 | 48 | 29 | 3 | 16 | 20 | 31 |
| btl95 UAS-btl | 90 | 70 | 40 | 20 | 0 | 30 | 20 | 45 | 20 | 25 | 10 | 20 | 10 | 5 | 10 | 20 | 20 |
| btl95btl-GAL4/btl95UAS-btl | 100 | 100 | 100 | 90 | 70 | 96 | 90 | 80 | 100 | 100 | 96 | 100 | 83 | 100 | 100 | 100 | 30 |
| btl95btl-GAL4/btl95UAS-knrl | 89 | 100 | 100 | 44 | 33 | 83 | 72 | 89 | 33 | 33 | 0 | 17 | 5 | 0 | 17 | 17 | 18 |
| btl95btl-GAl4/btl95UAS-kni | 91 | 85 | 76 | 67 | 24 | 27 | 15 | 36 | 79 | 51 | 12 | 9 | 3 | 0 | 0 | 0 | 33 |
| btl95btl-GAL4/btl95UAS-tkv | 69 | 78 | 28 | 14 | 0 | 7 | 17 | 33 | 76 | 45 | 17 | 40 | 12 | 5 | 0 | 14 | 42 |
| btl95btl-GAL4/btl95UAS-act-tkv | 90 | 100 | 60 | 70 | 40 | 40 | 40 | 70 | 60 | 40 | 10 | 20 | 20 | 10 | 20 | 30 | 10 |
n = number of homozygous embryos scored; numbers = percent of successful migration events within individual metameres.
Sequence analysis of the btl ORF from btl95 genomic DNA revealed two nucleotide changes that result in residue substitutions in the N-terminal extracellular domain. The first residue substitution D50N maps near the N terminus, replacing the negatively charged aspartic acid residue (D) with a neutrally charged asparagine (N). The second residue substitution N181I maps within the second immunoglobulin-like domain, replacing a neutrally charged hydrophilic residue with a hydrophobic residue. Given the nature of the substitutions and their position in the protein, either of the mutations could easily affect ligand binding. We also sequenced one of the null btl alleles identified in the screen (btl469) and discovered a 13-nucleotide deletion in the ORF (nucleotides 2515–2527). This deletion results in a loss of the C-terminal 256 residues, which includes ~70% of the essential intracellular tyrosine kinase domain, consistent with a complete loss of btl function.
In the mutagenesis screen, we also isolated a weak allele of pointed (pnt). pnt encodes an ETS-type transcription factor that was recently shown to function downstream of Btl activation to mediate btl autoregulation (Brunner et al., 1994; Klaes et al., 1994; Klambt, 1993; Ohshiro et al., 2002; O’Neill et al., 1994). Embryos homozygous for pnt737 also had defects in dorsal–ventral tracheal migration (Figs. 3K and L, arrows and left arrowhead). In addition to the DB and Lta defects seen in btl95 embryos, however, the GB was also affected in pnt737 mutants (Fig. 3K, right arrowhead). Previous studies with the pnt null allele, pntΔ88, had suggested roles for pnt in both secondary branching and in maintaining the continuity of the DT (Beitel and Krasnow, 2000; Samakovlis et al., 1996). pnt737 in trans to pntΔ88, which in our hands has severe migration defects in nearly all branches (Figs. 3M and N), results in an intermediate phenotype (Fig. 3O), suggesting that pnt737 is a hypomorphic allele of pointed. Although both btl95 and pnt737 mutations affected DB migration, more DBs per embryo were affected in btl95 mutants than in pnt737 mutants. Thus, in our mutagenesis screen, we generated hypomorphic alleles of btl and pnt, with defects suggesting differential requirements for these genes in the migration of specific tracheal branches.
Migration of tracheal branches in the dorsal–ventral direction requires components of the Dpp-signaling pathway as well as localized expression of the Btl ligand, Bnl (reviewed by Uv et al., 2003). To determine if the DB and Lta fail to migrate in btl95 embryos because of altered expression of bnl or components of the Dpp-signaling pathway, we performed whole mount RNA in situ hybridization experiments with probes for bnl, btl, dpp, thick veins (tkv), knirps (kni), and knirps-related (knrl), as well as antibody staining with αKni (Chen et al., 1998; Vincent et al., 1997). Expression of bnl, btl, dpp, and tkv was not detectably altered in btl95 homozygous embryos (data not shown); expression of kni and knrl, however, was either reduced or absent in the DB cells of btl95 embryos. In the early tracheal placode, Kni protein is normally observed in all tracheal cells; however, by embryonic stage 13, Kni protein is restricted to the DB, VB, Lta, and GB (Chen et al., 1998; Vincent et al., 1997; Fig. 4A). In stage 13 btl95 homozygotes, αKni staining was either completely absent or significantly reduced in the DB precursors of many tracheal metameres (Fig. 4B, arrowheads and arrows). The DBs most affected in their migration in btl95 embryos, specifically those of metameres five to nine (Table 4), were often completely lacking detectable Kni. Kni levels in both the Lta and the GB were only slightly reduced; whereas levels of Kni in the VB were similar to levels in wild type and btl heterozygous embryos. As observed with Kni protein, kni mRNA was either absent or significantly reduced in the DBs of the tracheal metameres most affected in btl95 mutants (Figs. 4C and D; Table 4). kni mRNA accumulation in the visceral and ventral tracheal branches appeared relatively normal (Figs. 4C and D; data not shown).
Fig. 4.

Dorsal expression of kni and knrl is reduced in breathless and pointed mutants. By stage 14, Kni expression in wild-type embryos (A) is restricted to the migrating dorsal branches (arrow), visceral branch (slightly out of focus in this image), and the ventral branches. In btl95 embryos (B), dorsal KNI expression is absent in metameres five to nine (arrows) and reduced in metameres two to four (arrowheads). In pnt737 embryos (C), Kni expression is reduced in the dorsal cells (arrows), whereas in pntΔ88 embryos (D), Kni is present only in dorsal cells of metameres three and four (arrows). In contrast to their heterozygous siblings (E), dorsal kni RNA is either absent (arrows) or reduced (arrowheads) in btl95 homozygous embryos (F). In pnt737 homozygous embryos (H), kni RNA is reduced in the dorsal cells of some tracheal metameres (arrows) compared to the same metameres of their heterozygous siblings (G). knrl RNA was undetectable in dorsal cells of btl95 mutants (J) compared with their heterozygous siblings (I). knrl RNA levels were variably reduced in dorsal cells of pnt737 mutants (L) compared with their heterozygous siblings (K). Dorsal knrl expression was missing in several metameres in pntΔ88 mutants (N, arrow) compared with their heterozygous siblings (M, arrow). All embryos shown are at stage 14. Embryos in A to D were stained for Kni protein and β-gal, whereas embryos in E to N were processed for in situ hybridization to kni (E–H) or knrl (I–N) (blue) and lacZ (pink) RNA.
Similar but less severe changes in kni expression were observed in the pnt737 homozygotes. Kni expression was observed in dorsal cells of all tracheal metameres in stage 13 pnt737 embryos (Fig. 4C, arrows); the number of Kni-positive cells was reduced, however, with most DBs having only one to three Kni-positive cells compared to five to seven Kni-positive cells in WT embryos (Fig. 4A, arrow). In embryos homozygous for the pntΔ88 allele, Kni was detected in the dorsal cells of only a few tracheal metameres, correlating with the more severe defects in dorsal–ventral branch migration observed in embryos mutant for this allele of pnt (Fig. 4D, arrows). Thus, the degree of failure of the DB to migrate in btl95 and pnt mutant embryos correlates with reduced levels of kni expression (Figs. 4A–H).
Since kni and the nearby knirps-related (knrl) gene share spatiotemporal patterns of expression and redundant functions during tracheal development (Chen et al., 1998), we also analyzed knrl expression in btl95 and pnt mutant embryos. knrl mRNA was absent in the DB cells and slightly reduced in the ventral branch cells of btl95 stage 13 embryos (Figs. 4I and J, arrows). knrl expression was also slightly reduced in the DB cells of pnt737 mutants compared to their heterozygous siblings (Figs. 4K and L, arrow). knrl was mostly absent from most dorsal cells of pntΔ88 embryos, again correlating with the more severe defects in dorsal–ventral branch migration observed in these mutants (Figs. 4M and N, arrow). Overall, the expression studies reveal consistently reduced kni and knrl accumulation in the DB tracheal precursors, which are among the cells most affected by the hypomorphic mutations in btl and pnt.
knirps and knirps-related are sufficient to rescue dorsal branch migration
Since both kni and knrl mRNA levels were reduced in the DB cells of btl95 mutants, we asked if expression of either of these genes throughout the trachea using the btl-GAL4 driver could rescue the migration defects of btl95 homozygotes. Indeed, expressing knrl in all tracheal cells rescued DB migration in some segments almost to the same levels as expression of btl itself (Fig. 6F, Table 4). Expression of kni also rescued DB migration, although not as many DBs were rescued as with expression of knrl, perhaps because knrl expression was more severely diminished in the btl95 mutants (Fig. 6F; Table 4). The rescue experiments suggest that it is only the loss of kni and knrl expression that leads to the failure of DB migration in the btl95 mutants. It is possible, however, that reduced btl function results in a failure of the DB precursors to migrate sufficiently dorsally to detect the local source of DPP, which has been shown to be required to work through its receptors to activate kni/knrl expression and DB dorsal migration. The ability of kni and knrl expression to rescue the dorsal migration of DB cells may be a consequence of kni or knrl expression throughout the trachea causing more cells to take on a DB fate (Chen et al., 1998). With more DB cells attempting to migrate dorsally, enough cells could succeed to give an apparent rescue of dorsal DB migration. Indeed, expression of kni or knrl throughout the trachea not only rescues DB migration but also causes defects in DT formation (Figs. 5E and F). The same phenotypes were observed in rescue experiments using an activated form of the DPP receptor (act-TKV), which is known to induce kni and knrl expression and convert DT cells to a DB cell fate (Fig. 5D, Table 4). Thus, although expression of kni and knrl rescues the migration defects in the btl95 mutants, the results are inconclusive with regards to the primary cause of DB migration failure.
Fig. 6.
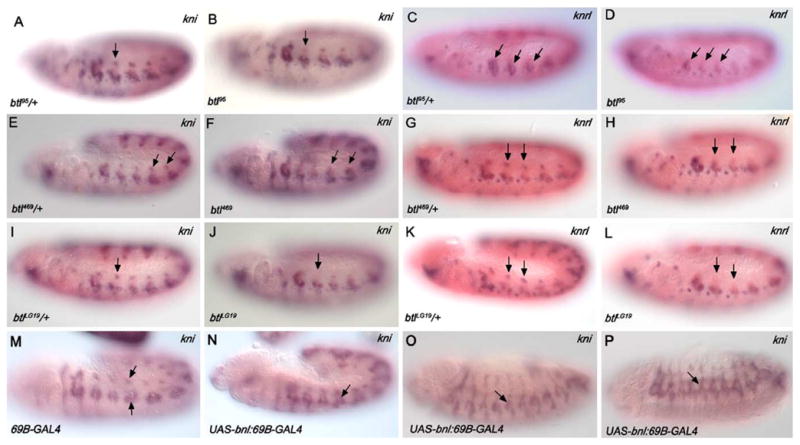
Expression of kni and knrl is altered in breathless mutant embryos and in embryos in which the Btl-signaling pathway is activated ectopically. Dorsal kni expression in the trachea is reduced in btl homozygotes (arrows in B, F, and J) compared with expression in their heterozygous siblings (arrows in A, E, and I). Dorsal knrl expression in the trachea is also reduced in btl homozygotes (arrows in D, H, and L) compared with expression in their heterozygous siblings (arrows in C, G, and K). Embryos carrying the 69B-GAL4 have normal levels of kni expression in the DB and ventral branch precursors (arrows, M), whereas embryos carrying 69B-GAL4 driver and the UAS-bnl constructs have ectopic domains of kni expression in the DT and TC (arrow in N) and regions dorsal to the ventral branches (O and P). Embryos in panels A–N are stage 11 and were processed for RNA in situ hybridization with probes for kni (purple in A, B, E, F, I, J, M–O) or knrl (blue in C, D, G, H, K, and L). Embryos in A–L were also hybridized with probes for trh (pink) and lacZ (pink). Embryos shown in O and P are stage 13.
The Lta migration defect of btl95 mutants was not rescued by tracheal expression of kni, knrl, or activated-TKV (Fig. 6 and Table 4), consistent with our findings that levels of kni and knrl were not significantly affected in ventral branch precursors of the btl95 mutants. Our findings support earlier studies suggesting that knrl and, to a lesser extent, kni are sufficient to promote DB migration but not ventral branch migration (Chen, 1998), which most likely requires additional factors controlled by DPP and BTL signaling.
To test the hypothesis that in the btl mutants DB cells are failing to migrate and, as a consequence, fail to express kni and knrl versus the alternative hypothesis that DB cells fail to express kni and knrl and, as a consequence, fail to migrate, we examined expression of kni and knrl at very early stages, prior to primary branch formation in both btl mutants and wild-type embryos. For these experiments, we did triple RNA in situ hybridizations with either kni, trh, and lacZ probes or knrl, trh, and lacZ probes. trh is expressed in the tracheal system from the time the cells are first specified and throughout embryogenesis and serves as a useful marker for the entire population of tracheal cells (Isaac and Andrew, 1996). lacZ probes were included to distinguish the homozygous mutant animals from their heterozygous siblings, which serve as internal controls for staining levels. Interestingly, we observed reduced levels of kni and knrl expression in dorsal cells of btl mutants compared to their heterozygous siblings as early as embryonic stage 11, prior to the initiation of DB dorsal migration (Fig. 6). kni and knrl expression in this cell population was also diminished during embryonic stages 12 and 13 in the btl mutants relative to their heterozygous siblings. The early loss of dorsal kni and knrl expression was observed in mutants for the weak allele, btl95, and two null alleles, btl469 and btlLG19. Thus, the earliest defect observed in the DB precursors of btl mutants is the loss of kni and knrl expression, not the failure to undergo dorsal migration.
As a further test of a role for Btl-signaling in kni and knrl tracheal expression, we induced Btl signaling in additional tracheal cells by expressing the Bnl ligand at ectopic sites using the 69B-GAL4 driver to express a UAS-bnl transgene (Sutherland et al., 1996). Ectopic kni and knrl expression was observed in stage 11 embryos in the both the DT and TC progenitors, the subset of tracheal branches that normally separate the DB from the ventral branches and that normally do not express kni and knrl at this stage (Figs. 6M and N; data not shown). Similarly, in later stage embryos, ectopic kni expression was observed in regions of the trachea that correspond to the DT and TC. Thus, Btl-signaling is not only necessary for kni/knrl expression, but Btl pathway activation is sufficient to induce kni/knrl expression in additional tracheal cells.
Our studies indicate that Btl and its downstream effector, Pnt, are required to achieve full level transcriptional activation of two genes previously known to be transcriptional targets of Dpp signaling, kni and knrl. This finding is the first identified molecular link between the FGF- and Dpp-signaling pathways, both of which are required for DB migration. Since kni and knrl are initially expressed in the entire tracheal placode and are also required for the migration of most tracheal branches, we asked if simultaneous reduction of signaling from both Btl and Dpp or Btl and kni could affect migration. Surprisingly, simultaneous heterozygosity for btl or pnt and either one of the Dpp-receptor genes punt or kni itself led to significant defects in migration of all branches in approximately 25–30% of the embryos. In embryos heterozygous for null alleles of btlLG19 and punt, most of the primary branches failed to migrate (Fig. 7A). An identical tracheal defect was observed in embryos heterozygous for kni6 and either btlLG19 or btl469, null alleles of btl (Fig. 7B and data ot shown). Similar, albeit somewhat milder migration defects were observed in embryos heterozygous for the null pnt allele, pntΔ88, and kni (Fig. 7D). kni6 in trans to the btl hypomorph btl95 showed no defects in tracheal development (data not shown), whereas kni6 in trans to a btl deficiency led to a range of tracheal defects (Figs. 7E and F). Since simultaneous reduction of the Btl- and Dpp-signaling pathways results in the frequent failure of all the primary branches to migrate, a phenotype more severe than that of punt or kni homozygous mutants alone, interactions between these pathways must occur earlier in the tracheal placode. Since at least one of these mutant combinations results in a significant reduction in kni expression (Figs. 7G and H), these findings further support transcriptional activation of kni and knrl as a link between the FGF- and DPP-signaling pathways.
Fig. 7.

Genetic interactions are observed between breathless, pointed, and components of the Dpp-signaling pathway. Migration of tracheal branches is disrupted in embryos carrying punt10460 in trans to btlLG19 (A) and in trans to pntΔ88 (C). Similarly, embryos carrying kni6 in trans to btlLG19 (B) or pntΔ88 (D) form rudimentary tracheal branches. Embryos carrying kni6 in trans to Df(3L)fz-Gf3b have a range of tracheal phenotypes, with migration of all primary branches being variably affected (E and F). Embryos carrying kni6 in trans to btl95 have normal tracheal branching (not shown). Embryos heterozygous for both btlLG19 and kni6 show a reduction in kni expression in dorsal tracheal cells (G) when compared to kni expression levels in embryos heterozygous for only kni6 (H). Embryos in A, C, and F are at stage 14, whereas those in B, C, and D are at stage 13. Embryos in A and B were stained for Trh, Crb, and β-gal. Embryos in C–F were stained for Trh and β-gal. Embryos in G and H are stage 11 and were processed for in situs with probes for kni (purple), trh (pink), and lacZ (pink). All of the genotypes in this panel were also hybridized with probes to detect bnl expression. In all cases, the levels of bnl in the trans-heterozygotes were equivalent to levels observed in each of the individual heterozygotes, indicating that reduction in levels of Bnl is not contributing to the migration defects.
Discussion
We carried out a chemical mutagenesis screen as a first step toward obtaining a better understanding of the molecular and cellular basis for tracheal tube morphogenesis. From the screen, we identified new functions for genes previously not known to be required for tracheal development, generated additional alleles of genes known to be required and discovered new genes that affect migration of all or some of the tracheal branches. Interestingly, mutations in three of the four new genes also affect salivary gland morphogenesis, suggesting that these genes may have a general role in epithelial tube formation. The mutagenesis screen does not appear to be saturating since we did not obtain new alleles of all third chromosome genes known to affect tracheal development, including trh, drifter, dally, and dally-like. We also did not recover new mutations in kni or knrl, which is not surprising given their redundant functions in the trachea (Chen et al., 1998). In contrast to P-element or X-ray mutagenesis and targeted gene knock-outs, EMS mutagenesis most often creates single nucleotide changes, which can lead to subtler defects that often reveal novel function for genes that have been characterized previously. Our isolation of the new EMS alleles of btl and pnt, which were already known to affect tracheal development, provide examples of the subtler phenotypes that can be observed with EMS mutations. Moreover, further characterization of these alleles revealed a mechanism by which Btl and Dpp-signals are integrated to regulate dorsal branch migration, specifically through transcriptional regulation of kni and knrl (Fig. 8).
Fig. 8.

A model is presented for the regulation of kni and knrl by DPP- and BTL-signaling pathways. Bnl/Btl signaling through RAS/MAPK activates pnt, which leads to positive-autoregulation of btl expression as well as activation of kni and knrl expression. Dpp-signaling through its receptors, Punt and Thick veins, and its intracellular effectors, Mad and Medea, is also required to maintain kni and knrl expression in the dorsal and ventral branches.
btl and pnt play branch-specific roles during tracheal migration
In the tracheal placode, ubiquitous expression of btl is controlled by the transcription factors, Trachealess and Tango (reviewed in Affolter and Shilo, 2000). Early BTL protein is required for the migration of the invaginated tracheal cells to form the initial outgrowths of the six primary branches; in btl null mutants, only rudimentary branches are formed in all metameres (Klambt et al., 1992). Btl also acts through a positive feed-back mechanism to maintain its own expression specifically in the dorsal, ventral, and visceral branches (Ohshiro et al., 2002; Fig. 8). The positive-feedback regulation of btl expression requires the ETS-domain transcription factors, Pnt and Anterior-open (Aop), which act positively and negatively, respectively. In this study, reduction of either btl or pnt activity results in similar dorsal and ventral migration defects, concomitant with a reduction of kni and knrl expression, especially in the dorsal branch precursors. These findings suggest that btl signaling through pnt also activates expression of kni and knrl, in addition to btl autoregulation (Fig. 8). When Btl signaling is compromised, as in btl95 homozygous embryos, overexpression of knrl, kni, or a transgene encoding an activated form of the TKV receptor is sufficient to rescue the DB migration defects. Thus, our studies indicate that DB formation requires induction of kni and knrl expression by both the Btl and Dpp signaling pathways.
Although the DB migration defects in btl95 mutants were rescued by tracheal expression of kni, knrl, and activated Tkv, the Lta migration defects were not. This finding is not surprising since the effects of btl95 on expression of kni and knrl were quite minor in the ventral branches; nonetheless, the failure of kni or knrl to rescue btl95ventral branch phenotypes suggests that additional factors may be required in ventral cells. One molecular difference between dorsal and ventral trachea is early sal expression, which is observed only in dorsal and not ventral cells, later becoming restricted to the DT precursors (Franch-Marro and Casanova, 2002). Loss of sal function appears to convert dorsal tracheal cells to ventral tracheal fates, providing a regulatory basis for differential molecular requirements for directed cell migration of dorsal and ventral cells. The finding that at least one of the ventral branches, Lta, is also quite sensitive to loss of btl suggests that Btl- and Dpp-signaling pathways may cooperate to activate expression of an as yet unidentified factor required for Lta migration. An alternative explanation would be that migration may be rescued by expression of kni or knrl but fusion of the Lta may fail, causing the branches to retract and giving a phenotype that appears to be a failure in migration. Live imaging would be required to distinguish between these possibilities.
BTL signaling and Kni/Knrl functions are required for the migration of most branches
kni and knrl are initially expressed ubiquitously in the early tracheal placode and only become branch-specific just prior to primary branch formation (Vincent et al., 1997; Chen et al., 1998; Fig. 6). Furthermore, in embryos lacking kni and knrl, all branches fail to migrate (Chen et al., 1998), resembling the bnl (Sutherland et al., 1996) and btl null phenotypes (Klambt et al., 1992). These data suggest that Kni/Knrl activity is essential for tracheal cells to migrate. In this study, we showed that 25–30% of embryos trans-heterozygous for null alleles of either btl or pnt, and of components of the dpp pathway, such as punt and kni, exhibit tracheal phenotypes reminiscent of null alleles of btl, bnl, hbr, and kni knrl double mutants, in which only rudimentary branches form. These data provide support for an early role for Dpp-signaling through kni and knrl in tracheal migration and suggest that the Btl and Dpp pathways interact prior to and during primary branch formation.
Although null btl EMS alleles in trans to either kni6 or put10460 formed only rudimentary branches, a deficiency that deletes btl, Df(3L)fz-GF3b, resulted in more variable and often less severe tracheal defects in trans to kni6, where many of the primary branches formed normally (Figs. 7E and F). These data are consistent with the Btl receptor functioning as a dimer, as do all known receptor tyrosine kinases. In the case of a deficiency in trans to a wild-type copy of the btl gene, functional Btl levels would be reduced by 50%. In the case of the EMS alleles, where a nonfunctional Btl protein is produced, functional Btl levels would be less than 50% and perhaps as low as 25% of wild-type levels. Moreover, in light of the Btl positive-feedback autoregulation reported by Ohshiro et al. (2002; Fig. 8), it is possible that in the btl EMS mutants, the amount of functional Btl would be reduced even further. We believe that it is in the background of this very low level of Btl function that a 50% reduction of Dpp-signaling components leads to such severe defects in early tracheal migration. Nonetheless, the findings add support to a model where critical levels of both Btl signaling and KNI/KNRL activities are required for migration. The dorsal branch is particularly sensitive to loss of btl function since there is the additional transcriptional link between Btl signaling and kni/knrl transcriptional activation.
Btl signaling and KNI/KNRL have distinct activities in tracheal migration
At a gross morphological level, loss of btl, bnl, and hbr have very similar tracheal migration defects to the loss of both kni and knrl. However, it has been shown that although Btl signaling is essential for the formation of filopodia at the tips of the growing tracheal buds, loss of dpp function does not affect filopodial formation (Ribeiro et al., 2002). In embryos where the dpp pathway is specifically inhibited through ectopic expression of Daughters against dpp (DAD), an inhibitory SMAD protein, dorsal cells form short bud-like structures, but do not form single-celled tubes and are eventually reintegrated into the DT (Ribeiro et al., 2002). Therefore, while Btl signaling mediates the first cellular response to the chemoattractant Bnl by producing filopodia in random directions, Dpp-signaling-dependent events may provide cells with the ability to coordinate their migration and subsequent tube elongation specifically in the dorsal (and ventral) direction. We would like to propose that much, if not all, of the directional response provided by DPP signaling is through transcriptional activation of Kni and Knrl.
Although kni and knrl are expressed uniformly in the early tracheal placode, at later stages, they are specifically excluded from the DT and transverse connectives (TC), the two tubes that migrate the least distance. One reason for kni/knrl exclusion from these specific branches may be that early kni/knrl expression is sufficient for the limited migration that this group of cells normally undergoes. Only in those branches that migrate greater distances is kni/knrl expression normally maintained. The distance the cells migrate correlates nicely with the size of the tube that forms. A morphometric analysis of tracheal tube sizes revealed that the DT and the TC tubes have the greatest diameter, whereas the DB and LTa tubes, which form from the cells that express the highest levels of kni and knrl, have the smallest diameter (Beitel and Krasnow, 2000). Furthermore, ubiquitous tracheal expression of either dpp or kni increases the number of cells that form the DB while reducing DT formation, the latter being accompanied by repression of spalt (sal; Chen et al., 1998; Kuhnlein and Schuh, 1996; Llimargas and Casanova, 1997; Wappner et al., 1997). In contrast, ubiquitous tracheal expression of sal disrupts DB migration and causes only rudimentary branches to form (Chen et al., 1998). Interestingly, these rudimentary dorsal tubes have wider lumena than normal. Thus, there is a direct link between the extent to which the cells migrate and the relative size of the tubes that form, with wide-bored DT tubes migrating shorter distances than fine-bored DB tubes. Thus, kni and knrl in response to Dpp- and Btl-signaling activates a genetic program that specifies the extent of migration and consequently the size of tube that is to form. In a recent paper, Ribeiro et al. (2004) uncover a cellular basis for the differences in kni/knrl expressing branches and those that express sal. sal-expressing branches form multicellular tubes with intercellular adherens junctions (AJs), whereas branches that do not express sal because of repression by Kni/Knrl, form unicellular tubes with auto-cellular AJs, consistent with the differences in bore size of the respective tubes. They show that sal expression prevents the formation of autocellullar AJs and the corresponding cell rearrangements required for DB cells to intercalate (i.e., slide past one another) for branch elongation. In support of a role for Sal in preventing formation of single cell tubes, the authors demonstrate that DB migration in the absence of DPP-signaling can be rescued if sal expression is also removed from the DB precursors. Overall, these findings suggest that the roles of the different signaling pathways that impinge on tracheal development play more of a role in controlling morphogenetic events than in specifying cell fates within the tracheal network.
Acknowledgments
We thank the Bloomington Stock Center, the Developmental Biology Hybridoma Bank, and our many generous colleagues for fly stocks, cDNAs, and antisera, including M. Affolter, S. Hayash, M. Krasnow, and R. Schuh for fly lines, D. Kosman for antisera, and P. Beachy, W. Gelbart, S. Hayashi, and D. Montell for cDNAs. We thank the following people who helped with the mutagenesis screen: B. Kerman, M. Strohminger, J. Chou, R. Lamour, J. Lee, C. Marcinkewiez, M. Schaffer, and especially, in fond memory, Xueqin Xia. We thank past and present members of the Andrew lab for their advice and support in the course of this study. We thank P. Bradley, A. Cheshire, M. Vining, and two anonymous reviewers for their critical comments on the manuscript. This work was supported by an NIH grant (NIH RO1 DE12873) to D.J.A., the Scholar Development and Faculty Transition Award (K22) from the NIDCR to M.M.M. H.L. was supported in part by a fellowship from Hopkins-PREP (Post-baccalaureate Research Education Program, NIH R25 GM 64124).
References
- Affolter M, Shilo BZ. Genetic control of branching morphogenesis during Drosophila tracheal development. Curr Opin Cell Biol. 2000;12:731–735. doi: 10.1016/s0955-0674(00)00160-5. [DOI] [PubMed] [Google Scholar]
- Affolter M, Nellen D, Nussbaumer U, Basler K. Multiple requirements for the receptor serine/threonine kinase Thick veins reveal novel functions of TGFb homologs during Drosophila embryogenesis. Development. 1994;120:3105–3117. doi: 10.1242/dev.120.11.3105. [DOI] [PubMed] [Google Scholar]
- Beitel GJ, Krasnow MA. Genetic control of epithelial tube size in the Drosophila tracheal system. Development. 2000;127:3271–3282. doi: 10.1242/dev.127.15.3271. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:469–515. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brunner D, Ducker KNO, Hafen E, Scholz H, Klambt C. The ETS domain protein Pointed-P2 is a target of MAP kinase in the Sevenless signal transduction pathway. Nature. 1994;370:386–389. doi: 10.1038/370386a0. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. The Embryonic Development of Drosophila melanogaster. Springer-Verlag; Berlin Heidelberg: 1997. [Google Scholar]
- Chen CK, Kuhnlein RP, Eulenberg KG, Vincent S, Affolter M, Schuh R. The transcription factors KNIRPS and KNIRPS RELATED control cell migration and branch morphogenesis during Drosophila tracheal development. Development. 1998;125:4959–4968. doi: 10.1242/dev.125.24.4959. [DOI] [PubMed] [Google Scholar]
- Franch-Marro X, Casanova J. spalt-induced specification of distinct dorsal and ventral domains is required for Drosophila tracheal patterning. Dev Biol. 2002;250:374–382. [PubMed] [Google Scholar]
- Grigliatti T. Mutagenesis. In: Roberts DB, editor. Drosophila: A Practical Approach. IRL Press; Oxford, England: 1986. pp. 39–58. [Google Scholar]
- Imam F, Sutherland D, Huang W, Krasnow MA. stumps, a Drosophila gene required for fibroblast growth factor (FGF)-directed migrations of tracheal and mesodermal cells. Genetics. 1999;152:307–318. doi: 10.1093/genetics/152.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac DD, Andrew DJ. Tubulogenesis in Drosophila: a requirement for trachealess gene product. Genes Dev. 1996;10:103–117. doi: 10.1101/gad.10.1.103. [DOI] [PubMed] [Google Scholar]
- Klaes A, Menne T, Stollewerk A, Scholz H, Klambt C. The Ets transcription factors encoded by the Drosophila gene pointed direct glial cell differentiation in the embryonic CNS. Cell. 1994;78:149–160. doi: 10.1016/0092-8674(94)90581-9. [DOI] [PubMed] [Google Scholar]
- Klambt C. The Drosophila gene pointed encodes two ETS-like proteins, which are involved in the development of the midline glial cells. Development. 1993;117:163–176. doi: 10.1242/dev.117.1.163. [DOI] [PubMed] [Google Scholar]
- Klambt C, Glazer L, Shilo BZ. Breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev. 1992;6:1668–1678. doi: 10.1101/gad.6.9.1668. [DOI] [PubMed] [Google Scholar]
- Kuhnlein RP, Schuh R. Dual function of the region-specific homeotic gene spalt during Drosophila tracheal system development. Development. 1996;122:2215–2223. doi: 10.1242/dev.122.7.2215. [DOI] [PubMed] [Google Scholar]
- Lehmann R, Tautz D. In situ hybridization to RNA. In: Goldstein LSB, Fyrberg EA, editors. Drosophila Melanogaster: Practical Uses in Cell and Molecular Biology. Academic Press; San Diego, CA: 1994. p. 755. [Google Scholar]
- Llimargas M, Casanova J. Ventral veinless, a POU domain transcription factor, regulates different transduction pathways required for tracheal branching in Drosophila. Development. 1997;124:3273–3281. doi: 10.1242/dev.124.17.3273. [DOI] [PubMed] [Google Scholar]
- Manning G, Krasnow M. Development of the Drosophila tracheal system. In: Martinez A, Bate M, editors. The Development of Drosophila Melanogaster. I. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1993. pp. 609–685. [Google Scholar]
- Michelson AM, Gisselbrecht S, Buff E, Skeath JB. Heartbroken is a specific downstream mediator of FGF receptor signalling in Drosophila. Development. 1998;125:4379–4389. doi: 10.1242/dev.125.22.4379. [DOI] [PubMed] [Google Scholar]
- Myat MM, Andrew DJ. Epithelial tube morphology is determined by the polarized growth and delivery of apical membrane. Cell. 2002;111:879–891. doi: 10.1016/s0092-8674(02)01140-6. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Emori Y, Saigo K. Ligand-dependent activation of breathless FGF receptor gene in Drosophila developing trachea. Mech Dev. 2002;114:3–11. doi: 10.1016/s0925-4773(02)00042-4. [DOI] [PubMed] [Google Scholar]
- O’Neill EM, Rebay I, Tijan R, Rubin GM. The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell. 1994;78:137–147. doi: 10.1016/0092-8674(94)90580-0. [DOI] [PubMed] [Google Scholar]
- Petit V, Nussbaumer U, Dossenbach C, Affolter M. Downstream-of-FGFR is a fibroblast growth factor-specific scaffolding protein and recruits Corkscrew upon receptor activation. Mol Cell Biol. 2004;9:3769–3781. doi: 10.1128/MCB.24.9.3769-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter R, Panganiban GE, Hoffmann FM, Scott MP. Homeotic genes regulate the spatial expression of putative growth factors in the visceral mesoderm of Drosophila embryos. Development. 1990;110:1031–1040. doi: 10.1242/dev.110.4.1031. [DOI] [PubMed] [Google Scholar]
- Ribeiro C, Ebner A, Affolter M. In vivo imaging reveals different cellular functions for FGF and DPP signaling in tracheal branching morphogenesis. Dev Cell. 2002;2:677–683. doi: 10.1016/s1534-5807(02)00171-5. [DOI] [PubMed] [Google Scholar]
- Ribeiro C, Neumann M, Affolter M. Genetic control of cell intercalation during tracheal morphogenesis in Drosophila. Curr Biol. 2004;14:2197–2207. doi: 10.1016/j.cub.2004.11.056. [DOI] [PubMed] [Google Scholar]
- Ruberte E, Nellen MT, Affolter M, Basler K. An absolute requirement for both the type II and type I receptors, Punt and Thick veins, for Dpp signaling in vivo. Cell. 1995;80:889–897. doi: 10.1016/0092-8674(95)90292-9. [DOI] [PubMed] [Google Scholar]
- Samakovlis C, Hacochen N, Manning G, Sutherland D, Guillemin K, Krasnow MA. Development of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development. 1996;122:1395–1407. doi: 10.1242/dev.122.5.1395. [DOI] [PubMed] [Google Scholar]
- Sutherland D, Samakovlis C, Krasnow MA. branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell. 1996;87:1091–1101. doi: 10.1016/s0092-8674(00)81803-6. [DOI] [PubMed] [Google Scholar]
- Tepass U, Theres C, Knust E. crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell. 1990;61:787–799. doi: 10.1016/0092-8674(90)90189-l. [DOI] [PubMed] [Google Scholar]
- Uv A, Canterra R, Samakovlis C. Drosophila tracheal morphogenesis: intricate cellular solutions to basic plumbing problems. Trends Cell Biol. 2003;13:301–309. doi: 10.1016/s0962-8924(03)00083-7. [DOI] [PubMed] [Google Scholar]
- Vincent S, Ruberte E, Grieder NC, Chen CK, Haerry T, Schuh R, Affolter M. DPP controls cell migration along the dorsoventral body axis of the Drosophila embryo. Development. 1997;124:2741–2750. doi: 10.1242/dev.124.14.2741. [DOI] [PubMed] [Google Scholar]
- Vincent S, Wilson R, Coelho C, Affolter M, Leptin M. The Drosophila protein Dof is specifically required for FGF signaling. Mol Cell. 1998;2:515–525. doi: 10.1016/s1097-2765(00)80151-3. [DOI] [PubMed] [Google Scholar]
- Wappner P, Gabay L, Shilo BZ. Interactions between the EGF receptor and DPP pathways establish distinct cell fates in the tracheal placodes. Development. 1997;124:4704–4716. doi: 10.1242/dev.124.22.4707. [DOI] [PubMed] [Google Scholar]
- Wolf C, Gerlach N, Schuh R. Drosophila tracheal system formation involves FGF-dependent cell extensions contacting bridge-cells. EMBO Rep. 2002;3:563–568. doi: 10.1093/embo-reports/kvf115. [DOI] [PMC free article] [PubMed] [Google Scholar]